
Is Autophagy Always a Barrier to Cisplatin Therapy?



Abstract
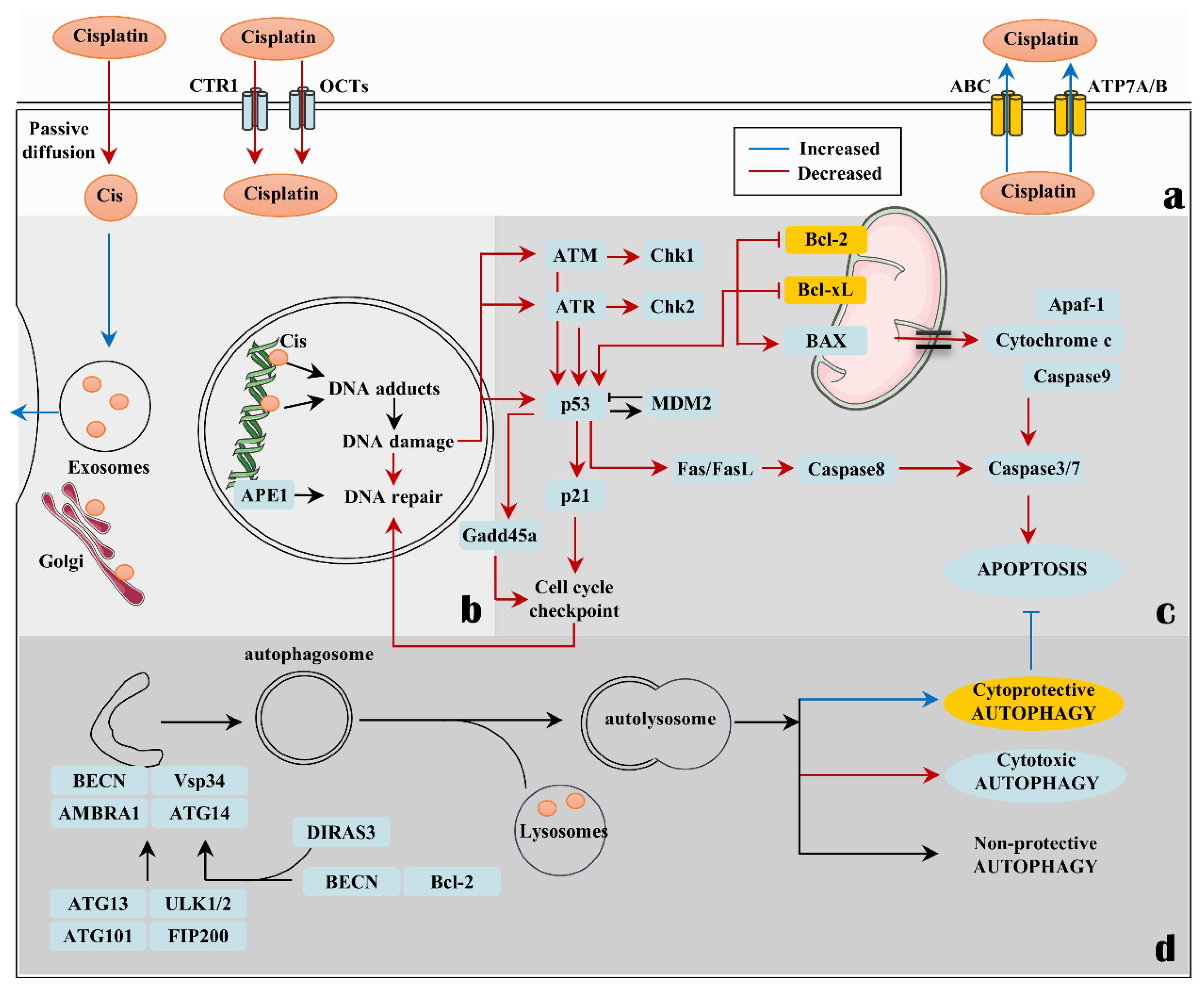
1. Introduction
2. Cisplatin-Induced Autophagy
2.1. Cisplatin Resistance
2.2. Factors That Affect the Role of Autophagy in Cisplatin-Sensitive Cells
2.3. Factors That Affect the Role of Autophagy in Cisplatin-Resistant Cells
2.4. The Non-Coding RNA That Affects the Role of Autophagy in Cisplatin-Treated Cells
2.5. Other Genes That Affect the Role of Autophagy in Cisplatin-Treated Tumor Cells
3. The Yin and Yang Faces of Autophagy Inhibition in Cisplatin Therapy
3.1. A Beneficial Treatment Strategy of Autophagy Inhibition Combined with Cisplatin Is Closely Related to Tumor Types
3.2. Does Autophagy Inhibition Have the Potential to Exacerbate the Toxicity of Cisplatin to Normal Tissue?
4. Autophagy in Cisplatin Combination with Immunotherapy
5. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
References
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef]
- Chen, S.H.; Chang, J.Y. New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment. Int. J. Mol. Sci. 2019, 20, 4136. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, B.; Van Camp, L.; Grimley, E.B.; Thomson, A.J. The inhibition of growth or cell division in Escherichia coli by different ionic species of platinum(IV) complexes. J. Biol. Chem. 1967, 242, 1347–1352. [Google Scholar] [CrossRef]
- Rosenberg, B.; VanCamp, L.; Trosko, J.E.; Mansour, V.H. Platinum compounds: A new class of potent antitumour agents. Nature 1969, 222, 385–386. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.M.; Loeb, E.; MacLellan, A.; Hill, N.O.; Khan, A.; King, J.J. Clinical studies of Platinum Coordination compounds in the treatment of various malignant diseases. Cancer Chemother. Rep. 1975, 59, 647–659. [Google Scholar] [PubMed]
- Santos, N.; Ferreira, R.S.; Santos, A.C.D. Overview of cisplatin-induced neurotoxicity and ototoxicity, and the protective agents. Food Chem. Toxicol. 2020, 136, 111079. [Google Scholar] [CrossRef]
- Johnson, S.W.; Laub, P.B.; Beesley, J.S.; Ozols, R.F.; Hamilton, T.C. Increased platinum-DNA damage tolerance is associated with cisplatin resistance and cross-resistance to various chemotherapeutic agents in unrelated human ovarian cancer cell lines. Cancer Res. 1997, 57, 850–856. [Google Scholar] [PubMed]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- Kartalou, M.; Essigmann, J.M. Recognition of cisplatin adducts by cellular proteins. Mutat. Res. 2001, 478, 1–21. [Google Scholar] [CrossRef]
- Danford, A.J.; Wang, D.; Wang, Q.; Tullius, T.D.; Lippard, S.J. Platinum anticancer drug damage enforces a particular rotational setting of DNA in nucleosomes. Proc. Natl. Acad. Sci. USA 2005, 102, 12311–12316. [Google Scholar] [CrossRef]
- Jung, Y.; Lippard, S.J. Direct cellular responses to platinum-induced DNA damage. Chem. Rev. 2007, 107, 1387–1407. [Google Scholar] [CrossRef]
- Wynne, P.; Newton, C.; Ledermann, J.A.; Olaitan, A.; Mould, T.A.; Hartley, J.A. Enhanced repair of DNA interstrand crosslinking in ovarian cancer cells from patients following treatment with platinum-based chemotherapy. Br. J. Cancer 2007, 97, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Makovec, T. Cisplatin and beyond: Molecular mechanisms of action and drug resistance development in cancer chemotherapy. Radiol. Oncol. 2019, 53, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Boulikas, T.; Vougiouka, M. Cisplatin and platinum drugs at the molecular level. (Review). Oncol. Rep. 2003, 10, 1663–1682. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Fuertes, M.A.; Castilla, J.; Alonso, C.; Perez, J.M. Cisplatin biochemical mechanism of action: From cytotoxicity to induction of cell death through interconnections between apoptotic and necrotic pathways. Curr. Med. Chem. 2003, 10, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Tanabe, K.; Uchida, Y.; Emi, M.; Inoue, H.; Toge, T. Current status of the molecular mechanisms of anticancer drug-induced apoptosis. The contribution of molecular-level analysis to cancer chemotherapy. Cancer Chemother. Pharm. 2002, 50, 343–352. [Google Scholar] [CrossRef]
- Bai, L.; Wang, S. Targeting apoptosis pathways for new cancer therapeutics. Annu. Rev. Med. 2014, 65, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Tchounwou, P.B.; Dasari, S.; Noubissi, F.K.; Ray, P.; Kumar, S. Advances in Our Understanding of the Molecular Mechanisms of Action of Cisplatin in Cancer Therapy. J. Exp. Pharmacol. 2021, 13, 303–328. [Google Scholar] [CrossRef] [PubMed]
- Koberle, B.; Grimaldi, K.A.; Sunters, A.; Hartley, J.A.; Kelland, L.R.; Masters, J.R. DNA repair capacity and cisplatin sensitivity of human testis tumour cells. Int. J. Cancer. J. Int. Du Cancer 1997, 70, 551–555. [Google Scholar] [CrossRef]
- Basu, A.; Krishnamurthy, S. Cellular responses to Cisplatin-induced DNA damage. J. Nucleic Acids 2010, 2010, 201367. [Google Scholar] [CrossRef]
- Zhou, J.; Kang, Y.; Chen, L.; Wang, H.; Liu, J.; Zeng, S.; Yu, L. The Drug-Resistance Mechanisms of Five Platinum-Based Antitumor Agents. Front. Pharmacol. 2020, 11, 343. [Google Scholar] [CrossRef] [PubMed]
- DeHaan, R.D.; Yazlovitskaya, E.M.; Persons, D.L. Regulation of p53 target gene expression by cisplatin-induced extracellular signal-regulated kinase. Cancer Chemother. Pharm. 2001, 48, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Tu, H. Activation of ERK during DNA damage-induced apoptosis involves protein kinase Cdelta. Biochem. Biophys. Res. Commun. 2005, 334, 1068–1073. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.J.; Wang, J.; Zhou, J.Y.; Wu, G.S. Role of the Akt/mTOR survival pathway in cisplatin resistance in ovarian cancer cells. Biochem. Biophys. Res. Commun. 2010, 394, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Gasiorkiewicz, B.M.; Koczurkiewicz-Adamczyk, P.; Piska, K.; Pekala, E. Autophagy modulating agents as chemosensitizers for cisplatin therapy in cancer. Investig. New Drugs 2021, 39, 538–563. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Gu, C.; Zhong, D.; Shi, L.; Kong, Y.; Zhou, Z.; Liu, S. Induction of autophagy counteracts the anticancer effect of cisplatin in human esophageal cancer cells with acquired drug resistance. Cancer Lett. 2014, 355, 34–45. [Google Scholar] [CrossRef]
- Gao, J.; Wang, W. Knockdown of galectin-1 facilitated cisplatin sensitivity by inhibiting autophagy in neuroblastoma cells. Chem.-Biol. Interact. 2019, 297, 50–56. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, G.; Jiang, J. Profiling of apoptosis- and autophagy-associated molecules in human lung cancer A549 cells in response to cisplatin treatment using stable isotope labeling with amino acids in cell culture. Int. J. Oncol. 2019, 54, 1071–1085. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Saleh, T.; Bloukh, S.; Carpenter, V.J.; Alwohoush, E.; Bakeer, J.; Darwish, S.; Azab, B.; Gewirtz, D.A. Therapy-Induced Senescence: An “Old” Friend Becomes the Enemy. Cancers 2020, 12, 822. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, K.; Fujiwara, Y.; Nishio, K.; Ohmori, T.; Sugimoto, Y.; Komiya, K.; Matsuda, T.; Saijo, N. Metallothionein content correlates with the sensitivity of human small cell lung cancer cell lines to cisplatin. Cancer Res. 1991, 51, 3237–3242. [Google Scholar] [PubMed]
- Safaei, R.; Katano, K.; Larson, B.J.; Samimi, G.; Holzer, A.K.; Naerdemann, W.; Tomioka, M.; Goodman, M.; Howell, S.B. Intracellular localization and trafficking of fluorescein-labeled cisplatin in human ovarian carcinoma cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 756–767. [Google Scholar]
- Sancho-Martinez, S.M.; Prieto-Garcia, L.; Prieto, M.; Lopez-Novoa, J.M.; Lopez-Hernandez, F.J. Subcellular targets of cisplatin cytotoxicity: An integrated view. Pharmacol. Ther. 2012, 136, 35–55. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.S.; Liang, X.J.; Su, A.W.; Pai-Panandiker, A.; Shen, D.W.; Hanover, J.A.; Gottesman, M.M. Reduced endocytosis and altered lysosome function in cisplatin-resistant cell lines. Br. J. Cancer 2003, 88, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Safaei, R.; Howell, S.B. Copper transporters regulate the cellular pharmacology and sensitivity to Pt drugs. Crit. Rev. Oncol./Hematol. 2005, 53, 13–23. [Google Scholar] [CrossRef]
- Liang, X.J.; Shen, D.W.; Garfield, S.; Gottesman, M.M. Mislocalization of membrane proteins associated with multidrug resistance in cisplatin-resistant cancer cell lines. Cancer Res. 2003, 63, 5909–5916. [Google Scholar] [PubMed]
- Petruzzelli, R.; Polishchuk, R.S. Activity and Trafficking of Copper-Transporting ATPases in Tumor Development and Defense against Platinum-Based Drugs. Cells 2019, 8, 1080. [Google Scholar] [CrossRef] [PubMed]
- Vaisman, A.; Varchenko, M.; Umar, A.; Kunkel, T.A.; Risinger, J.I.; Barrett, J.C.; Hamilton, T.C.; Chaney, S.G. The role of hMLH1, hMSH3, and hMSH6 defects in cisplatin and oxaliplatin resistance: Correlation with replicative bypass of platinum-DNA adducts. Cancer Res. 1998, 58, 3579–3585. [Google Scholar]
- Lowe, S.W.; Ruley, H.E.; Jacks, T.; Housman, D.E. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell 1993, 74, 957–967. [Google Scholar] [CrossRef]
- Bragado, P.; Armesilla, A.; Silva, A.; Porras, A. Apoptosis by cisplatin requires p53 mediated p38alpha MAPK activation through ROS generation. Apoptosis 2007, 12, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Crook, T.; Parker, G.A.; Rozycka, M.; Crossland, S.; Allday, M.J. A transforming p53 mutant, which binds DNA, transactivates and induces apoptosis reveals a nuclear:cytoplasmic shuttling defect. Oncogene 1998, 16, 1429–1441. [Google Scholar] [CrossRef]
- Soussi, T. The p53 tumor suppressor gene: From molecular biology to clinical investigation. Ann. N. Y. Acad. Sci. 2000, 910, 121–137; discussion 137–139. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Kawamata, H.; Fujimori, T.; Omotehara, F.; Horiuchi, H.; Ohkura, Y.; Igarashi, S.; Kotake, K.; Kubota, K. Dysfunction of p53 pathway in human colorectal cancer: Analysis of p53 gene mutation and the expression of the p53-associated factors p14ARF, p33ING1, p21WAF1 and MDM2. Int. J. Oncol. 2004, 25, 913–920. [Google Scholar] [PubMed]
- Jahnson, S.; Karlsson, M.G. Tumor mapping of regional immunostaining for p21, p53, and mdm2 in locally advanced bladder carcinoma. Cancer 2000, 89, 619–629. [Google Scholar] [CrossRef]
- Gewirtz, D.A. The four faces of autophagy: Implications for cancer therapy. Cancer Res. 2014, 74, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhu, Y.; Han, S.; Chen, M.; Song, P.; Dai, D.; Xu, W.; Jiang, T.; Feng, L.; Shin, V.Y.; et al. Impaired autophagic degradation of lncRNA ARHGAP5-AS1 promotes chemoresistance in gastric cancer. Cell Death Dis. 2019, 10, 383. [Google Scholar] [CrossRef]
- Patel, N.H.; Xu, J.; Saleh, T.; Wu, Y.; Lima, S.; Gewirtz, D.A. Influence of nonprotective autophagy and the autophagic switch on sensitivity to cisplatin in non-small cell lung cancer cells. Biochem Pharm. 2020, 175, 113896. [Google Scholar] [CrossRef] [PubMed]
- Gewirtz, D.A. An autophagic switch in the response of tumor cells to radiation and chemotherapy. Biochem Pharm. 2014, 90, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Chakradeo, S.; Sharma, K.; Alhaddad, A.; Bakhshwin, D.; Le, N.; Harada, H.; Nakajima, W.; Yeudall, W.A.; Torti, S.V.; Torti, F.M.; et al. Yet another function of p53--the switch that determines whether radiation-induced autophagy will be cytoprotective or nonprotective: Implications for autophagy inhibition as a therapeutic strategy. Mol. Pharmacol. 2015, 87, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.N.; Bristol, M.L.; Di, X.; Maltese, W.A.; Koterba, K.; Beckman, M.J.; Gewirtz, D.A. A switch between cytoprotective and cytotoxic autophagy in the radiosensitization of breast tumor cells by chloroquine and vitamin D. Horm. Cancer 2011, 2, 272–285. [Google Scholar] [CrossRef]
- Shen, P.; Chen, M.; He, M.; Chen, L.; Song, Y.; Xiao, P.; Wan, X.; Dai, F.; Pan, T.; Wang, Q. Inhibition of ERalpha/ERK/P62 cascades induces “autophagic switch” in the estrogen receptor-positive breast cancer cells exposed to gemcitabine. Oncotarget 2016, 7, 48501–48516. [Google Scholar] [CrossRef]
- Reinhardt, H.C.; Aslanian, A.S.; Lees, J.A.; Yaffe, M.B. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 2007, 11, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, J.; Liang, G.; Geng, G.; Zhao, F.; Yin, P.; Nowsheen, S.; Wu, C.; Li, Y.; Li, L.; et al. CHK2-FOXK axis promotes transcriptional control of autophagy programs. Sci. Adv. 2020, 6, eaax5819. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.R.; Rocha, C.R.R.; Martins, D.J.; Fiore, A.; Kinker, G.S.; Bruni-Cardoso, A.; Menck, C.F.M. ATR mediates cisplatin resistance in 3D-cultured breast cancer cells via translesion DNA synthesis modulation. Cell Death Dis. 2019, 10, 459. [Google Scholar] [CrossRef] [PubMed]
- Cianfanelli, V.; Fuoco, C.; Lorente, M.; Salazar, M.; Quondamatteo, F.; Gherardini, P.F.; De Zio, D.; Nazio, F.; Antonioli, M.; D’Orazio, M.; et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat. Cell Biol. 2015, 17, 20–30. [Google Scholar] [CrossRef]
- Antonioli, M.; Di Rienzo, M.; Piacentini, M.; Fimia, G.M. Emerging Mechanisms in Initiating and Terminating Autophagy. Trends Biochem. Sci. 2017, 42, 28–41. [Google Scholar] [CrossRef]
- Liu, J.; Chen, Z.; Guo, J.; Wang, L.; Liu, X. Ambra1 induces autophagy and desensitizes human prostate cancer cells to cisplatin. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, L.; Yu, L.; Wei, W.; Lin, X.; Hou, X.; Tian, Y. shRNA-mediated AMBRA1 knockdown reduces the cisplatin-induced autophagy and sensitizes ovarian cancer cells to cisplatin. J. Toxicol. Sci. 2016, 41, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, M.; Pagni, B.; Vescovo, T.; Ellis, R.; Cosway, B.; Rollo, F.; Bordoni, V.; Agrati, C.; Labus, M.; Covello, R.; et al. HPV sensitizes OPSCC cells to cisplatin-induced apoptosis by inhibiting autophagy through E7-mediated degradation of AMBRA1. Autophagy 2021, 17, 2842–2855. [Google Scholar] [CrossRef] [PubMed]
- Shih, T.C.; Fan, Y.; Kiss, S.; Li, X.; Deng, X.N.; Liu, R.; Chen, X.J.; Carney, R.; Chen, A.; Ghosh, P.M.; et al. Galectin-1 inhibition induces cell apoptosis through dual suppression of CXCR4 and Ras pathways in human malignant peripheral nerve sheath tumors. Neuro-oncology 2019, 21, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Seyrek, K.; Richter, M.; Lavrik, I.N. Decoding the sweet regulation of apoptosis: The role of glycosylation and galectins in apoptotic signaling pathways. Cell Death Differ. 2019, 26, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Vladoiu, M.C.; Labrie, M.; St-Pierre, Y. Intracellular galectins in cancer cells: Potential new targets for therapy (Review). Int. J. Oncol. 2014, 44, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.Y.; Tang, S.J.; Sun, G.H.; Chou, T.Y.; Yeh, T.S.; Yu, S.L.; Sun, K.H. Galectin-1 promotes lung cancer progression and chemoresistance by upregulating p38 MAPK, ERK, and cyclooxygenase-2. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 4037–4047. [Google Scholar] [CrossRef]
- Zhu, H.; Chen, A.; Li, S.; Tao, X.; Sheng, B.; Chetry, M.; Zhu, X. Predictive role of galectin-1 and integrin alpha5beta1 in cisplatin-based neoadjuvant chemotherapy of bulky squamous cervical cancer. Biosci. Rep. 2017, 37, BSR20170958. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhang, P.; Shi, B.; Zhou, M.; Jiang, H.; Zhang, H.; Pan, X.; Gao, H.; Sun, H.; Li, Z. Galectin-1 overexpression promotes progression and chemoresistance to cisplatin in epithelial ovarian cancer. Cell Death Dis. 2014, 5, e991. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.C.; Davuluri, G.V.; Chen, C.H.; Shiau, D.C.; Chen, C.C.; Chen, C.L.; Lin, Y.S.; Chang, C.P. Galectin-1-Induced Autophagy Facilitates Cisplatin Resistance of Hepatocellular Carcinoma. PLoS ONE 2016, 11, e0148408. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Marquez, R.T.; Lu, Z.; Liu, J.; Lu, K.H.; Issa, J.P.; Fishman, D.M.; Yu, Y.; Bast, R.C., Jr. Imprinted tumor suppressor genes ARHI and PEG3 are the most frequently down-regulated in human ovarian cancers by loss of heterozygosity and promoter methylation. Cancer 2008, 112, 1489–1502. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Chang, I.S.; Lin, W.; Ye, W.; Luo, R.Z.; Lu, Z.; Lu, Y.; Zhang, K.; Liao, W.S.; Tao, T.; et al. ARHI (DIRAS3), an imprinted tumour suppressor gene, binds to importins and blocks nuclear import of cargo proteins. Biosci. Rep. 2009, 30, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.Z.; Fang, X.; Marquez, R.; Liu, S.Y.; Mills, G.B.; Liao, W.S.; Yu, Y.; Bast, R.C. ARHI is a Ras-related small G-protein with a novel N-terminal extension that inhibits growth of ovarian and breast cancers. Oncogene 2003, 22, 2897–2909. [Google Scholar] [CrossRef] [PubMed]
- Badgwell, D.B.; Lu, Z.; Le, K.; Gao, F.; Yang, M.; Suh, G.K.; Bao, J.J.; Das, P.; Andreeff, M.; Chen, W.; et al. The tumor-suppressor gene ARHI (DIRAS3) suppresses ovarian cancer cell migration through inhibition of the Stat3 and FAK/Rho signaling pathways. Oncogene 2012, 31, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Yang, H.; Sutton, M.N.; Yang, M.; Clarke, C.H.; Liao, W.S.; Bast, R.C., Jr. ARHI (DIRAS3) induces autophagy in ovarian cancer cells by downregulating the epidermal growth factor receptor, inhibiting PI3K and Ras/MAP signaling and activating the FOXo3a-mediated induction of Rab7. Cell Death Differ. 2014, 21, 1275–1289. [Google Scholar] [CrossRef]
- Lu, Z.; Luo, R.Z.; Lu, Y.; Zhang, X.; Yu, Q.; Khare, S.; Kondo, S.; Kondo, Y.; Yu, Y.; Mills, G.B.; et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J. Clin. Investig. 2008, 118, 3917–3929. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cui, G.; Sun, L.; Wang, S.J.; Tian, S.; Guan, Z.; Fan, W.S.; Yan, Z.F.; Yang, Y.Z.; You, Y.Q.; et al. ARHI overexpression induces epithelial ovarian cancer cell apoptosis and excessive autophagy. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2014, 24, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Washington, M.N.; Suh, G.; Orozco, A.F.; Sutton, M.N.; Yang, H.; Wang, Y.; Mao, W.; Millward, S.; Ornelas, A.; Atkinson, N.; et al. ARHI (DIRAS3)-mediated autophagy-associated cell death enhances chemosensitivity to cisplatin in ovarian cancer cell lines and xenografts. Cell Death Dis. 2015, 6, e1836. [Google Scholar] [CrossRef] [PubMed]
- Gotze, S.; Feldhaus, V.; Traska, T.; Wolter, M.; Reifenberger, G.; Tannapfel, A.; Kuhnen, C.; Martin, D.; Muller, O.; Sievers, S. ECRG4 is a candidate tumor suppressor gene frequently hypermethylated in colorectal carcinoma and glioma. BMC Cancer 2009, 9, 447. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Gao, J.; Wang, Q.; Hou, S.; Wu, C. ECRG4 Represses Cell Proliferation and Invasiveness via NFIC/OGN/NF-kappaB Signaling Pathway in Bladder Cancer. Front. Genet. 2020, 11, 846. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Wu, X.; Hong, C.Q.; Chen, J.; Wei, X.L.; Zhou, L.; Zhang, H.X.; Huang, Y.T.; Peng, L. Downregulated ECRG4 is correlated with lymph node metastasis and predicts poor outcome for nasopharyngeal carcinoma patients. Clin. Transl. Oncol. Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2017, 19, 84–90. [Google Scholar] [CrossRef]
- Lee, J.; Dang, X.; Borboa, A.; Coimbra, R.; Baird, A.; Eliceiri, B.P. Thrombin-processed Ecrg4 recruits myeloid cells and induces antitumorigenic inflammation. Neuro-Oncol. 2015, 17, 685–696. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Yang, W.; Qin, X.; Wang, F.; Li, H.; Lin, C.; Li, W.; Gu, C.; Zhang, Y.; Ran, Y. ECRG4 acts as a tumor suppressor and as a determinant of chemotherapy resistance in human nasopharyngeal carcinoma. Cell. Oncol. 2015, 38, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, A.F.; Mahmoud, W.; Al-Harizy, R.M. Targeting glucose metabolism to suppress cancer progression: Prospective of anti-glycolytic cancer therapy. Pharmacol. Res. Off. J. Ital. Pharmacol. Soc. 2019, 150, 104511. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, T.; Chesney, J.; Metz, C.; Leng, L.; Donnelly, S.; Makita, Z.; Mitchell, R.; Bucala, R. High expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (iPFK-2; PFKFB3) in human cancers. Cancer Res. 2002, 62, 5881–5887. [Google Scholar] [PubMed]
- Li, H.M.; Yang, J.G.; Liu, Z.J.; Wang, W.M.; Yu, Z.L.; Ren, J.G.; Chen, G.; Zhang, W.; Jia, J. Blockage of glycolysis by targeting PFKFB3 suppresses tumor growth and metastasis in head and neck squamous cell carcinoma. J. Exp. Clin. Cancer Res. CR 2017, 36, 7. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.K.; Zhu, X.D.; Wang, C.H.; Zhang, Y.Y.; Cai, H.; Li, X.L.; Cao, M.Q.; Zhang, S.Z.; Li, K.S.; Sun, H.C. PFKFB3 blockade inhibits hepatocellular carcinoma growth by impairing DNA repair through AKT. Cell Death Dis. 2018, 9, 428. [Google Scholar] [CrossRef]
- Minchenko, O.H.; Ochiai, A.; Opentanova, I.L.; Ogura, T.; Minchenko, D.O.; Caro, J.; Komisarenko, S.V.; Esumi, H. Overexpression of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-4 in the human breast and colon malignant tumors. Biochimie 2005, 87, 1005–1010. [Google Scholar] [CrossRef]
- Bobarykina, A.Y.; Minchenko, D.O.; Opentanova, I.L.; Moenner, M.; Caro, J.; Esumi, H.; Minchenko, O.H. Hypoxic regulation of PFKFB-3 and PFKFB-4 gene expression in gastric and pancreatic cancer cell lines and expression of PFKFB genes in gastric cancers. Acta Biochim. Pol. 2006, 53, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Roy, D.; Sarkar Bhattacharya, S.; Jin, L.; Jung, D.; Zhang, S.; Kalogera, E.; Staub, J.; Wang, Y.; Xuyang, W.; et al. Therapeutic targeting of PFKFB3 with a novel glycolytic inhibitor PFK158 promotes lipophagy and chemosensitivity in gynecologic cancers. Int. J. Cancer. J. Int. Du Cancer 2019, 144, 178–189. [Google Scholar] [CrossRef]
- Cantelmo, A.R.; Conradi, L.C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.A.; et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968–985. [Google Scholar] [CrossRef]
- Li, F.L.; Liu, J.P.; Bao, R.X.; Yan, G.; Feng, X.; Xu, Y.P.; Sun, Y.P.; Yan, W.; Ling, Z.Q.; Xiong, Y.; et al. Acetylation accumulates PFKFB3 in cytoplasm to promote glycolysis and protects cells from cisplatin-induced apoptosis. Nat. Commun. 2018, 9, 508. [Google Scholar] [CrossRef]
- Xiao, Y.; Jin, L.; Deng, C.; Guan, Y.; Kalogera, E.; Ray, U.; Thirusangu, P.; Staub, J.; Sarkar Bhattacharya, S.; Xu, H.; et al. Inhibition of PFKFB3 induces cell death and synergistically enhances chemosensitivity in endometrial cancer. Oncogene 2021, 40, 1409–1424. [Google Scholar] [CrossRef] [PubMed]
- Tung, M.C.; Lin, P.L.; Wang, Y.C.; He, T.Y.; Lee, M.C.; Yeh, S.D.; Chen, C.Y.; Lee, H. Mutant p53 confers chemoresistance in non-small cell lung cancer by upregulating Nrf2. Oncotarget 2015, 6, 41692–41705. [Google Scholar] [CrossRef] [PubMed]
- Lavarino, C.; Pilotti, S.; Oggionni, M.; Gatti, L.; Perego, P.; Bresciani, G.; Pierotti, M.A.; Scambia, G.; Ferrandina, G.; Fagotti, A.; et al. p53 gene status and response to platinum/paclitaxel-based chemotherapy in advanced ovarian carcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2000, 18, 3936–3945. [Google Scholar] [CrossRef] [PubMed]
- Baltaci, S.; Orhan, D.; Turkolmez, K.; Yesilli, C.; Beduk, Y.; Tulunay, O. P53, bcl-2 and bax immunoreactivity as predictors of response and outcome after chemotherapy for metastatic germ cell testicular tumours. BJU Int. 2001, 87, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Qi, Z.; Yin, H.; Yang, G. Interaction between p53 and Ras signaling controls cisplatin resistance via HDAC4- and HIF-1alpha-mediated regulation of apoptosis and autophagy. Theranostics 2019, 9, 1096–1114. [Google Scholar] [CrossRef]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.; Ash, D.; Shaha, C. Beclin-1-p53 interaction is crucial for cell fate determination in embryonal carcinoma cells. J. Cell. Mol. Med. 2014, 18, 2275–2286. [Google Scholar] [CrossRef]
- Maycotte, P.; Aryal, S.; Cummings, C.T.; Thorburn, J.; Morgan, M.J.; Thorburn, A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy 2012, 8, 200–212. [Google Scholar] [CrossRef]
- Yee, K.S.; Grochola, L.; Hamilton, G.; Grawenda, A.; Bond, E.E.; Taubert, H.; Wurl, P.; Bond, G.L.; O’Neill, E. A RASSF1A polymorphism restricts p53/p73 activation and associates with poor survival and accelerated age of onset of soft tissue sarcoma. Cancer Res. 2012, 72, 2206–2217. [Google Scholar] [CrossRef] [PubMed]
- Koul, S.; McKiernan, J.M.; Narayan, G.; Houldsworth, J.; Bacik, J.; Dobrzynski, D.L.; Assaad, A.M.; Mansukhani, M.; Reuter, V.E.; Bosl, G.J.; et al. Role of promoter hypermethylation in Cisplatin treatment response of male germ cell tumors. Mol Cancer 2004, 3, 16. [Google Scholar] [CrossRef] [PubMed]
- de Fraipont, F.; Levallet, G.; Creveuil, C.; Bergot, E.; Beau-Faller, M.; Mounawar, M.; Richard, N.; Antoine, M.; Rouquette, I.; Favrot, M.C.; et al. An apoptosis methylation prognostic signature for early lung cancer in the IFCT-0002 trial. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 2976–2986. [Google Scholar] [CrossRef] [PubMed]
- Levesley, J.; Lusher, M.E.; Lindsey, J.C.; Clifford, S.C.; Grundy, R.; Coyle, B. RASSF1A and the BH3-only mimetic ABT-737 promote apoptosis in pediatric medulloblastoma cell lines. Neuro-Oncol. 2011, 13, 1265–1276. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, X.; Yang, F.; Yang, Y.; Wang, T.; Liu, W.; Zhou, H.; Zhao, W. RASSF1A Enhances Chemosensitivity of NSCLC Cells Through Activating Autophagy by Regulating MAP1S to Inactivate Keap1-Nrf2 Pathway. Drug Des. Dev. Ther. 2021, 15, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Hirao, S.; Kabe, Y.; Ogura, Y.; Sato, I.; Yamaguchi, Y.; Wada, T.; Handa, H. A new APE1/Ref-1-dependent pathway leading to reduction of NF-kappaB and AP-1, and activation of their DNA-binding activity. Nucleic Acids Res. 2008, 36, 4327–4336. [Google Scholar] [CrossRef] [PubMed]
- Lando, D.; Pongratz, I.; Poellinger, L.; Whitelaw, M.L. A redox mechanism controls differential DNA binding activities of hypoxia-inducible factor (HIF) 1alpha and the HIF-like factor. J. Biol. Chem. 2000, 275, 4618–4627. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, A.; Chattopadhyay, R.; Burnette, B.R.; Cross, J.V.; Mitra, S.; Ernst, P.B.; Bhakat, K.K.; Crowe, S.E. Acetylation of apurinic/apyrimidinic endonuclease-1 regulates Helicobacter pylori-mediated gastric epithelial cell apoptosis. Gastroenterology 2009, 136, 2258–2269. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, A.A.; Jiang, Y.; Luo, M.; Reed, A.M.; Shahda, S.; He, Y.; Maitra, A.; Kelley, M.R.; Fishel, M.L. APE1/Ref-1 regulates STAT3 transcriptional activity and APE1/Ref-1-STAT3 dual-targeting effectively inhibits pancreatic cancer cell survival. PLoS ONE 2012, 7, e47462. [Google Scholar] [CrossRef] [PubMed]
- Fishel, M.L.; Wu, X.; Devlin, C.M.; Logsdon, D.P.; Jiang, Y.; Luo, M.; He, Y.; Yu, Z.; Tong, Y.; Lipking, K.P.; et al. Apurinic/apyrimidinic endonuclease/redox factor-1 (APE1/Ref-1) redox function negatively regulates NRF2. J. Biol. Chem. 2015, 290, 3057–3068. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Xiang, D.B.; Yang, X.Q.; Chen, L.S.; Li, M.X.; Zhong, Z.Y.; Zhang, Y.S. APE1 overexpression is associated with cisplatin resistance in non-small cell lung cancer and targeted inhibition of APE1 enhances the activity of cisplatin in A549 cells. Lung Cancer 2009, 66, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Manguinhas, R.; Fernandes, A.S.; Costa, J.G.; Saraiva, N.; Camoes, S.P.; Gil, N.; Rosell, R.; Castro, M.; Miranda, J.P.; Oliveira, N.G. Impact of the APE1 Redox Function Inhibitor E3330 in Non-small Cell Lung Cancer Cells Exposed to Cisplatin: Increased Cytotoxicity and Impairment of Cell Migration and Invasion. Antioxidants 2020, 9, 550. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Li, Z.; Zhang, S.; Xiong, Y.; Cun, Y.; Qian, C.; Li, M.; Ren, T.; Xia, L.; Cheng, Y.; et al. Association of DNA base excision repair genes (OGG1, APE1 and XRCC1) polymorphisms with outcome to platinum-based chemotherapy in advanced nonsmall-cell lung cancer patients. Int. J. Cancer. J. Int. Du Cancer 2014, 135, 2687–2696. [Google Scholar] [CrossRef] [PubMed]
- Tell, G.; Fantini, D.; Quadrifoglio, F. Understanding different functions of mammalian AP endonuclease (APE1) as a promising tool for cancer treatment. Cell. Mol. Life Sci. CMLS 2010, 67, 3589–3608. [Google Scholar] [CrossRef]
- Li, Z.; Wang, Y.; Wu, L.; Dong, Y.; Zhang, J.; Chen, F.; Xie, W.; Huang, J.; Lu, N. Apurinic endonuclease 1 promotes the cisplatin resistance of lung cancer cells by inducing Parkinmediated mitophagy. Oncol. Rep. 2019, 42, 2245–2254. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.T.; Zhou, J.; Yang, F.; Zhou, S.F.; Ren, T. Proteomics reveals a therapeutic vulnerability via the combined blockade of APE1 and autophagy in lung cancer A549 cells. BMC Cancer 2020, 20, 634. [Google Scholar] [CrossRef] [PubMed]
- Latorre, E.; Carelli, S.; Raimondi, I.; D’Agostino, V.; Castiglioni, I.; Zucal, C.; Moro, G.; Luciani, A.; Ghilardi, G.; Monti, E.; et al. The Ribonucleic Complex HuR-MALAT1 Represses CD133 Expression and Suppresses Epithelial-Mesenchymal Transition in Breast Cancer. Cancer Res. 2016, 76, 2626–2636. [Google Scholar] [CrossRef] [PubMed]
- YiRen, H.; YingCong, Y.; Sunwu, Y.; Keqin, L.; Xiaochun, T.; Senrui, C.; Ende, C.; XiZhou, L.; Yanfan, C. Long noncoding RNA MALAT1 regulates autophagy associated chemoresistance via miR-23b-3p sequestration in gastric cancer. Mol. Cancer 2017, 16, 174. [Google Scholar] [CrossRef]
- Xi, Z.; Si, J.; Nan, J. LncRNA MALAT1 potentiates autophagyassociated cisplatin resistance by regulating the microRNA30b/autophagyrelated gene 5 axis in gastric cancer. Int. J. Oncol. 2019, 54, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.F.; Li, C.S.; Zhou, Y.; Lu, X.H. Propofol facilitates cisplatin sensitivity via lncRNA MALAT1/miR-30e/ATG5 axis through suppressing autophagy in gastric cancer. Life Sci. 2020, 244, 117280. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Sang, H.; Wei, S.; Li, Y.; Jin, D.; Zhu, X.; Li, X.; Dang, Y.; Zhang, G. circCUL2 regulates gastric cancer malignant transformation and cisplatin resistance by modulating autophagy activation via miR-142-3p/ROCK2. Mol. Cancer 2020, 19, 156. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wang, W.; Li, Z.; Chen, Z.; Zhi, X.; Xu, J.; Li, Q.; Wang, L.; Huang, X.; Wang, L.; et al. MicroRNA-148a-3p enhances cisplatin cytotoxicity in gastric cancer through mitochondrial fission induction and cyto-protective autophagy suppression. Cancer Lett. 2017, 410, 212–227. [Google Scholar] [CrossRef]
- Zhao, R.; Zhang, X.; Zhang, Y.; Zhang, Y.; Yang, Y.; Sun, Y.; Zheng, X.; Qu, A.; Umwali, Y.; Zhang, Y. HOTTIP Predicts Poor Survival in Gastric Cancer Patients and Contributes to Cisplatin Resistance by Sponging miR-216a-5p. Front. Cell Dev. Biol. 2020, 8, 348. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.X.; Chen, H.J.; Zheng, F.X.; Gao, Z.Y.; Sun, P.F.; Peng, Q.; Liu, Y.; Deng, X.; Huang, Y.H.; Zhao, C.; et al. LncRNA BLACAT1 is involved in chemoresistance of nonsmall cell lung cancer cells by regulating autophagy. Int. J. Oncol. 2019, 54, 339–347. [Google Scholar] [CrossRef]
- Hua, L.; Zhu, G.; Wei, J. MicroRNA-1 overexpression increases chemosensitivity of non-small cell lung cancer cells by inhibiting autophagy related 3-mediated autophagy. Cell Biol. Int. 2018, 42, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xing, Y.; Rong, L. miR-181 regulates cisplatin-resistant non-small cell lung cancer via downregulation of autophagy through the PTEN/PI3K/AKT pathway. Oncol. Rep. 2018, 39, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, J.; Zhang, S.; Zheng, X.; Xie, S.; Mao, J.; Cai, Y.; Lu, X.; Hu, L.; Shen, J.; et al. MiR-223 regulates autophagy associated with cisplatin resistance by targeting FBXW7 in human non-small cell lung cancer. Cancer Cell Int. 2020, 20, 258. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yuwen, D.; Chen, J.; Zheng, B.; Gao, J.; Fan, M.; Xue, W.; Wang, Y.; Li, W.; Shu, Y.; et al. Exosomal Transfer of Cisplatin-Induced miR-425-3p Confers Cisplatin Resistance in NSCLC Through Activating Autophagy. Int. J. Nanomed. 2019, 14, 8121–8132. [Google Scholar] [CrossRef]
- Wang, S.; Li, M.Y.; Liu, Y.; Vlantis, A.C.; Chan, J.Y.; Xue, L.; Hu, B.G.; Yang, S.; Chen, M.X.; Zhou, S.; et al. The role of microRNA in cisplatin resistance or sensitivity. Expert Opin. Ther. Targets 2020, 24, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, J.; Ren, J.; Vlantis, A.C.; Li, M.Y.; Liu, S.Y.W.; Ng, E.K.W.; Chan, A.B.W.; Luo, D.C.; Liu, Z.; et al. MicroRNA-125b Interacts with Foxp3 to Induce Autophagy in Thyroid Cancer. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 2295–2303. [Google Scholar] [CrossRef]
- Li, Y.; Hu, T.; Chen, T.; Yang, T.; Ren, H.; Chen, M. Combination treatment of FTY720 and cisplatin exhibits enhanced antitumour effects on cisplatin-resistant non-small lung cancer cells. Oncol. Rep. 2018, 39, 565–572. [Google Scholar] [CrossRef]
- Luo, L.; Sun, W.; Zhu, W.; Li, S.; Zhang, W.; Xu, X.; Fang, D.; Grahn, T.H.M.; Jiang, L.; Zheng, Y. BCAT1 decreases the sensitivity of cancer cells to cisplatin by regulating mTOR-mediated autophagy via branched-chain amino acid metabolism. Cell Death Dis. 2021, 12, 169. [Google Scholar] [CrossRef]
- Bai, Y.; Liu, X.; Qi, X.; Liu, X.; Peng, F.; Li, H.; Fu, H.; Pei, S.; Chen, L.; Chi, X.; et al. PDIA6 modulates apoptosis and autophagy of non-small cell lung cancer cells via the MAP4K1/JNK signaling pathway. EBioMedicine 2019, 42, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Ranzuglia, V.; Lorenzon, I.; Pellarin, I.; Sonego, M.; Dall’Acqua, A.; D’Andrea, S.; Lovisa, S.; Segatto, I.; Coan, M.; Polesel, J.; et al. Serum- and glucocorticoid- inducible kinase 2, SGK2, is a novel autophagy regulator and modulates platinum drugs response in cancer cells. Oncogene 2020, 39, 6370–6386. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Liu, K.; Shao, Y.; Feng, X.; Ji, Z.; Chang, B.; Wang, Y.; Xu, L.; Yang, G. ID1 confers cancer cell chemoresistance through STAT3/ATF6-mediated induction of autophagy. Cell Death Dis. 2020, 11, 137. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Tang, L.; Hu, J.; Wang, S.; Liu, Y.; Yang, M.; Zhang, J.; Tang, B. Inhibition of MGMT-mediated autophagy suppression decreases cisplatin chemosensitivity in gastric cancer. Biomed. Pharmacother. Biomed. Pharmacother. 2020, 125, 109896. [Google Scholar] [CrossRef]
- Ma, H.; Li, Y.; Wang, X.; Wu, H.; Qi, G.; Li, R.; Yang, N.; Gao, M.; Yan, S.; Yuan, C.; et al. PBK, targeted by EVI1, promotes metastasis and confers cisplatin resistance through inducing autophagy in high-grade serous ovarian carcinoma. Cell Death Dis. 2019, 10, 166. [Google Scholar] [CrossRef]
- Takeda, T.; Komatsu, M.; Chiwaki, F.; Komatsuzaki, R.; Nakamura, K.; Tsuji, K.; Kobayashi, Y.; Tominaga, E.; Ono, M.; Banno, K.; et al. Upregulation of IGF2R evades lysosomal dysfunction-induced apoptosis of cervical cancer cells via transport of cathepsins. Cell Death Dis. 2019, 10, 876. [Google Scholar] [CrossRef]
- Liu, Y.; Fu, Y.; Hu, X.; Chen, S.; Miao, J.; Wang, Y.; Zhou, Y.; Zhang, Y. Caveolin-1 knockdown increases the therapeutic sensitivity of lung cancer to cisplatin-induced apoptosis by repressing Parkin-related mitophagy and activating the ROCK1 pathway. J. Cell. Physiol. 2020, 235, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Wang, W.; Li, Y.; Yang, D.; Li, X.; Shen, C.; Liu, Y.; Ke, X.; Guo, S.; Guo, Z. HSP90AA1-mediated autophagy promotes drug resistance in osteosarcoma. J. Exp. Clin. Cancer Res. CR 2018, 37, 201. [Google Scholar] [CrossRef]
- Zhou, F.; Yang, X.; Zhao, H.; Liu, Y.; Feng, Y.; An, R.; Lv, X.; Li, J.; Chen, B. Down-regulation of OGT promotes cisplatin resistance by inducing autophagy in ovarian cancer. Theranostics 2018, 8, 5200–5212. [Google Scholar] [CrossRef]
- Monisha, J.; Roy, N.K.; Padmavathi, G.; Banik, K.; Bordoloi, D.; Khwairakpam, A.D.; Arfuso, F.; Chinnathambi, A.; Alahmadi, T.A.; Alharbi, S.A.; et al. NGAL is Downregulated in Oral Squamous Cell Carcinoma and Leads to Increased Survival, Proliferation, Migration and Chemoresistance. Cancers 2018, 10, 228. [Google Scholar] [CrossRef]
- Kim, M.; Jung, J.Y.; Choi, S.; Lee, H.; Morales, L.D.; Koh, J.T.; Kim, S.H.; Choi, Y.D.; Choi, C.; Slaga, T.J.; et al. GFRA1 promotes cisplatin-induced chemoresistance in osteosarcoma by inducing autophagy. Autophagy 2017, 13, 149–168. [Google Scholar] [CrossRef]
- Homewood, C.A.; Warhurst, D.C.; Peters, W.; Baggaley, V.C. Lysosomes, pH and the anti-malarial action of chloroquine. Nature 1972, 235, 50–52. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Isaka, Y. Chloroquine in cancer therapy: A double-edged sword of autophagy. Cancer Res. 2013, 73, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.; Cardelli, J.; Barr, M.P.; O’Byrne, K.; Mills, G.; El-Osta, H. Modulating lysosomal function through lysosome membrane permeabilization or autophagy suppression restores sensitivity to cisplatin in refractory non-small-cell lung cancer cells. PLoS ONE 2017, 12, e0184922. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Oda, K.; Wada-Hiraike, O.; Sone, K.; Inaba, K.; Ikeda, Y.; Miyasaka, A.; Kashiyama, T.; Tanikawa, M.; Arimoto, T.; et al. The anti-malarial chloroquine suppresses proliferation and overcomes cisplatin resistance of endometrial cancer cells via autophagy inhibition. Gynecol. Oncol. 2015, 137, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Schlutermann, D.; Skowron, M.A.; Berleth, N.; Bohler, P.; Deitersen, J.; Stuhldreier, F.; Wallot-Hieke, N.; Wu, W.; Peter, C.; Hoffmann, M.J.; et al. Targeting urothelial carcinoma cells by combining cisplatin with a specific inhibitor of the autophagy-inducing class III PtdIns3K complex. Urol. Oncol. 2018, 36, 160.e1–160.e13. [Google Scholar] [CrossRef]
- Hwang, J.R.; Kim, W.Y.; Cho, Y.J.; Ryu, J.Y.; Choi, J.J.; Jeong, S.Y.; Kim, M.S.; Kim, J.H.; Paik, E.S.; Lee, Y.Y.; et al. Chloroquine reverses chemoresistance via upregulation of p21(WAF1/CIP1) and autophagy inhibition in ovarian cancer. Cell Death Dis. 2020, 11, 1034. [Google Scholar] [CrossRef] [PubMed]
- Gunda, V.; Pathania, A.S.; Chava, S.; Prathipati, P.; Chaturvedi, N.K.; Coulter, D.W.; Pandey, M.K.; Durden, D.L.; Challagundla, K.B. Amino Acids Regulate Cisplatin Insensitivity in Neuroblastoma. Cancers 2020, 12, 2576. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Feng, Y.; Cui, X.; Wang, Q.; Pan, X. Autophagy inhibition induces the repolarisation of tumour-associated macrophages and enhances chemosensitivity of laryngeal cancer cells to cisplatin in mice. Cancer Immunol. Immunother. CII 2019, 68, 1909–1920. [Google Scholar] [CrossRef] [PubMed]
- Magnano, S.; Hannon Barroeta, P.; Duffy, R.; O’Sullivan, J.; Zisterer, D.M. Cisplatin induces autophagy-associated apoptosis in human oral squamous cell carcinoma (OSCC) mediated in part through reactive oxygen species. Toxicol. Appl. Pharm. 2021, 427, 115646. [Google Scholar] [CrossRef]
- Levy, J.M.; Thorburn, A. Modulation of pediatric brain tumor autophagy and chemosensitivity. J. Neuro-Oncol. 2012, 106, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Phoo, N.L.L.; Dejkriengkraikul, P.; Khaw-On, P.; Yodkeeree, S. Transcriptomic Profiling Reveals AKR1C1 and AKR1C3 Mediate Cisplatin Resistance in Signet Ring Cell Gastric Carcinoma via Autophagic Cell Death. Int. J. Mol. Sci. 2021, 22, 12512. [Google Scholar] [CrossRef]
- Wang, J.; Wu, G.S. Role of autophagy in cisplatin resistance in ovarian cancer cells. J. Biol. Chem. 2014, 289, 17163–17173. [Google Scholar] [CrossRef]
- Ko, J.C.; Tsai, M.S.; Chiu, Y.F.; Weng, S.H.; Kuo, Y.H.; Lin, Y.W. Up-regulation of extracellular signal-regulated kinase 1/2-dependent thymidylate synthase and thymidine phosphorylase contributes to cisplatin resistance in human non-small-cell lung cancer cells. J. Pharmacol. Exp. Ther. 2011, 338, 184–194. [Google Scholar] [CrossRef]
- Lai, S.T.; Wang, Y.; Peng, F. Astragaloside IV sensitizes non-small cell lung cancer cells to cisplatin by suppressing endoplasmic reticulum stress and autophagy. J. Thorac. Dis. 2020, 12, 3715–3724. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.I.; Hong, S.H.; Ku, J.M.; Kim, M.J.; Ju, S.W.; Chang, S.W.; Cheon, C.; Ko, S.G. Gardenia jasminoides Enhances CDDP-Induced Apoptosis of Glioblastoma Cells via AKT/mTOR Pathway While Protecting Death of Astrocytes. Nutrients 2020, 12, 196. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.X.; Wang, Y.; An, H.W.; Qi, B.; Wang, J.; Wang, L.; Shi, J.; Mei, L.; Wang, H. Peptide-Based Autophagic Gene and Cisplatin Co-delivery Systems Enable Improved Chemotherapy Resistance. Nano Lett. 2019, 19, 2968–2978. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Liu, X.; Yan, W.; Chen, Y. Antitumor effect of poly lactic acid nanoparticles loaded with cisplatin and chloroquine on the oral squamous cell carcinoma. Aging 2020, 13, 2593–2603. [Google Scholar] [CrossRef]
- Wang, K.; Liu, X.; Liu, Q.; Ho, I.H.; Wei, X.; Yin, T.; Zhan, Y.; Zhang, W.; Zhang, W.; Chen, B.; et al. Hederagenin potentiated cisplatin- and paclitaxel-mediated cytotoxicity by impairing autophagy in lung cancer cells. Cell Death Dis. 2020, 11, 611. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Zhuang, X.; Zhang, Q.; Cheng, Y.; Wu, D.; Wang, X.; Qiao, T. Acetyl-11-keto-beta-boswellic acid enhances the cisplatin sensitivity of non-small cell lung cancer cells through cell cycle arrest, apoptosis induction, and autophagy suppression via p21-dependent signaling pathway. Cell Biol. Toxicol. 2021, 37, 209–228. [Google Scholar] [CrossRef]
- Mi, S.; Xiang, G.; Yuwen, D.; Gao, J.; Guo, W.; Wu, X.; Wu, X.; Sun, Y.; Su, Y.; Shen, Y.; et al. Inhibition of autophagy by andrographolide resensitizes cisplatin-resistant non-small cell lung carcinoma cells via activation of the Akt/mTOR pathway. Toxicol. Appl. Pharm. 2016, 310, 78–86. [Google Scholar] [CrossRef]
- Zhou, J.; Hu, S.E.; Tan, S.H.; Cao, R.; Chen, Y.; Xia, D.; Zhu, X.; Yang, X.F.; Ong, C.N.; Shen, H.M. Andrographolide sensitizes cisplatin-induced apoptosis via suppression of autophagosome-lysosome fusion in human cancer cells. Autophagy 2012, 8, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Pal Singh, M.; Pal Khaket, T.; Bajpai, V.K.; Alfarraj, S.; Kim, S.G.; Chen, L.; Huh, Y.S.; Han, Y.K.; Kang, S.C. Morin Hydrate Sensitizes Hepatoma Cells and Xenograft Tumor towards Cisplatin by Downregulating PARP-1-HMGB1 Mediated Autophagy. Int. J. Mol. Sci. 2020, 21, 8253. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.P.; Cho, H.J.; Kim, J.T.; Baek, K.E.; Lee, H.G.; Kang, S.C. Morin Hydrate Reverses Cisplatin Resistance by Impairing PARP1/HMGB1-Dependent Autophagy in Hepatocellular Carcinoma. Cancers 2019, 11, 986. [Google Scholar] [CrossRef] [PubMed]
- de Jonge, M.J.; Verweij, J. Renal toxicities of chemotherapy. Semin. Oncol. 2006, 33, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.L.; Kellum, J.A.; Shah, S.V.; Molitoris, B.A.; Ronco, C.; Warnock, D.G.; Levin, A.; Acute Kidney Injury, N. Acute Kidney Injury Network: Report of an initiative to improve outcomes in acute kidney injury. Crit. Care 2007, 11, R31. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Kimura, T.; Takabatake, Y.; Namba, T.; Kaimori, J.; Kitamura, H.; Matsui, I.; Niimura, F.; Matsusaka, T.; Fujita, N.; et al. Autophagy guards against cisplatin-induced acute kidney injury. Am. J. Pathol. 2012, 180, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Kaimori, J.Y.; Matsui, I.; Namba, T.; Kitamura, H.; Niimura, F.; Matsusaka, T.; Soga, T.; et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J. Am. Soc. Nephrol. JASN 2011, 22, 902–913. [Google Scholar] [CrossRef]
- Zhang, D.; Pan, J.; Xiang, X.; Liu, Y.; Dong, G.; Livingston, M.J.; Chen, J.K.; Yin, X.M.; Dong, Z. Protein Kinase Cdelta Suppresses Autophagy to Induce Kidney Cell Apoptosis in Cisplatin Nephrotoxicity. J. Am. Soc. Nephrol. JASN 2017, 28, 1131–1144. [Google Scholar] [CrossRef]
- Lee, D.; Kang, K.B.; Kim, H.W.; Park, J.S.; Hwang, G.S.; Kang, K.S.; Choi, S.; Yamabe, N.; Kim, K.H. Unique Triterpenoid of Jujube Root Protects Cisplatin-induced Damage in Kidney Epithelial LLC-PK1 Cells via Autophagy Regulation. Nutrients 2020, 12, 677. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zheng, C.; Wan, X.; Shi, M.; McMillan, K.; Maique, J.; Cao, C. Retinoic Acid Alleviates Cisplatin-Induced Acute Kidney Injury Through Activation of Autophagy. Front. Pharmacol. 2020, 11, 987. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Z.; Shu, S.; Cai, J.; Tang, C.; Dong, Z. AMPK/mTOR Signaling in Autophagy Regulation During Cisplatin-Induced Acute Kidney Injury. Front. Physiol. 2020, 11, 619730. [Google Scholar] [CrossRef] [PubMed]
- Minocha, E.; Sinha, R.A.; Jain, M.; Chaturvedi, C.P.; Nityanand, S. Amniotic fluid stem cells ameliorate cisplatin-induced acute renal failure through induction of autophagy and inhibition of apoptosis. Stem Cell Res. Ther. 2019, 10, 370. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Gu, J.; Chen, Y.; Kang, W.; Wang, X.; Wu, H. Current Strategies to Combat Cisplatin-Induced Ototoxicity. Front. Pharmacol. 2020, 11, 999. [Google Scholar] [CrossRef] [PubMed]
- El Nashar, E.M.; Alghamdi, M.A.; Alasmari, W.A.; Hussein, M.M.A.; Hamza, E.; Taha, R.I.; Ahmed, M.M.; Al-Khater, K.M.; Abdelfattah-Hassan, A. Autophagy Promotes the Survival of Adipose Mesenchymal Stem/Stromal Cells and Enhances Their Therapeutic Effects in Cisplatin-Induced Liver Injury via Modulating TGF-beta1/Smad and PI3K/AKT Signaling Pathways. Cells 2021, 10, 2475. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hahn, T.; Garrison, K.; Cui, Z.H.; Thorburn, A.; Thorburn, J.; Hu, H.M.; Akporiaye, E.T. The vitamin E analogue alpha-TEA stimulates tumor autophagy and enhances antigen cross-presentation. Cancer Res. 2012, 72, 3535–3545. [Google Scholar] [CrossRef] [PubMed]
- Santanam, U.; Banach-Petrosky, W.; Abate-Shen, C.; Shen, M.M.; White, E.; DiPaola, R.S. Atg7 cooperates with Pten loss to drive prostate cancer tumor growth. Genes Dev. 2016, 30, 399–407. [Google Scholar] [CrossRef]
- Levy, J.; Cacheux, W.; Bara, M.A.; L’Hermitte, A.; Lepage, P.; Fraudeau, M.; Trentesaux, C.; Lemarchand, J.; Durand, A.; Crain, A.M.; et al. Intestinal inhibition of Atg7 prevents tumour initiation through a microbiome-influenced immune response and suppresses tumour growth. Nat. Cell Biol. 2015, 17, 1062–1073. [Google Scholar] [CrossRef]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef]
- Ko, A.; Kanehisa, A.; Martins, I.; Senovilla, L.; Chargari, C.; Dugue, D.; Marino, G.; Kepp, O.; Michaud, M.; Perfettini, J.L.; et al. Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signalling. Cell Death Differ. 2014, 21, 92–99. [Google Scholar] [CrossRef]
- Solari, J.I.G.; Filippi-Chiela, E.; Pilar, E.S.; Nunes, V.; Gonzalez, E.A.; Figueiro, F.; Andrade, C.F.; Klamt, F. Damage-associated molecular patterns (DAMPs) related to immunogenic cell death are differentially triggered by clinically relevant chemotherapeutics in lung adenocarcinoma cells. BMC Cancer 2020, 20, 474. [Google Scholar] [CrossRef] [PubMed]
- Wahba, J.; Natoli, M.; Whilding, L.M.; Parente-Pereira, A.C.; Jung, Y.; Zona, S.; Lam, E.W.; Smith, J.R.; Maher, J.; Ghaem-Maghami, S. Chemotherapy-induced apoptosis, autophagy and cell cycle arrest are key drivers of synergy in chemo-immunotherapy of epithelial ovarian cancer. Cancer Immunol. Immunother. CII 2018, 67, 1753–1765. [Google Scholar] [CrossRef] [PubMed]
- Heckmann, B.L.; Green, D.R. LC3-associated phagocytosis at a glance. J. Cell Sci. 2019, 132, jcs222984. [Google Scholar] [CrossRef] [PubMed]
- Cunha, L.D.; Yang, M.; Carter, R.; Guy, C.; Harris, L.; Crawford, J.C.; Quarato, G.; Boada-Romero, E.; Kalkavan, H.; Johnson, M.D.L.; et al. LC3-Associated Phagocytosis in Myeloid Cells Promotes Tumor Immune Tolerance. Cell 2018, 175, 429–441 e416. [Google Scholar] [CrossRef]


{kind=link}
| Cancer Types | In Vitro Study Models | In Vivo Study Models | Effect of CQ or HCQ on Cisplatin Sensitivity | Reference |
|---|---|---|---|---|
| NSCLC | A549/cisplatin cells | - | Increased | [146] |
| H460 cells | - | No effect | [50] | |
| Endometrial cancer cells | Ishikawa/cisplatin cells | - | Increased | [147] |
| Urothelial carcinoma cells | RT-112/cisplatin cells | - | Increased | [148] |
| Ovarian cancer | A2780-CP20/cisplatin cells | An orthotopic mouse model established with A2780-CP20 cells and a drug-resistant patient-derived xenograft model | Increased | [149] |
| ARHI-low expressed SKOV3 cells | - | No effect | [77] | |
| Esophageal cancers | EC109/cisplatin cells | Nude mice xenografted with EC109/cisplatin cells | Increased | [29] |
| Neuroblastoma cells | Cisplatin-resistant model SK-N-BE(2)Cres cells | - | Increased | [150] |
| Oral squamous cell carcinoma | SCC-4 cells and SCC-4/cisplatin cells | - | Increased in SCC-4 cells, no effect in SCC-4/cisplatin cells | [152] |
| Pediatric medulloblastoma cells | DAOY and ONS76 cells | - | no effect | [153] |
| Breast cancer cells | 67NR and 4T1 cells | - | no effect | [100] |
| Compound | In Vitro Study Models | In Vivo Study Models | The Role of Autophagy in Cisplatin Only-Treated Models | Effect of Combination Treatment on Autophagy | Reference |
|---|---|---|---|---|---|
| Astragaloside IV (AS-IV) derived from Astragalus membranaceus | Cisplatin-resistant NSCLC cell lines | - | Unknown | Decreased autophagy levels | [157] |
| Hederagenin, a triterpenoid derived from Hedera helix | NSCLC cell lines NCI-H1299 and NCI-H1975 | NCI-H1299 cells xenograft model | Unknown | Decreased autophagy levels | [161] |
| Acetyl-11-keto-β-boswellic acid (AKBA), a pentacyclic triterpenes, from Boswellia serrata | NSCLC cell lines A549 | - | Unknown | Decreased autophagy levels | [162] |
| Andrographolide (Andro), one of the major active components in Andrographis paniculata | Cisplatin-resistant A549 cells | A549/cisplatin cells xenograft model | Unknown | Decreased autophagy levels | [163] |
| Colon cancer cells HCT-116 (p53 wild type and p53-null) | - | Cytoprotective autophagy (both cell lines) | Decreased autophagy levels | [164] | |
| Morin hydrate, a bioflavonoid, isolated from the Moraceae family | HepG2 cell | HepG2 xenograft nude mice | Unknown | Decreased autophagy levels | [165] |
| Cisplatin-resistant HepG2 cells | Cisplatin-resistant HepG2 xenograft nude mice | Unknown | Decreased autophagy levels | [166] | |
| Gardenia jasminoides (GJ) is a medicinal herb abundant with flavonoids | Glioblastoma multiform U87MG and U373MG cells | - | Unknown, but induced cytotoxic autophagy when combined with GJ | Increased cytotoxic autophagy levels | [158] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Gewirtz, D.A. Is Autophagy Always a Barrier to Cisplatin Therapy? Biomolecules 2022, 12, 463. https://doi.org/10.3390/biom12030463
Xu J, Gewirtz DA. Is Autophagy Always a Barrier to Cisplatin Therapy? Biomolecules. 2022; 12(3):463. https://doi.org/10.3390/biom12030463
Chicago/Turabian StyleXu, Jingwen, and David A. Gewirtz. 2022. "Is Autophagy Always a Barrier to Cisplatin Therapy?" Biomolecules 12, no. 3: 463. https://doi.org/10.3390/biom12030463
APA StyleXu, J., & Gewirtz, D. A. (2022). Is Autophagy Always a Barrier to Cisplatin Therapy? Biomolecules, 12(3), 463. https://doi.org/10.3390/biom12030463