
Discovery of APL-1030, a Novel, High-Affinity Nanofitin Inhibitor of C3-Mediated Complement Activation

 , ,
, ,
Abstract
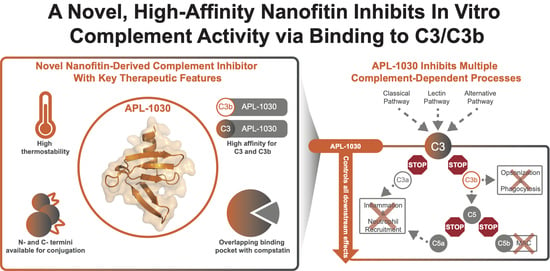
1. Introduction
2. Materials and Methods
2.1. Biotinylation of Antigens
2.2. Ribosome Display Selection Rounds and Isolation of Clones
2.3. ELISA Methods
2.4. Cloning in pQE30 Vector
2.5. Expression and Purification of Nanofitins
2.6. Biolayer Interferometry
2.7. Surface Plasmon Resonance
2.8. Isothermal Titration Calorimetry
2.9. The Wieslab Assay for Alternative and Classical Pathway Inhibition
2.10. Differential Scanning Calorimetry
2.11. Crystallization, Data Collection, and Structure Determination
3. Results
3.1. Discovery of APL-1030
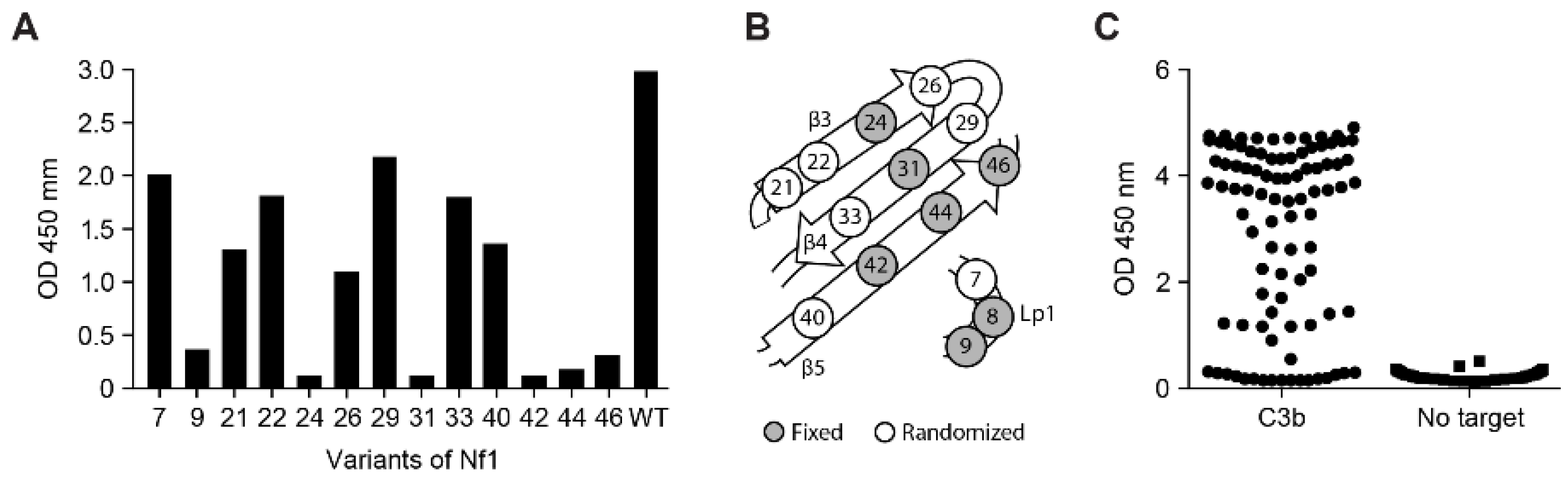
3.1.1. Isolation of Nanofitins Targeting C3b
3.1.2. Identification of Compstatin-like Nanofitins
3.1.3. Affinity Maturation of Nf1
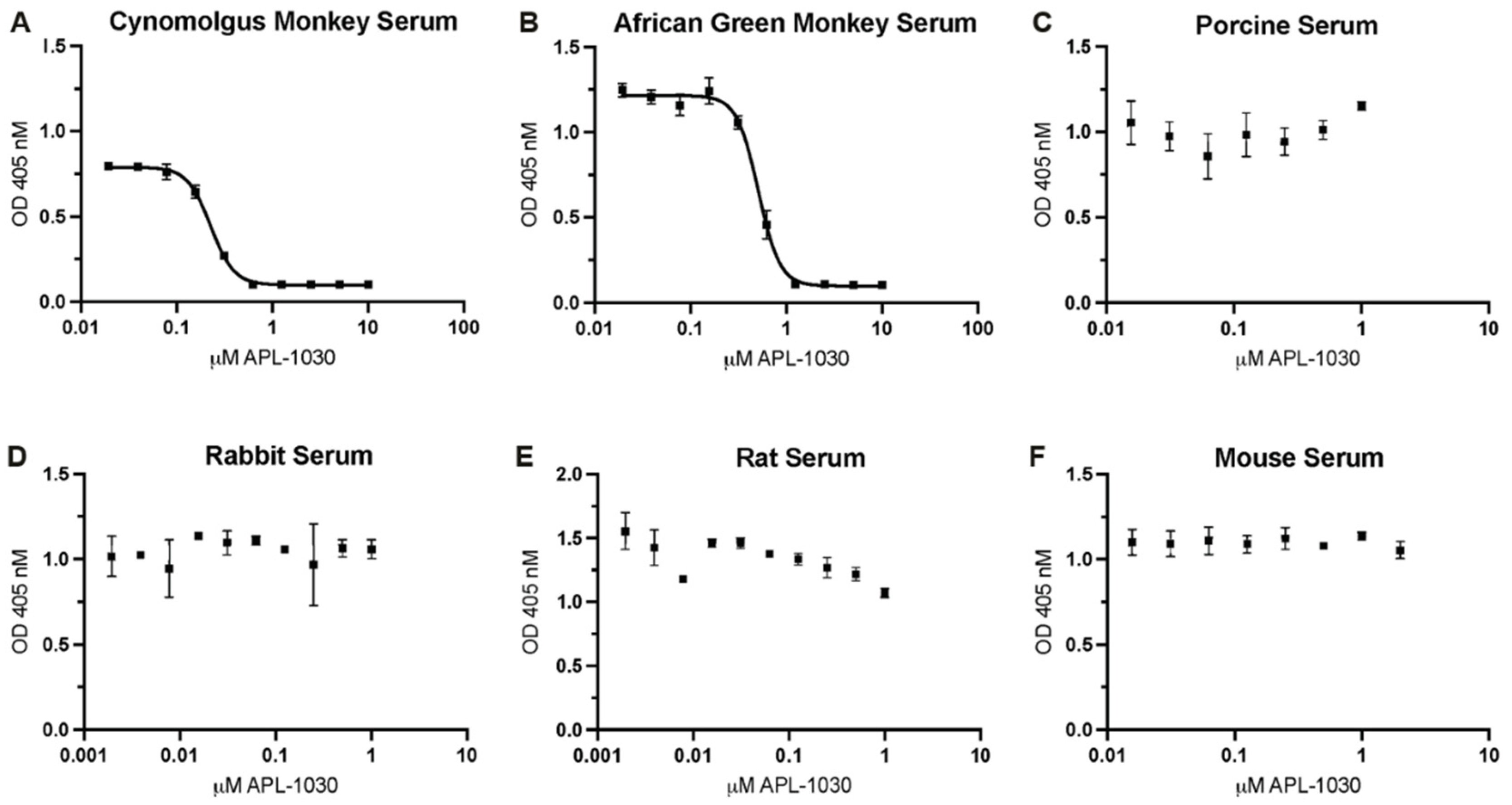
3.1.4. In Vitro Characterization of APL-1030
3.1.5. Structure of the C3b-APL-1030 Complex
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| APL-1030 Binding to: | N | KD nM | ΔG kcal/mol | ΔH kcal/mol | −TΔS kcal/mol |
|---|---|---|---|---|---|
| C3 | 1.18 | 1.36 ± 1.13 | −12.1 | −19.3 | 7.2 |
| C3b | 1.21 | 4.33 ± 0.68 | −11.4 | −20.2 | 8.8 |
| Space group | P 21 |
| Unit cell parameters | a = 99.28 Å, b = 104.46 Å, c = 102.89 Å α = γ = 90°, β = 97.44° |
| Resolution (Å) | 102–3.4 (3.6–3.4) * |
| Measured reflections | 92,835 |
| Unique reflections | 28,812 |
| Rsym (%) | 15.4 (149.2) * |
| Rpim (%) | 10.2 (100.3) |
| Completeness (%) | 99.2 (98.1) * |
| I/σI | 6.3 (0.9) * |
| CC(1/2) | 0.994 (0.460) * |
| Redundancy | 3.2 (3.1) * |
| Resolution range | 102–3.4 Å | ||
| Rworke | 24.0% | ||
| Rfree | 32.8% | ||
| Protomer details (per asymmetric unit): | |||
| C3b | 1e | ||
| APL-1030 | 1 | ||
| rms bond lengths | 0.007 Å | ||
| rms bond angles | 1.032° | ||
| Ramachandran plot | preferred | allowed | outliers |
| 76.9% | 22.8% | 0.4% | |




References
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement system part I—Molecular mechanisms of activation and regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed]
- Baines, A.C.; Brodsky, R.A. Complementopathies. Blood Rev. 2017, 31, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Dunkelberger, J.R.; Song, W.C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef]
- Bajic, G.; Degn, S.E.; Thiel, S.; Andersen, G.R. Complement activation, regulation, and molecular basis for complement-related diseases. EMBO J. 2015, 34, 2735–2757. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Noe, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement system part II: Role in immunity. Front. Immunol. 2015, 6, 257. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C.; Alexopoulos, H.; Spaeth, P.J. Complement in neurological disorders and emerging complement-targeted therapeutics. Nat. Rev. Neurol. 2020, 16, 601–617. [Google Scholar] [CrossRef] [PubMed]
- Mantegazza, R.; Vanoli, F.; Frangiamore, R.; Cavalcante, P. Complement inhibition for the treatment of myasthenia gravis. Immunotargets Ther. 2020, 9, 317–331. [Google Scholar] [CrossRef] [PubMed]
- McCombe, P.A.; Lee, J.D.; Woodruff, T.M.; Henderson, R.D. The peripheral immune system and amyotrophic lateral sclerosis. Front. Neurol. 2020, 11, 279. [Google Scholar] [CrossRef]
- Park, D.H.; Connor, K.M.; Lambris, J.D. The challenges and promise of complement therapeutics for ocular diseases. Front. Immunol. 2019, 10, 1007. [Google Scholar] [CrossRef] [PubMed]
- Poppelaars, F.; Thurman, J.M. Complement-mediated kidney diseases. Mol. Immunol. 2020, 128, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Risitano, A.M.; Rotoli, B. Paroxysmal nocturnal hemoglobinuria: Pathophysiology, natural history and treatment options in the era of biological agents. Biologics 2008, 2, 205–222. [Google Scholar] [CrossRef] [PubMed]
- Sahu, A.; Lambris, J.D. Complement inhibitors: A resurgent concept in anti-inflammatory therapeutics. Immunopharmacology 2000, 49, 133–148. [Google Scholar] [CrossRef]
- Tenner, A.J. Complement-Mediated events in Alzheimer’s disease: Mechanisms and potential therapeutic targets. J. Immunol. 2020, 204, 306–315. [Google Scholar] [CrossRef]
- Ricklin, D.; Lambris, J.D. Compstatin: A complement inhibitor on its way to clinical application. Adv. Exp. Med. Biol. 2008, 632, 273–292. [Google Scholar] [PubMed]
- Risitano, A.M.; Marotta, S.; Ricci, P.; Marano, L.; Frieri, C.; Cacace, F.; Sica, M.; Kulasekararaj, A.; Calado, R.T.; Scheinberg, P.; et al. Anti-complement treatment for paroxysmal nocturnal hemoglobinuria: Time for proximal complement inhibition? A position paper from the SAAWP of the EBMT. Front. Immunol. 2019, 10, 1157. [Google Scholar] [CrossRef] [PubMed]
- Mastellos, D.C.; Reis, E.S.; Yancopoulou, D.; Risitano, A.M.; Lambris, J.D. Expanding complement therapeutics for the treatment of paroxysmal nocturnal hemoglobinuria. Semin. Hematol. 2018, 55, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Lambris, J.D. Therapeutic control of complement activation at the level of the central component C3. Immunobiology 2016, 221, 740–746. [Google Scholar] [CrossRef]
- DiLillo, D.J.; Pawluczkowycz, A.W.; Peng, W.; Kennedy, A.D.; Beum, P.V.; Lindorfer, M.A.; Taylor, R.P. Selective and efficient inhibition of the alternative pathway of complement by a mAb that recognizes C3b/iC3b. Mol. Immunol. 2006, 43, 1010–1019. [Google Scholar] [CrossRef]
- Katschke, K.J., Jr.; Stawicki, S.; Yin, J.; Steffek, M.; Xi, H.; Sturgeon, L.; Hass, P.E.; Loyet, K.M.; DeForge, L.; Wu, Y.; et al. Structural and functional analysis of a C3b-specific antibody that selectively inhibits the alternative pathway of complement. J. Biol. Chem. 2009, 284, 10473–10479. [Google Scholar] [CrossRef] [PubMed]
- Paixão-Cavalcante, D.; Torreira, E.; Lindorfer, M.A.; de Cordoba, S.R.; Morgan, B.P.; Taylor, R.P.; Llorca, O.; Harris, C.L. A humanized antibody that regulates the alternative pathway convertase: Potential for therapy of renal disease associated with nephritic factors. J. Immunol. 2014, 192, 4844–4851. [Google Scholar] [CrossRef] [PubMed]
- Carpanini, S.M.; Torvell, M.; Morgan, B.P. Therapeutic inhibition of the complement system in diseases of the central nervous system. Front. Immunol. 2019, 10, 362. [Google Scholar] [CrossRef] [PubMed]
- Zelek, W.M.; Xie, L.; Morgan, B.P.; Harris, C.L. Compendium of current complement therapeutics. Mol. Immunol. 2019, 114, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Sahu, A.; Kay, B.K.; Lambris, J.D. Inhibition of human complement by a C3-binding peptide isolated from a phage-displayed random peptide library. J. Immunol. 1996, 157, 884–891. [Google Scholar] [PubMed]
- Janssen, B.J.; Halff, E.F.; Lambris, J.D.; Gros, P. Structure of compstatin in complex with complement component C3c reveals a new mechanism of complement inhibition. J. Biol. Chem. 2007, 282, 29241–29247. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.S.; Grossi, F.V.; El Mehdi, D.; Gerber, M.R.; Brown, D.M.; Heier, J.S.; Wykoff, C.C.; Singerman, L.J.; Abraham, P.; Grassmann, F.; et al. Complement C3 inhibitor pegcetacoplan for geographic atrophy secondary to age-related macular degeneration: A randomized Phase 2 trial. Ophthalmology 2020, 127, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Hillmen, P.; Szer, J.; Weitz, I.; Roth, A.; Hochsmann, B.; Panse, J.; Usuki, K.; Griffin, M.; Kiladjian, J.J.; de Castro, C.; et al. Pegcetacoplan versus Eculizumab in Paroxysmal Nocturnal Hemoglobinuria. N. Engl. J. Med. 2021, 384, 1028–1037. [Google Scholar] [CrossRef] [PubMed]
- Apellis Pharmaceuticals. Pipeline. Available online: https://apellis.com/our-science/our-pipeline/ (accessed on 31 January 2022).
- US Food and Drug Administration FDA Approves New Treatment for Adults with Serious Rare Blood Disease. 2021. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-approves-new-treatment-adults-serious-rare-blood-disease (accessed on 31 January 2022).
- Mouratou, B.; Schaeffer, F.; Guilvout, I.; Tello-Manigne, D.; Pugsley, A.P.; Alzari, P.M.; Pecorari, F. Remodeling a DNA-binding protein as a specific in vivo inhibitor of bacterial secretin PulD. Proc. Natl. Acad. Sci. USA 2007, 104, 17983–17988. [Google Scholar] [CrossRef] [PubMed]
- Goux, M.; Becker, G.; Gorré, H.; Dammicco, S.; Desselle, A.; Egrise, D.; Leroi, N.; Lallemand, F.; Bahri, M.A.; Doumont, G.; et al. Nanofitin as a new molecular-imaging agent for the diagnosis of epidermal growth factor receptor over-expressing tumors. Bioconj. Chem. 2017, 28, 2361–2371. [Google Scholar] [CrossRef] [PubMed]
- Marcion, G.; Hermetet, F.; Neiers, F.; Uyanik, B.; Dondaine, L.; Dias, A.M.; da Costa, L.; Moreau, M.; Bellaye, P.; Collin, B.; et al. Nanofitins targeting heat shock protein 110: An innovative immunotherapeutic modality in cancer. Int. J. Cancer 2021, 148, 3019–3031. [Google Scholar] [CrossRef]
- Affilogic. SADEL: Developing the 1st Generation of Oral Biotherapeutics in Inflammatory Bowel Diseases. Available online: https://www.affilogic.com/sadel (accessed on 31 January 2022).
- Loussouarn, A.; Behar, G.; Pecorari, F.; Croyal, M.; Renodon-Corniere, A. Characterization of Affitin proteolytic digestion in biorelevant media and improvement of their stabilities via protein engineering. Sci. Rep. 2020, 10, 19703. [Google Scholar] [CrossRef]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral vector platforms within the gene therapy landscape. Signal Transduct. Target. Ther. 2021, 6, 53. [Google Scholar] [CrossRef] [PubMed]
- Mouratou, B.; Behar, G.; Paillard-Laurance, L.; Colinet, S.; Pecorari, F. Ribosome display for the selection of Sac7d scaffolds. Methods Mol. Biol. 2012, 805, 315–331. [Google Scholar]
- Huet, S.; Gorre, H.; Perrocheau, A.; Picot, J.; Cinier, M. Use of the nanofitin alternative scaffold as a GFP-ready fusion tag. PLoS ONE 2015, 10, e0142304. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Smith, H.O.; Hutchison, C.A., 3rd; Venter, J.C.; Merryman, C. Chemical synthesis of the mouse mitochondrial genome. Nat. Methods 2010, 7, 901–903. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Vagin, A.A.; Steiner, R.A.; Lebedev, A.A.; Potterton, L.; McNicholas, S.; Long, F.; Murshudov, G.N. REFMAC5 dictionary: Organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2184–2195. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Sahu, A.; Morikis, D.; Lambris, J.D. Compstatin, a peptide inhibitor of complement, exhibits species-specific binding to complement component C3. Mol. Immunol. 2003, 39, 557–566. [Google Scholar] [CrossRef]
- Dammicco, S.; Goux, M.; Lemaire, C.; Becker, G.; Bahri, M.A.; Plenevaux, A.; Cinier, M.; Luxena, A. Regiospecific radiolabelling of Nanofitin on Ni magnetic beads with [(18)F]FBEM and in vivo PET studies. Nucl. Med. Biol. 2017, 51, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Heskamp, S.; Raave, R.; Boerman, O.; Rijpkema, M.; Goncalves, V.; Denat, F. (89)Zr-Immuno-Positron emission tomography in oncology: State-of-the-art (89)Zr radiochemistry. Bioconj. Chem. 2017, 28, 2211–2223. [Google Scholar] [CrossRef] [PubMed]
- McCrary, B.S.; Edmondson, S.P.; Shriver, J.W. Hyperthermophile protein folding thermodynamics: Differential scanning calorimetry and chemical denaturation of Sac7d. J. Mol. Biol. 1996, 264, 784–805. [Google Scholar] [CrossRef] [PubMed]








| APL-1030 Binding to: | ka [1/(M·s)] | kd [1/s] | Rmax | KD | X2 [RU2] |
|---|---|---|---|---|---|
| C3 | 8.94 × 104 | 1.42 × 10−4 | NA | 1.59 nM | 20.15 |
| C3b | 8.59 × 104 | 9.55 × 10−5 | NA | 1.11 nM | 14.57 |
| APL-1030 | C3b/C3c | Compstatin |
|---|---|---|
| Gly A345 O | Trp 4 N | |
| Lys 5 Nζ | Asp A373 O | |
| Lys 48 Nζ | Asp A373 Oδ1 | |
| Asp 7 Oδ1 | Thr A374 Oγ | |
| Asp 7 Oδ1 | Val A375 N | |
| Tyr 44 N | Asn A390 Oδ | Ile 1 N, Cys 2 N |
| Tyr 44 O | Asn A390 Nδ | |
| Glu 11 Oε1 | His A392 Nε2 | |
| Trp 42 Nε1 | Leu A455 O | |
| Ile 40 N | Met A457 O | Trp 7 Nε1 |
| Lys 39 Nζ | Asp A458 Oδ2 | |
| Gly 38 O | Arg A459 N | |
| Ser 21 Oγ, Asp 35 Oδ2 | Arg A459 Nη1 | |
| Tyr 24 OH | Asp A491 N | |
| Ser 33 Oγ | Asp A491 Oδ2 | Gln 5 Nε2 |
| Asp A491 Oδ1 | His 10 N |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garlich, J.; Cinier, M.; Chevrel, A.; Perrocheau, A.; Eyerman, D.J.; Orme, M.; Kitten, O.; Scheibler, L. Discovery of APL-1030, a Novel, High-Affinity Nanofitin Inhibitor of C3-Mediated Complement Activation. Biomolecules 2022, 12, 432. https://doi.org/10.3390/biom12030432
Garlich J, Cinier M, Chevrel A, Perrocheau A, Eyerman DJ, Orme M, Kitten O, Scheibler L. Discovery of APL-1030, a Novel, High-Affinity Nanofitin Inhibitor of C3-Mediated Complement Activation. Biomolecules. 2022; 12(3):432. https://doi.org/10.3390/biom12030432
Chicago/Turabian StyleGarlich, Joshua, Mathieu Cinier, Anne Chevrel, Anaëlle Perrocheau, David J. Eyerman, Mark Orme, Olivier Kitten, and Lukas Scheibler. 2022. "Discovery of APL-1030, a Novel, High-Affinity Nanofitin Inhibitor of C3-Mediated Complement Activation" Biomolecules 12, no. 3: 432. https://doi.org/10.3390/biom12030432
APA StyleGarlich, J., Cinier, M., Chevrel, A., Perrocheau, A., Eyerman, D. J., Orme, M., Kitten, O., & Scheibler, L. (2022). Discovery of APL-1030, a Novel, High-Affinity Nanofitin Inhibitor of C3-Mediated Complement Activation. Biomolecules, 12(3), 432. https://doi.org/10.3390/biom12030432