
CNN-XG: A Hybrid Framework for sgRNA On-Target Prediction


Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Sources
2.1.1. Benchmark Dataset
2.1.2. Four Cell Line Independent Datasets
2.1.3. Three Independent Datasets for Generalization Test
2.1.4. SpCas9 Variant Datasets
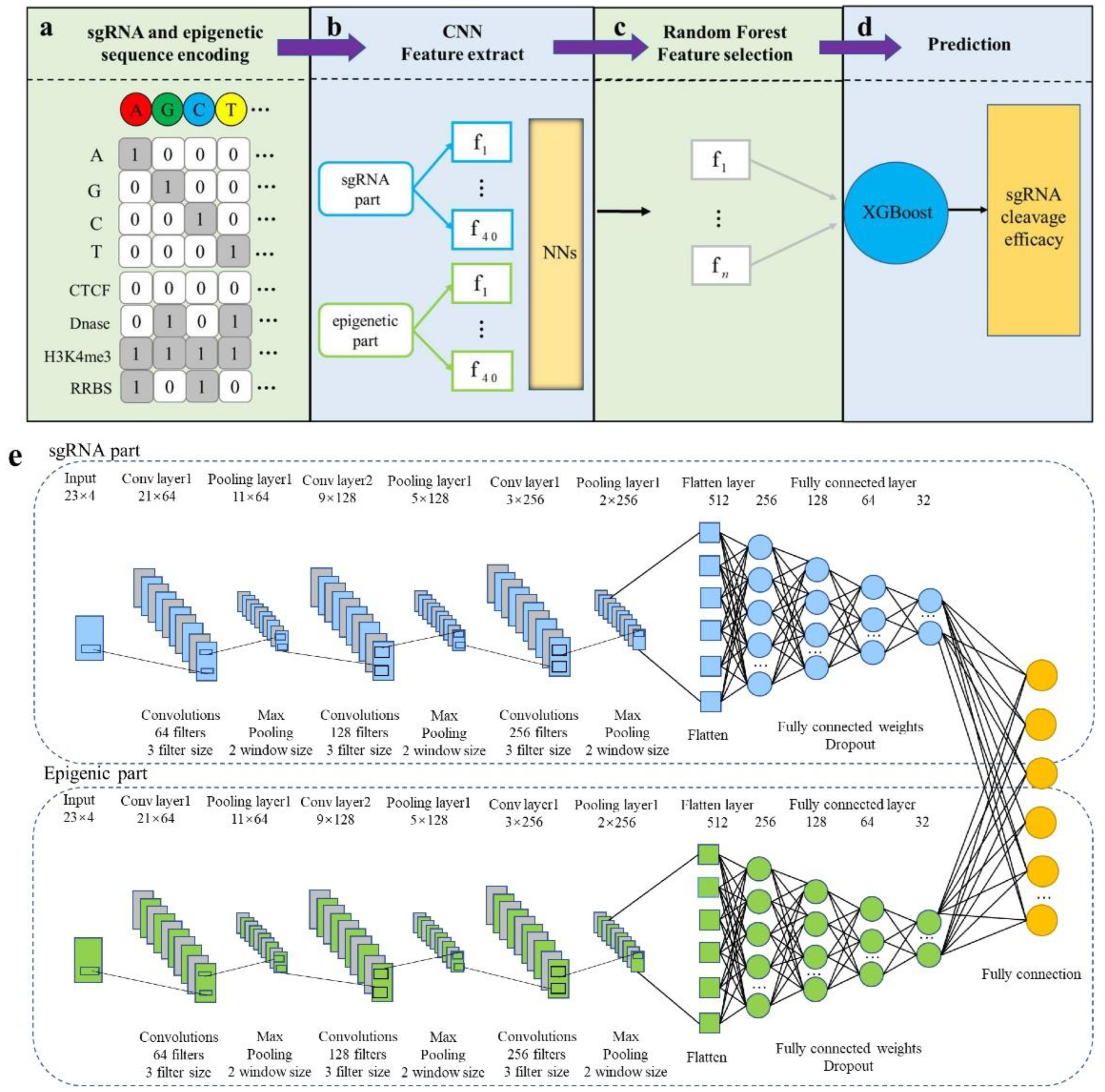
2.2. Design of CNN-XG
2.3. Sequence Encoding
2.4. CNN Model Establishing
2.5. Pre-Training of CNN
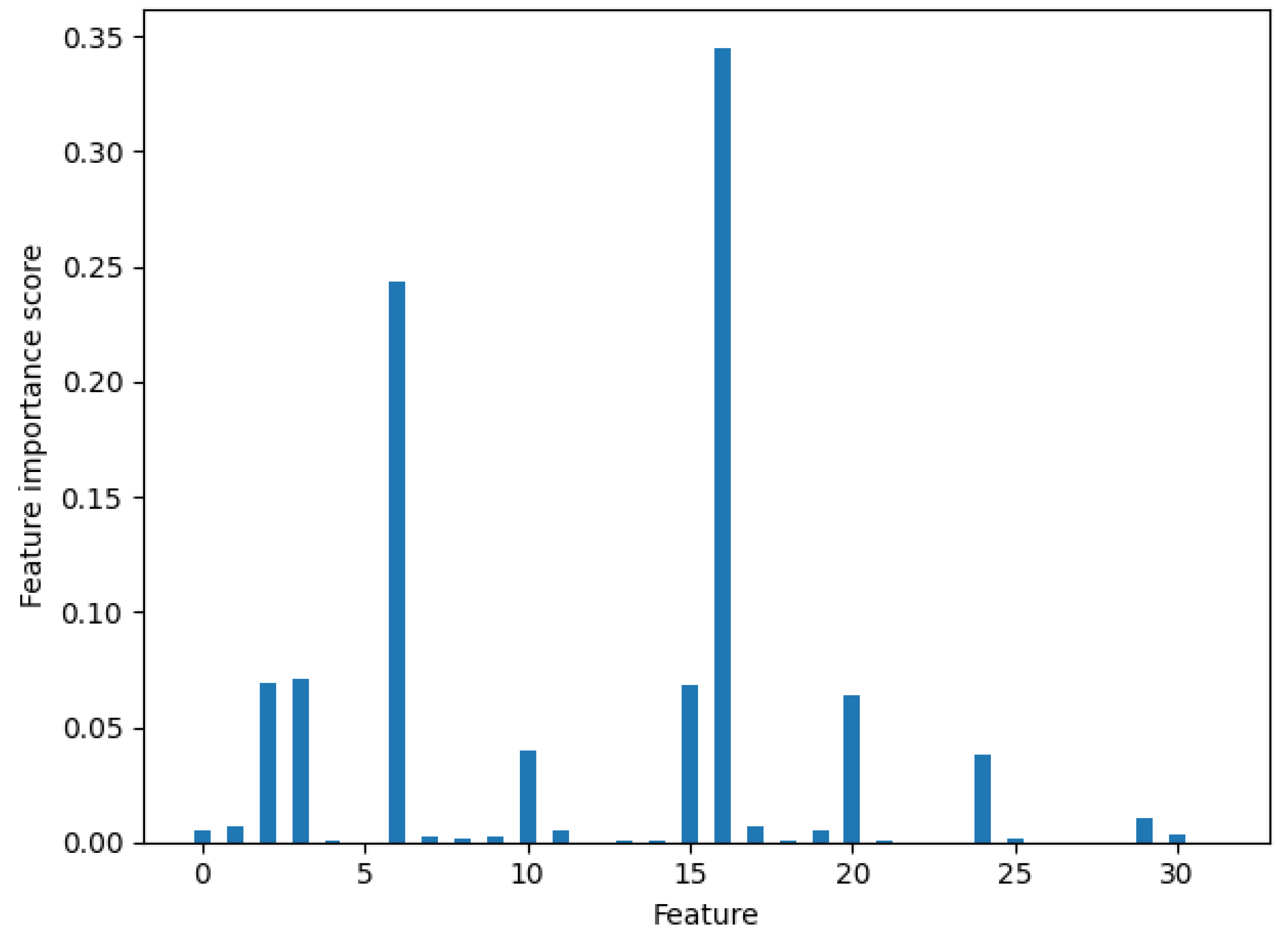
2.6. Feature Representation Optimization
2.7. XGBoost Model Training
3. Results
3.1. Overview of CNN-XG Model Architecture
3.2. Comparison with CNN Model and XGBoost Model
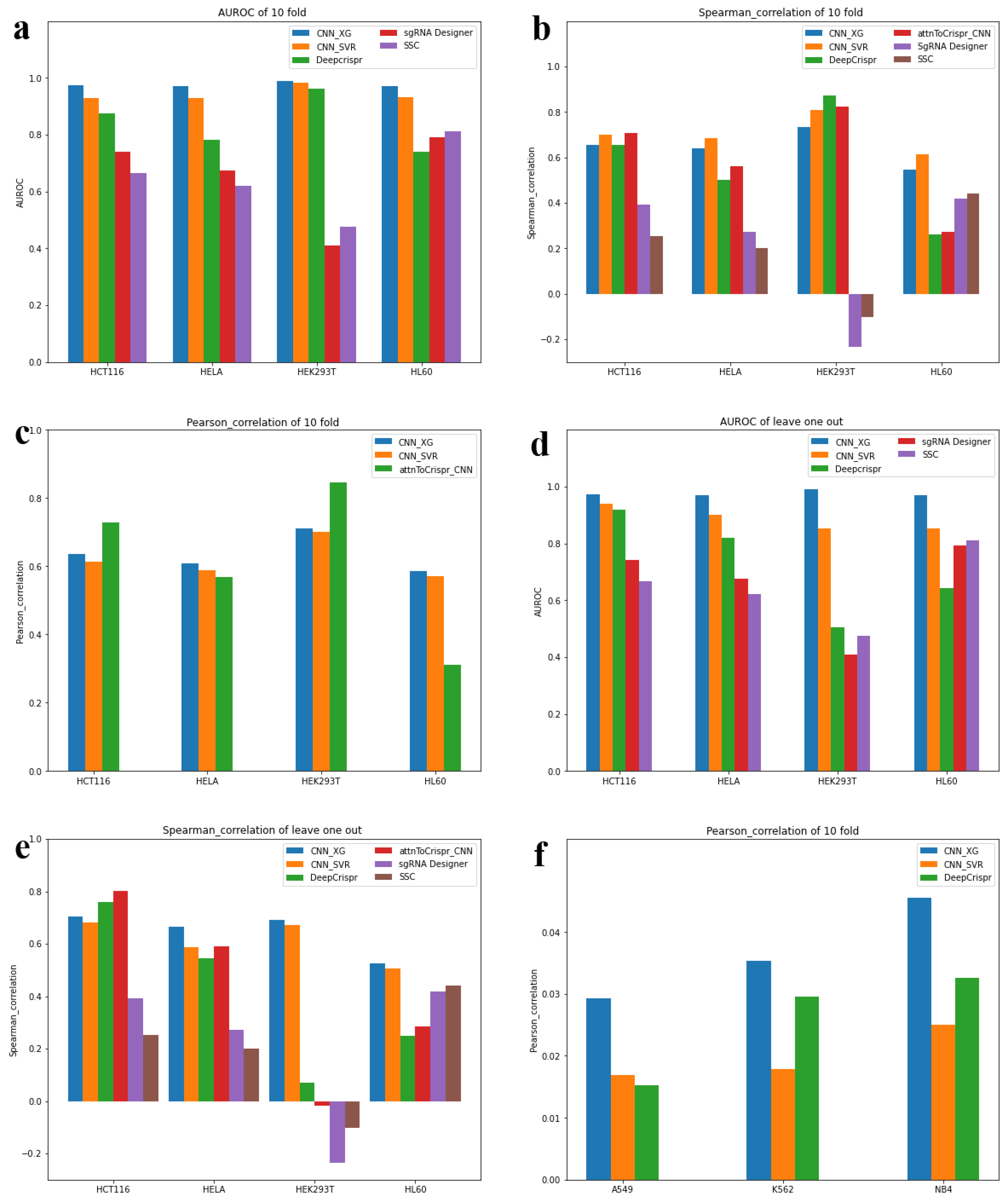
3.3. Model Comparison with State-of-the-Art Methods
3.3.1. Testing Scenario 1—Classification Schema
3.3.2. Testing Scenario 2—Classification Schema
3.3.3. Testing Scenario 3—Regression Schema
3.3.4. Testing Scenario 4—Regression Schema
3.4. Generalization Ability Test on Independent Datasets
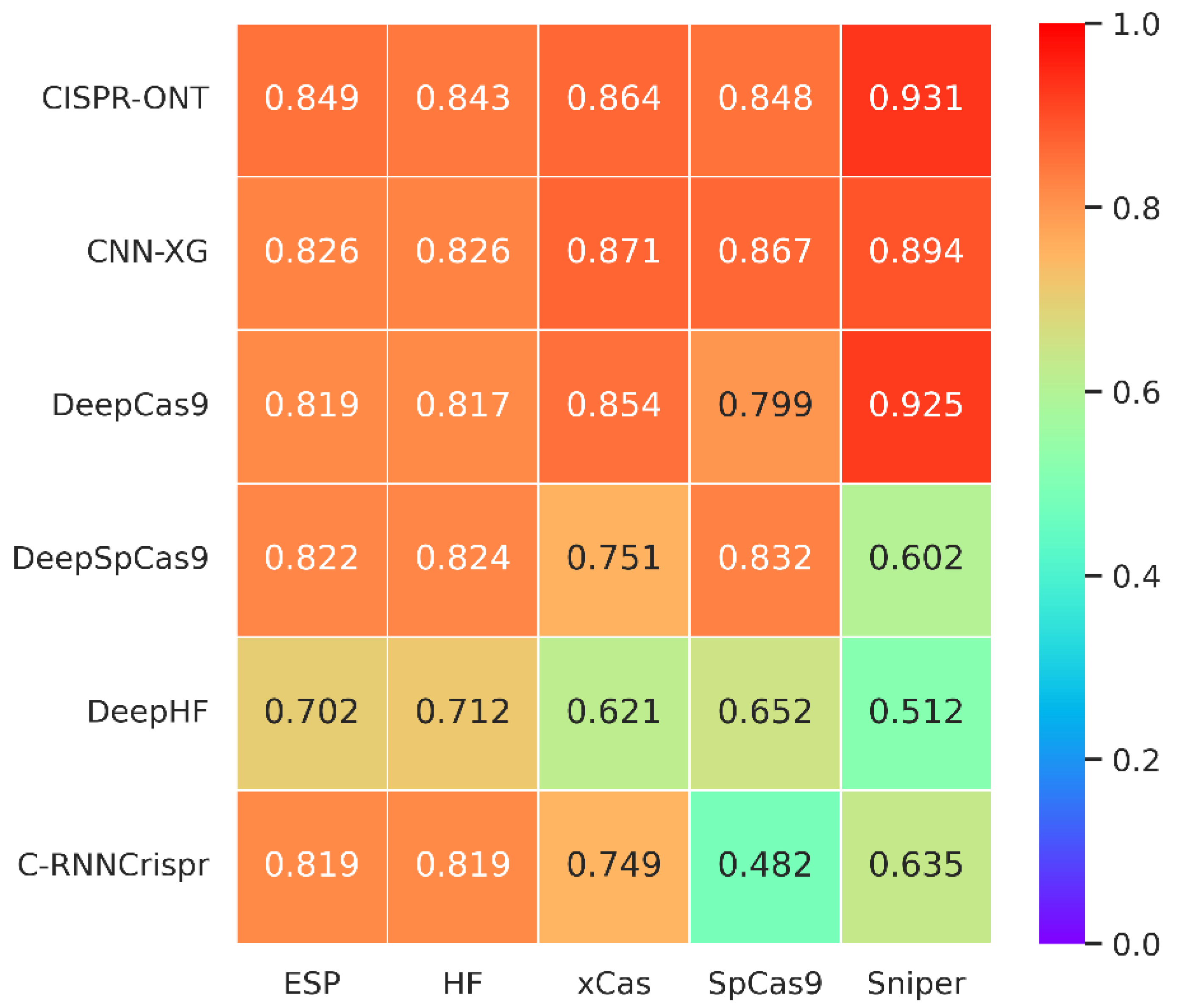
3.5. Comparison in SpCas9 Variant Data
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jansen, R.; Van Embden, J.D.A.; Gaastra, W.; Schouls, L.M. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 2002, 43, 1565–1575. [Google Scholar] [CrossRef]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [Green Version]
- Mojica, F.J.M.; Díez-Villaseñor, C.; García-Martínez, J.; Almendros, C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 2009, 155, 733–740. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Guilinger, J.P.; Thompson, D.B.; Liu, D.R. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat. Biotechnol. 2014, 32, 577–582. [Google Scholar] [CrossRef]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014, 32, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Chuai, G.; Ma, H.; Yan, J.; Chen, M.; Hong, N.; Xue, D.; Zhou, C.; Zhu, C.; Chen, K.; Duan, B.; et al. DeepCRISPR: Optimized CRISPR guide RNA design by deep learning. Genome Biol. 2018, 19, 1–18. [Google Scholar] [CrossRef]
- Kim, H.K.; Min, S.; Song, M.; Jung, S.; Choi, J.W.; Kim, Y.; Lee, S.; Yoon, S.; Kim, H.H. Deep learning improves prediction of CRISPR–Cpf1 guide RNA activity. Nat. Biotechnol. 2018, 36, 239–241. [Google Scholar] [CrossRef]
- Liu, Q.; He, D.; Xie, L. Prediction of off-target specificity and cell-specific fitness of CRISPR-Cas System using attention boosted deep learning and network-based gene feature. PLOS Comput. Biol. 2019, 15, e1007480. [Google Scholar] [CrossRef]
- Liu, Q.; Cheng, X.; Liu, G.; Li, B.; Liu, X. Deep learning improves the ability of sgRNA off-target propensity prediction. BMC Bioinform. 2020, 21, 51. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Dai, Z.; Dai, X. A Novel Hybrid CNN-SVR for CRISPR/Cas9 Guide RNA Activity Prediction. Front. Genet. 2020, 10, 1303. [Google Scholar] [CrossRef]
- Rahman, K.; Rahman, M.S. CRISPRpred: A flexible and efficient tool for sgRNAs on-target activity prediction in CRISPR/Cas9 systems. PLoS ONE 2017, 12, e0181943. [Google Scholar] [CrossRef]
- Lin, J.; Wong, K. Off-target predictions in CRISPR-Cas9 gene editing using deep learning. Bioinformatics 2018, 34, i656–i663. [Google Scholar] [CrossRef] [Green Version]
- Xue, L.; Tang, B.; Chen, W.; Luo, J. Prediction of CRISPR sgRNA Activity Using a Deep Convolutional Neural Network. J. Chem. Inf. Model. 2018, 59, 615–624. [Google Scholar] [CrossRef]
- Listgarten, J.; Weinstein, M.; Kleinstiver, B.; Sousa, A.A.; Joung, J.K.; Crawford, J.; Gao, K.; Hoang, L.; Elibol, M.; Doench, J.G.; et al. Prediction of off-target activities for the end-to-end design of CRISPR guide RNAs. Nat. Biomed. Eng. 2018, 2, 38–47. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, J. Prediction of sgRNA on-target activity in bacteria by deep learning. BMC Bioinform. 2019, 20, 517. [Google Scholar] [CrossRef] [Green Version]
- Lecun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436. [Google Scholar] [CrossRef]
- Huang, F.J.; LeCun, Y. Large-scale Learning with SVM and Convolutional for Generic Object Categorization. In Proceedings of the IEEE Computer Society Conference on Computer Vision and Pattern Recognition, New York, NY, USA, 17–22 June 2006. [Google Scholar]
- Chen, T.; Guestrin, C.E. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar]
- Schroff, F.; Kalenichenko, D.; Philbin, J. FaceNet: A unified embedding for face recognition and clustering. In Proceedings of the 2015 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Boston, MA, USA, 7–12 June 2015; pp. 815–823. [Google Scholar]
- Ma, X.; Sha, J.; Wang, D.; Yu, Y.; Yang, Q.; Niu, X. Study on a prediction of P2P network loan default based on the machine learning LightGBM and XGboost algorithms according to different high dimensional data cleaning. Electron. Commer. Res. Appl. 2018, 31, 24–39. [Google Scholar] [CrossRef]
- Ren, X.; Guo, H.; Li, S.; Wang, S.; Li, J. A Novel Image Classification Method with CNN-XGBoost Model. In Proceedings of the International Workshop on Digital Watermarking, Magdeburg, Germany, 23–25 August 2017. [Google Scholar]
- Doench, J.G.; Hartenian, E.; Graham, D.B.; Tothova, Z.; Hegde, M.; Smith, I.; Sullender, M.; Ebert, B.L.; Xavier, R.J.; Root, D.E. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat. Biotechnol. 2014, 32, 1262–1267. [Google Scholar] [CrossRef] [Green Version]
- ENCODE Project Consortium. The ENCODE (ENCyclopedia of DNA Elements) Project. Science 2004, 306, 636–640. [Google Scholar] [CrossRef] [Green Version]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, A.J.; Meyers, R.M.; Weir, B.A.; Vazquez, F.; Zhang, C.-Z.; Ben-David, U.; Cook, A.; Ha, G.; Harrington, W.F.; Doshi, M.B.; et al. Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov. 2016, 6, 914–929. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zhang, C.; Wang, B.; Li, B.; Wang, Q.; Liu, D.; Wang, H.; Zhou, Y.; Shi, L.; Lan, F.; et al. Optimized CRISPR guide RNA design for two high-fidelity Cas9 variants by deep learning. Nat. Commun. 2019, 10, 4284. [Google Scholar] [CrossRef]
- Kim, N.; Kim, H.K.; Lee, S.; Seo, J.H.; Choi, J.W.; Park, J.; Min, S.; Yoon, S.; Cho, S.-R.; Kim, H.H. Prediction of the sequence-specific cleavage activity of Cas9 variants. Nat. Biotechnol. 2020, 38, 1328–1336. [Google Scholar] [CrossRef]
- Whitney, A.W. A Direct Method of Nonparametric Measurement Selection. IEEE Trans. Comput. 1971, -20, 1100–1103. [Google Scholar] [CrossRef]
- Xu, H.; Xiao, T.; Chen, C.-H.; Li, W.; Meyer, C.A.; Wu, Q.; Wu, D.; Cong, L.; Zhang, F.; Liu, J.S.; et al. Sequence determinants of improved CRISPR sgRNA design. Genome Res. 2015, 25, 1147–1157. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef] [Green Version]
- Kearns, M.; Valiant, L.G. Cryptographic limitations on learning Boolean formulae and finite automata. J. Assoc. Comput. Mach. 1989, 41, 67–95. [Google Scholar]
- Zhang, G.; Zeng, T.; Dai, Z.; Dai, X. Prediction of CRISPR/Cas9 single guide RNA cleavage efficiency and specificity by attention-based convolutional neural networks. Comput. Struct. Biotechnol. J. 2021, 19, 1445–1457. [Google Scholar] [CrossRef]
- Zhang, G.; Dai, Z.; Dai, X. C-RNNCrispr: Prediction of CRISPR/Cas9 sgRNA activity using convolutional and recurrent neural networks. Comput. Struct. Biotechnol. J. 2020, 18, 344–354. [Google Scholar] [CrossRef]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | CNN-XG | CNN | XGBoost | CNN-XG | CNN | XGBoost |
|---|---|---|---|---|---|---|
| Spearman | AUROC | |||||
| HCT116 | 0.6548 | 0.6453 | 0.3112 | 0.9732 | 0.9208 | 0.6231 |
| HEK293T | 0.7352 | 0.7252 | 0.1557 | 0.9905 | 0.9716 | 0.5213 |
| HELA | 0.6397 | 0.6308 | 0.3273 | 0.9714 | 0.9163 | 0.6377 |
| HL60 | 0.5473 | 0.5470 | 0.3664 | 0.9706 | 0.9197 | 0.7110 |
| Model | CNN-XG | CNN-SVR | DeepCrispr | SgRNA Designer | SSC |
|---|---|---|---|---|---|
| HCT116 | 0.9732 | 0.9304 | 0.874 | 0.741 | 0.666 |
| HEK293T | 0.9905 | 0.9834 | 0.961 | 0.41 | 0.476 |
| HELA | 0.9714 | 0.9296 | 0.782 | 0.675 | 0.621 |
| HL60 | 0.9706 | 0.9309 | 0.739 | 0.792 | 0.811 |
| Model | CNN-XG | CNN-SVR | DeepCrispr | SgRNA Designer | SSC |
|---|---|---|---|---|---|
| HCT116 | 0.9721 | 0.939 | 0.919 | 0.741 | 0.666 |
| HEK293T | 0.992 | 0.852 | 0.506 | 0.41 | 0.476 |
| HELA | 0.9695 | 0.9024 | 0.82 | 0.675 | 0.621 |
| HL60 | 0.9708 | 0.8526 | 0.643 | 0.792 | 0.811 |
| Model | CNN-XG | CNN-SVR | DeepCrispr | SgRNA Designer | attnToCrispr _CNN | SSC |
|---|---|---|---|---|---|---|
| HCT116 | 0.6548 | 0.6998 | 0.654 | 0.391 | 0.707 | 0.252 |
| HEK293T | 0.7352 | 0.8095 | 0.874 | −0.24 | 0.824 | −0.10 |
| HELA | 0.6397 | 0.6843 | 0.501 | 0.273 | 0.561 | 0.2 |
| HL60 | 0.5473 | 0.6136 | 0.262 | 0.418 | 0.274 | 0.441 |
| Model | CNN-XG | CNN-SVR | AttnToCrispr_CNN |
|---|---|---|---|
| HCT116 | 0.6370 | 0.6125 | 0.728 |
| HEK293T | 0.7098 | 0.7000 | 0.846 |
| HELA | 0.6079 | 0.5878 | 0.568 |
| HL60 | 0.5849 | 0.5719 | 0.311 |
| Model | CNN-XG | CNN-SVR | DeepCrispr | SgRNA Designer | attnToCrispr _CNN | SSC |
|---|---|---|---|---|---|---|
| HCT116 | 0.703 | 0.683 | 0.761 | 0.391 | 0.801 | 0.252 |
| HEK293T | 0.691 | 0.673 | 0.069 | −0.24 | −0.02 | −0.10 |
| HELA | 0.665 | 0.586 | 0.544 | 0.273 | 0.591 | 0.2 |
| HL60 | 0.527 | 0.505 | 0.250 | 0.418 | 0.286 | 0.441 |
| Model | CNN-XG | CNN-SVR | DeepCrispr |
|---|---|---|---|
| A549 | 0.0293 1.405 × 10−15 | 0.0169 4.377 × 10−6 | 0.0152 3.737 × 10−5 |
| K562 | 0.0353 2.247 × 10−30 | 0.0178 8.242 × 10−9 | 0.0295 1.002 × 10−21 |
| NB4 | 0.0455 2.671 × 10−35 | 0.0250 1.020 × 10−11 | 0.0326 6.512 × 10−19 |
| Model | ESP | HF | xCas | SpCas9 | Sniper |
|---|---|---|---|---|---|
| CNN-XG | 694 | 601 | 470 | 456 | 725 |
| CRISPR-ONT | 4123 | 2700 | 2616 | 2170 | 2640 |
| C-RNNCrispr | 161,200 | 15,463 | 10,340 | 9266 | 10,410 |
| DeepHF | 37,860 | 36,287 | 24,289 | 19,320 | 24,018 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, B.; Ai, D.; Liu, X. CNN-XG: A Hybrid Framework for sgRNA On-Target Prediction. Biomolecules 2022, 12, 409. https://doi.org/10.3390/biom12030409
Li B, Ai D, Liu X. CNN-XG: A Hybrid Framework for sgRNA On-Target Prediction. Biomolecules. 2022; 12(3):409. https://doi.org/10.3390/biom12030409
Chicago/Turabian StyleLi, Bohao, Dongmei Ai, and Xiuqin Liu. 2022. "CNN-XG: A Hybrid Framework for sgRNA On-Target Prediction" Biomolecules 12, no. 3: 409. https://doi.org/10.3390/biom12030409
APA StyleLi, B., Ai, D., & Liu, X. (2022). CNN-XG: A Hybrid Framework for sgRNA On-Target Prediction. Biomolecules, 12(3), 409. https://doi.org/10.3390/biom12030409