
The Role of Lipids in CRAC Channel Function



Abstract
1. Introduction
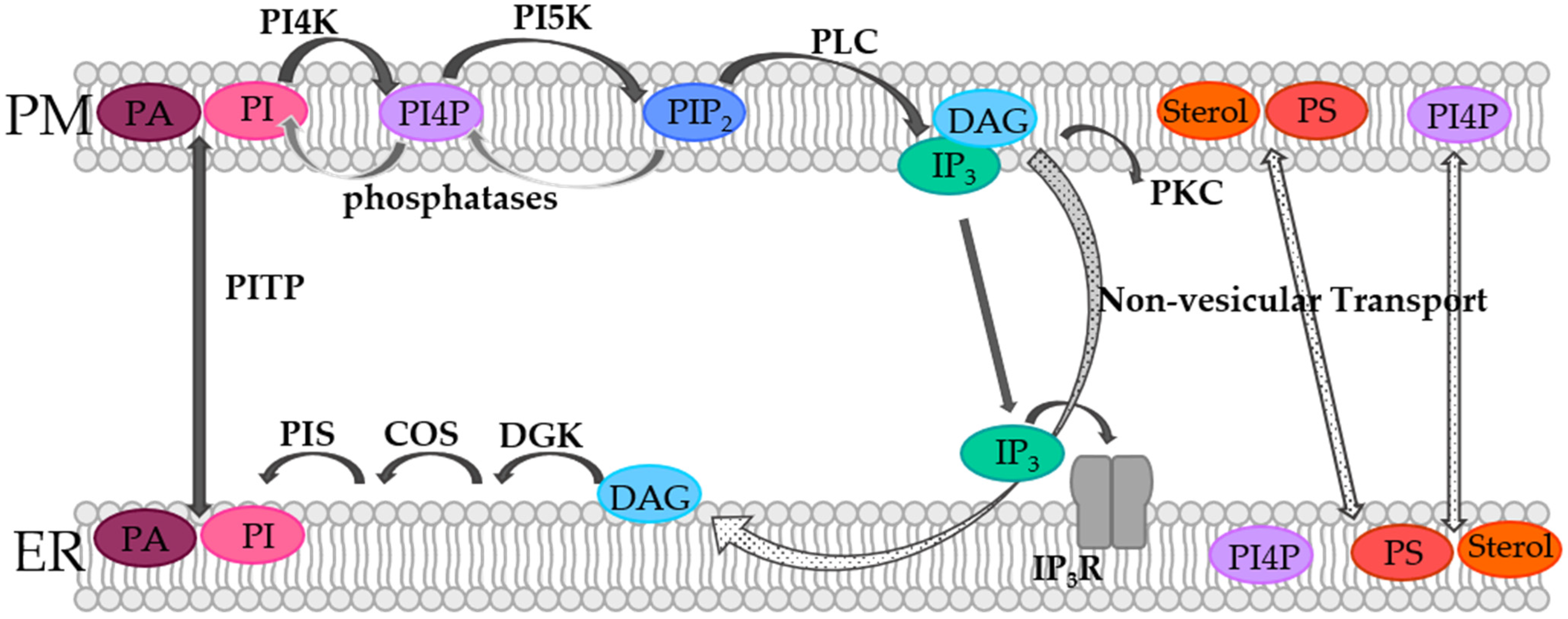
2. Lipid Compositions and Dynamics in the Membranes of the ER and the Cell
3. CRAC Channels
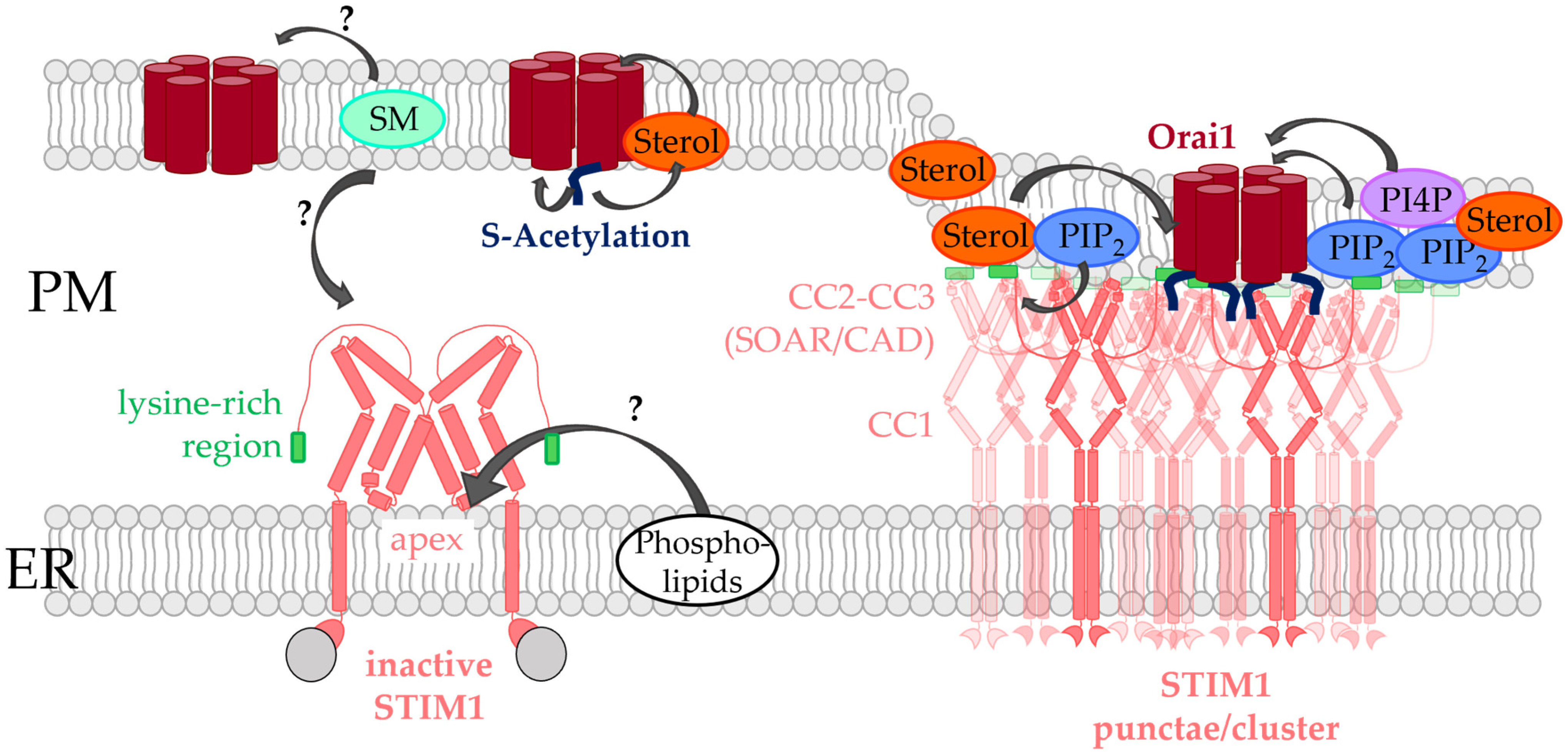
4. Lipids Directly Regulated CRAC Channel Components
4.1. Phosphoinositides
4.2. Cholesterol
4.3. Sphingomyelin
4.4. S-Acylation
4.5. Potential Role of Phospholipids in Controlling STIM1 Function
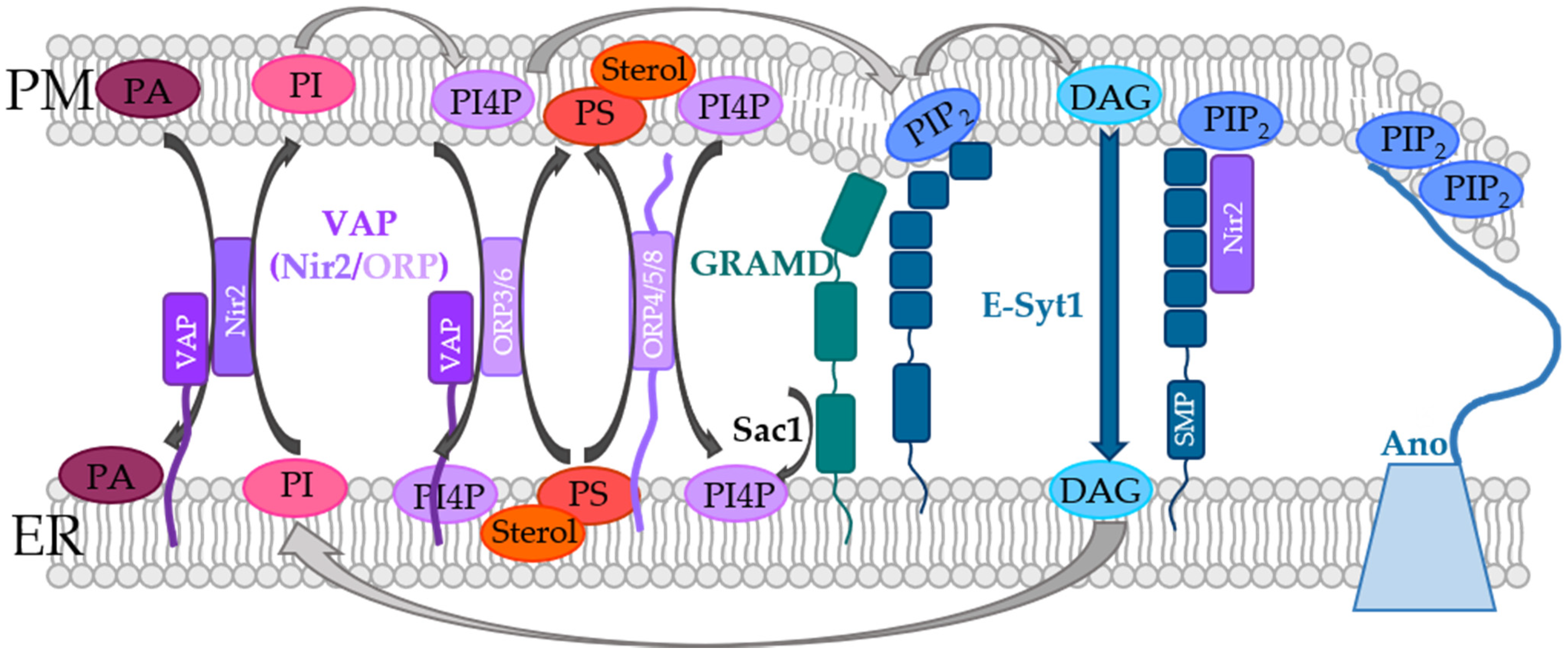
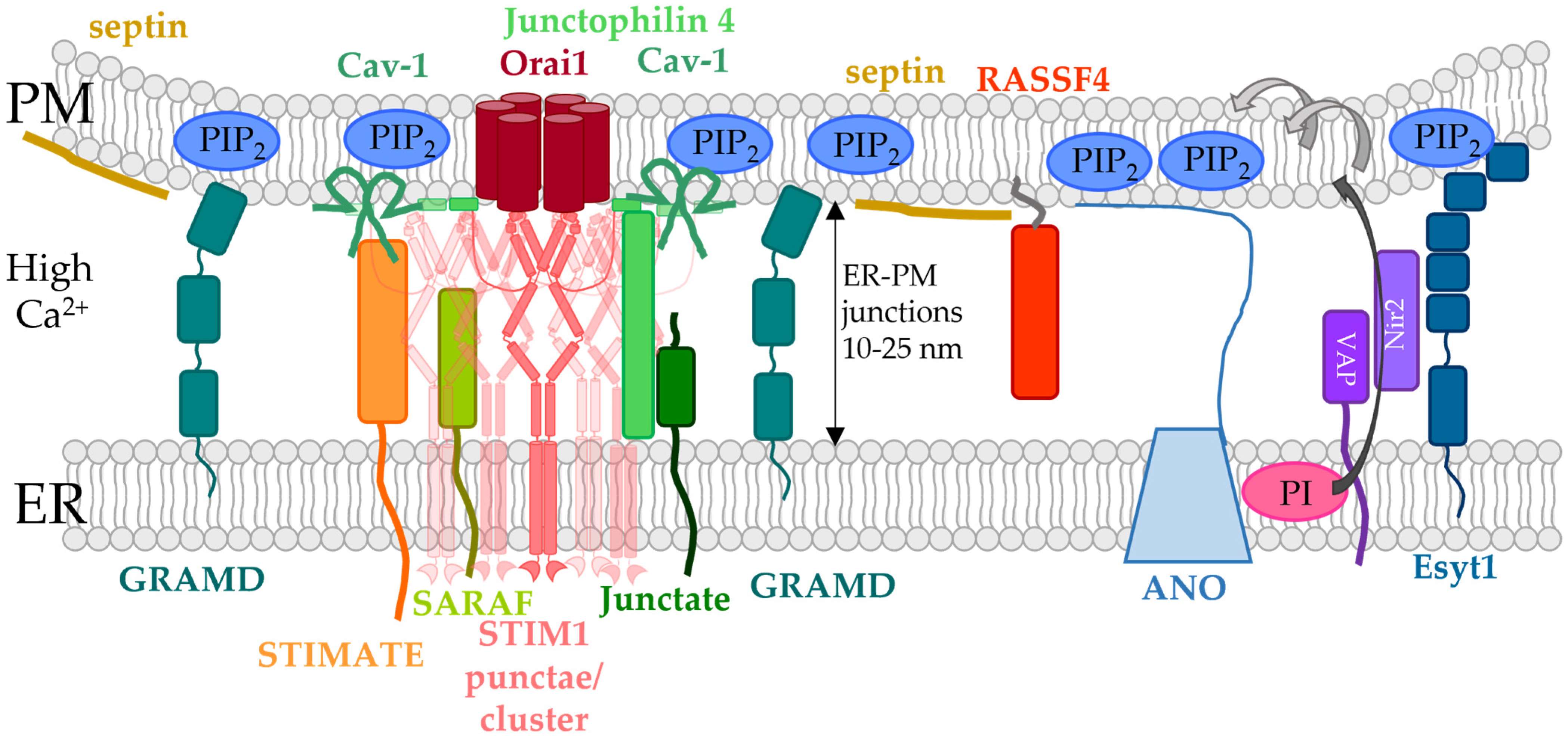
5. Indirect Control of STIM1/Orai1 Machinery at ER-PM Membrane Contact Sites
5.1. ER-PM Constact Sites and Methods for Their Characterization
5.2. Critical Factors Determining STIM1/Orai1 Co-Regulation at the ER-PM Contact Sites
5.3. ER-PM Spanning Proteins Involved in the Modulation of STIM1/Orai1 Function
5.3.1. Extended Synaptotagmins
5.3.2. VAP Proteins
5.3.3. GRAMD2a
5.3.4. ANO8
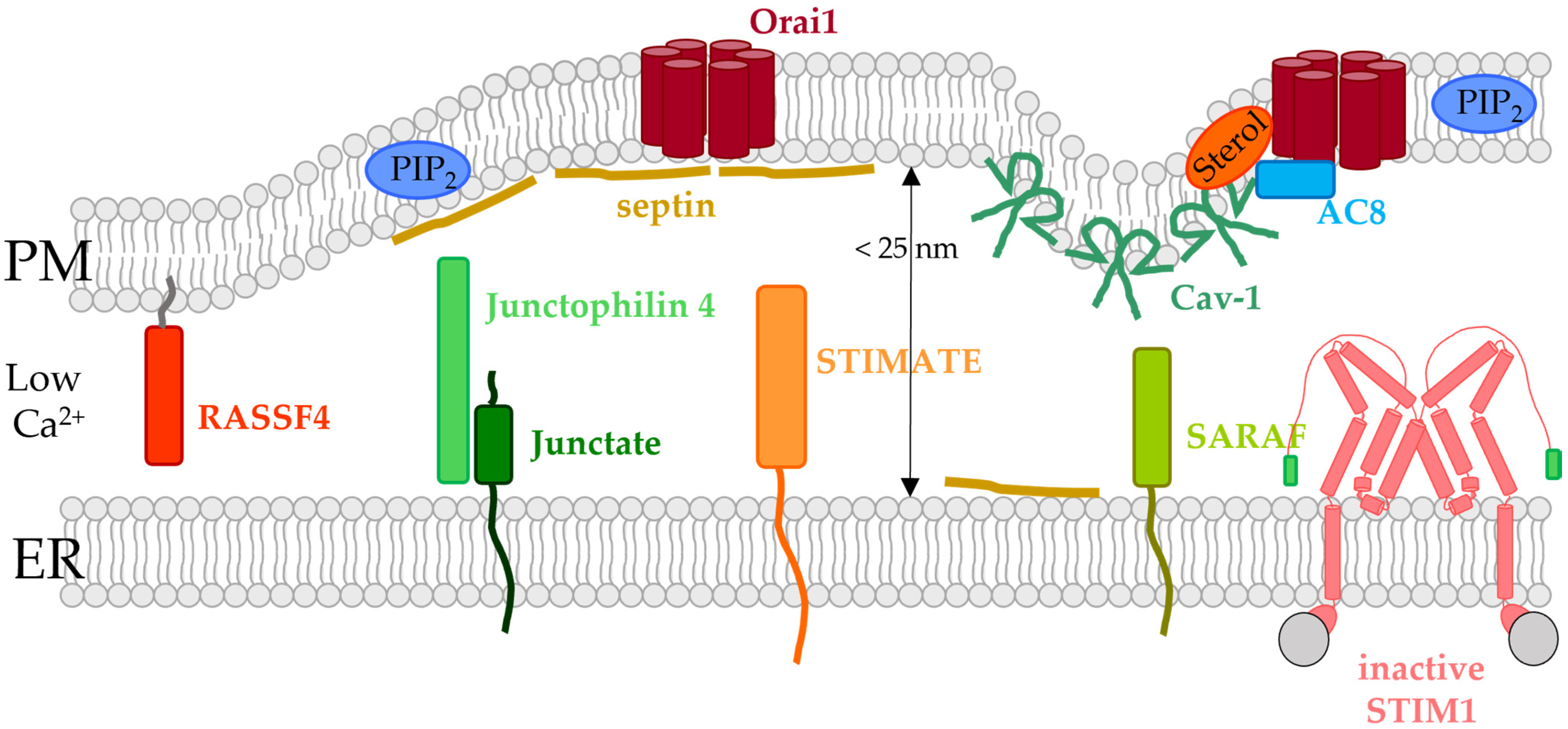
5.4. ER- or PM-Tethered Proteins Located in the ER-PM Junctions and Modulating the STIM1/Orai1 Interplay
5.4.1. Caveolin
5.4.2. Junctate and Junctophilin-4
5.4.3. SARAF
5.4.4. Septin
5.4.5. STIMATE/TMEM110
5.4.6. RASSF4
5.4.7. Adenylyl Cyclase 8 (AC8)
6. Lipid-Mediated Modulation of the Co-Regulation of CRAC Channel Components with Other Ion Channels
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Å | angstrom (unit of length equal to 10−10 m) |
| aa | amino acid |
| AC8 | adenylyl cyclase 8 |
| AKAP | A-kinase anchoring protein |
| ARF6 | adenosine diphosphate ribosylation factor 6 |
| BiFC | bimolecular fluorescence complementation |
| Ca2+ | calcium ion |
| CAD | Ca2+ release-activated Ca2+ activating domain |
| CaM | calmodulin |
| CaMMUT | calmodulin mutant |
| cAMP | cyclic adenosine monophosphate |
| Cav-1 | Caveolin-1 |
| CB | cholesterol-binding |
| CC | coiled-coil |
| Ccb9 | coiled-coil domain containing region b9 |
| CDS | CDP-diacylglycerol synthase |
| CRAC | Ca2+ release-activated Ca2+ |
| cryo-ET | cryo-electron tomography |
| CTID | COOH-terminal inhibitory domain |
| DAG | diacylglycerol |
| DGK | diacylglycerol kinase |
| EM | electron microscopy |
| ER | endoplasmic reticulum |
| ERK | extracellular signaling-related kinase |
| ETON | extended transmembrane Orai1 N-terminal |
| FP | fluorescent proteins |
| GoF | gain of function |
| GTP | guanosine triphosphate |
| HEK | human embryonic kidney |
| hNPCs | human neurons and human neural progenitor cells |
| IL-2 | interleukin-2 |
| IP3 | inositol-triphosphate |
| IP3R | inositol-triphosphate receptor |
| K+ | potassium ion |
| Kir | inward-rectifier potassium channels |
| Kv | voltage-gated K+ |
| LoF | loss of function |
| MßCD | methyl-β-cyclodextrin |
| MCS | membrane contact site |
| MD simulations | molecular dynamics simulations |
| MscL | large-conductance mechanosensitive channel |
| Na+ | sodium ion |
| NFAT | nuclear factor of activated T-cells |
| Nir | PYK2 N-terminal domain-interacting |
| NMR | nuclear magnetic resonance |
| OASF | Orai-activating small fragment |
| ORP | oxysterol-binding proteins |
| PA | phosphatic acid |
| PALM | photoactivated localization microscopy |
| PC | phosphatidylcholine |
| PE | phosphatidylethanolamine |
| PI4K | phosphoinositide 4-kinases |
| PIP2 | phosphatidylinositol 4,5-bisphosphate |
| PKC | protein kinase C |
| PI | phosphatidylinositol |
| PIs | phosphoinositides |
| PIS | PI synthase |
| PIP5K | phosphatidylinositol-4-phosphate-5-kinase |
| PLC | phospholipase C |
| PM | plasma membrane |
| PMCA | plasma membrane Ca2+ pump |
| PS | phosphatidylserine |
| PITP | phosphatidylinositol transfer protein |
| RAS | (RA) association region |
| RBL | rat basophilic leukemia |
| SARAF | store-operated calcium entry-associated regulatory factor |
| SARAH | Salvador-RASSF-Hippo domain |
| SCID | severe combined immune deficiency |
| SERCA | sarcoplasmic/endoplasmic reticulum calcium ATPase |
| SigmaR1 | Sigma non-opioid intracellular receptor 1 |
| SK channels | small-conductance Ca(2+)-activated K(+) channels |
| SM | sphingomyelin |
| SMase | sphingomyelinase |
| SOAP | STIM-Orai association pocket |
| SOAR | STIM-Orai activating region |
| SOC | store-operated channel |
| SOCE | store-operated calcium entry |
| STED | stimulated emission depletion microscopy |
| STIM | stromal interaction molecule |
| STIMATE | STIM-activating enhancer |
| STORM | STochastic optical reconstruction microscopy |
| TIRFM | total internal reflection microscopy |
| TM | transmembrane helices |
| TcB1/2/2 | tricarbins |
| Tyr | tyrosine |
References
- Bondar, A.-N. Introduction: Biomembrane Structure, Dynamics, and Reactions. Chem. Rev. 2019, 119, 5535–5536. [Google Scholar] [CrossRef]
- Stoeckenius, W. Structure of the Plasma Membrane. Circulation 1962, 26, 1066–1069. [Google Scholar] [CrossRef]
- Bangham, A.D. Lipid Bilayers and Biomembranes. Annu. Rev. Biochem. 1972, 41, 753–776. [Google Scholar] [CrossRef]
- Watson, H. Biological membranes. Essays Biochem. 2015, 59, 43–69. [Google Scholar] [CrossRef]
- Divecha, N.; Irvine, R.F. Phospholipid signaling. Cell 1995, 80, 269–278. [Google Scholar] [CrossRef]
- Mitra, K.; Ubarretxena-Belandia, I.; Taguchi, T.; Warren, G.; Engelman, D.M. Modulation of the bilayer thickness of exocytic pathway membranes by membrane proteins rather than cholesterol. Proc. Natl. Acad. Sci. USA 2004, 101, 4083–4088. [Google Scholar] [CrossRef]
- Eddin, L.; Jha, N.; Meeran, M.; Kesari, K.; Beiram, R.; Ojha, S. Neuroprotective Potential of Limonene and Limonene Containing Natural Products. Molecules 2021, 26, 4535. [Google Scholar] [CrossRef]
- Andersen, O.S.; Koeppe, R.E., II. Bilayer Thickness and Membrane Protein Function: An Energetic Perspective. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 107–130. [Google Scholar] [CrossRef]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Griffiths, W.J.; Abdel-Khalik, J.; Yutuc, E.; Morgan, A.H.; Gilmore, I.; Hearn, T.; Wang, Y. Cholesterolomics: An update. Anal. Biochem. 2017, 524, 56–67. [Google Scholar] [CrossRef]
- Fantini, J.; Barrantes, F.J. How cholesterol interacts with membrane proteins: An exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front. Physiol. 2013, 4, 31. [Google Scholar] [CrossRef]
- Dufourc, E.J. Sterols and membrane dynamics. J. Chem. Biol. 2008, 1, 63–77. [Google Scholar] [CrossRef]
- Crul, T.; Maléth, J. Endoplasmic Reticulum-Plasma Membrane Contact Sites as an Organizing Principle for Compartmentalized Calcium and cAMP Signaling. Int. J. Mol. Sci. 2021, 22, 4703. [Google Scholar] [CrossRef]
- Bagheri, Y.; Ali, A.A.; You, M. Current Methods for Detecting Cell Membrane Transient Interactions. Front. Chem. 2020, 8, 603259. [Google Scholar] [CrossRef]
- Poveda, J.; Giudici, A.; Renart, M.; Molina, M.; Montoya, E.; Fernández-Carvajal, A.; Fernández-Ballester, G.; Encinar, J.; González-Ros, J. Lipid modulation of ion channels through specific binding sites. Biochim. Biophys. Acta Biomembr. 2014, 1838, 1560–1567. [Google Scholar] [CrossRef]
- Conrard, L.; Tyteca, D. Regulation of Membrane Calcium Transport Proteins by the Surrounding Lipid Environment. Biomolecules 2019, 9, 513. [Google Scholar] [CrossRef]
- Berkefeld, H.; Fakler, B.; Schulte, U. Ca2+-Activated K+Channels: From Protein Complexes to Function. Physiol. Rev. 2010, 90, 1437–1459. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Lipid rafts as major platforms for signaling regulation in cancer. Adv. Biol. Regul. 2015, 57, 130–146. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Lipid rafts as signaling hubs in cancer cell survival/death and invasion: Implications in tumor progression and therapy. J. Lipid Res. 2020, 61, 611–635. [Google Scholar] [CrossRef]
- Thompson, M.J.; Baenziger, J.E. Ion channels as lipid sensors: From structures to mechanisms. Nat. Chem. Biol. 2020, 16, 1331–1342. [Google Scholar] [CrossRef]
- Chang, G.; Spencer, R.H.; Lee, A.T.; Barclay, M.T.; Rees, D.C. Structure of the MscL Homolog from Mycobacterium tuberculosis: A Gated Mechanosensitive Ion Channel. Science 1998, 282, 2220–2226. [Google Scholar] [CrossRef]
- Guo, Y.R.; MacKinnon, R. Structure-based membrane dome mechanism for Piezo mechanosensitivity. eLife 2017, 6, e33660. [Google Scholar] [CrossRef]
- Brohawn, S.; Campbell, E.B.; MacKinnon, R. Physical mechanism for gating and mechanosensitivity of the human TRAAK K+ channel. Nature 2014, 516, 126–130. [Google Scholar] [CrossRef]
- Hansen, S.B.; Tao, X.; MacKinnon, R. Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature 2011, 477, 495–498. [Google Scholar] [CrossRef]
- Levitan, I.; Fang, Y.; Rosenhouse-Dantsker, A.; Romanenko, V. Cholesterol and Ion Channels. Cholest. Bind. Cholest. Transp. Proteins 2010, 51, 509–549. [Google Scholar] [CrossRef]
- Elevitan, I.; Singh, D.K.; Erosenhouse-Dantsker, A. Cholesterol binding to ion channels. Front. Physiol. 2014, 5, 65. [Google Scholar] [CrossRef]
- Romanenko, V.G.; Fang, Y.; Byfield, F.; Travis, A.J.; Vandenberg, C.A.; Rothblat, G.H.; Levitan, I. Cholesterol Sensitivity and Lipid Raft Targeting of Kir2.1 Channels. Biophys. J. 2004, 87, 3850–3861. [Google Scholar] [CrossRef]
- Barbera, N.; Ayee, M.; Akpa, B.S.; Levitan, I. Molecular Dynamics Simulations of Kir2.2 Interactions with an Ensemble of Cholesterol Molecules. Biophys. J. 2018, 115, 1264–1280. [Google Scholar] [CrossRef]
- Hughes, T.E.T.; Pumroy, R.A.; Yazici, A.T.; Kasimova, M.A.; Fluck, E.C.; Huynh, K.W.; Samanta, A.; Molugu, S.K.; Zhou, Z.H.; Carnevale, V.; et al. Structural insights on TRPV5 gating by endogenous modulators. Nat. Commun. 2018, 9, 4198. [Google Scholar] [CrossRef]
- McGoldrick, L.L.; Singh, A.K.; Saotome, K.; Yelshanskaya, M.V.; Twomey, E.; Grassucci, R.A.; Sobolevsky, A.I. Opening of the human epithelial calcium channel TRPV6. Nature 2017, 553, 233–237. [Google Scholar] [CrossRef]
- Zhang, Z.; Tóth, B.; Szollosi, A.; Chen, J.; Csanády, L. Structure of a TRPM2 channel in complex with Ca2+ explains unique gating regulation. eLife 2018, 7, e36409. [Google Scholar] [CrossRef]
- Yin, Y.; Le, S.C.; Hsu, A.L.; Borgnia, M.J.; Yang, H.; Lee, S.-Y. Structural basis of cooling agent and lipid sensing by the cold-activated TRPM8 channel. Science 2019, 363, eaav9334. [Google Scholar] [CrossRef]
- Zubcevic, L.; Herzik, M.A.; Chung, B.C.; Liu, Z.; Lander, G.C.; Lee, S.-Y. Cryo-electron microscopy structure of the TRPV2 ion channel. Nat. Struct. Mol. Biol. 2016, 23, 180–186. [Google Scholar] [CrossRef]
- Gao, Y.; Cao, E.; Julius, D.; Cheng, Y. TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 2016, 534, 347–351. [Google Scholar] [CrossRef]
- Fan, C.; Choi, W.; Sun, W.; Du, J.; Lü, W. Structure of the human lipid-gated cation channel TRPC3. eLife 2018, 7, e36852. [Google Scholar] [CrossRef]
- Lichtenegger, M.; Tiapko, O.; Svobodova, B.; Stockner, T.; Glasnov, T.; Schreibmayer, W.; Platzer, D.; De La Cruz, G.G.; Krenn, S.; Schober, R.; et al. An optically controlled probe identifies lipid-gating fenestrations within the TRPC3 channel. Nat. Chem. Biol. 2018, 14, 396–404. [Google Scholar] [CrossRef]
- Bai, Y.; Yu, X.; Chen, H.; Horne, D.; White, R.; Wu, X.; Lee, P.; Gu, Y.; Ghimire-Rijal, S.; Lin, D.C.-H.; et al. Structural basis for pharmacological modulation of the TRPC6 channel. eLife 2020, 9, e53311. [Google Scholar] [CrossRef]
- Balse, E.; Boycott, H.E. Ion Channel Trafficking: Control of Ion Channel Density as a Target for Arrhythmias? Front. Physiol. 2017, 8, 808. [Google Scholar] [CrossRef]
- Robinson, C.V.; Rohacs, T.; Hansen, S.B. Tools for Understanding Nanoscale Lipid Regulation of Ion Channels. Trends Biochem. Sci. 2019, 44, 795–806. [Google Scholar] [CrossRef]
- Rosenhouse-Dantsker, A.; Mehta, D.; Levitan, I. Regulation of Ion Channels by Membrane Lipids. Compr. Physiol. 2012, 2, 31–68. [Google Scholar] [CrossRef]
- Harraz, O.F.; Hill-Eubanks, D.; Nelson, M.T. PIP2: A critical regulator of vascular ion channels hiding in plain sight. Proc. Natl. Acad. Sci. USA 2020, 117, 20378–20389. [Google Scholar] [CrossRef]
- Antonny, B.; Vanni, S.; Shindou, H.; Ferreira, T. From zero to six double bonds: Phospholipid unsaturation and organelle function. Trends Cell Biol. 2015, 25, 427–436. [Google Scholar] [CrossRef]
- Lorent, J.H.; Levental, K.R.; Ganesan, L.; Rivera-Longsworth, G.; Sezgin, E.; Doktorova, M.D.; Lyman, E.; Levental, I. Plasma membranes are asymmetric in lipid unsaturation, packing and protein shape. Nat. Chem. Biol. 2020, 16, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Murate, M.; Abe, M.; Kasahara, K.; Iwabuchi, K.; Umeda, M.; Kobayashi, T. Transbilayer lipid distribution in nano scale. J. Cell Sci. 2015, 128, 1627–1638. [Google Scholar] [CrossRef]
- Bretscher, M.S. Asymmetrical Lipid Bilayer Structure for Biological Membranes. Nat. New Biol. 1972, 236, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Verkleij, A.; Zwaal, R.; Roelofsen, B.; Comfurius, P.; Kastelijn, D.; Van Deenen, L. The asymmetric distribution of phospholipids in the human red cell membrane. A combined study using phospholipases and freeze-etch electron microscopy. Biochim. Biophys. Acta Biomembr. 1973, 323, 178–193. [Google Scholar] [CrossRef]
- Schroeder, C. Cholesterol-Binding Viral Proteins in Virus Entry and Morphogenesis. Subcell Biochem. 2010, 51, 77–108. [Google Scholar] [CrossRef]
- Raffy, S.; Teissie, J. Control of Lipid Membrane Stability by Cholesterol Content. Biophys. J. 1999, 76, 2072–2080. [Google Scholar] [CrossRef]
- Doktorova, M.; Symons, J.L.; Levental, I. Structural and functional consequences of reversible lipid asymmetry in living membranes. Nat. Chem. Biol. 2020, 16, 1321–1330. [Google Scholar] [CrossRef]
- Castro, H.; Bermeo, K.; Arenas, I.; Garcia, D.E. Maintenance of CaV2.2 channel-current by PIP2 unveiled by neomycin in sympathetic neurons of the rat. Arch. Biochem. Biophys. 2020, 682, 108261. [Google Scholar] [CrossRef]
- Cheng, K.T.; Ong, H.L.; Liu, X.; Ambudkar, I.S. Contribution and regulation of TRPC channels in store-operated Ca2+ Entry. In Current Topics in Membranes; Academic Press Inc.: Cambridge, MA, USA, 2013; Volume 71, pp. 149–179. [Google Scholar]
- Zaydman, M.A.; Cui, J.; Zaydman, M.A.; Cui, J. PIP2 regulation of KCNQ channels: Biophysical and molecular mechanisms for lipid modulation of voltage-dependent gating. Front. Physiol. 2014, 5, 195. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Meng, X.-Y.; Cui, M.; Pascal, J.M.; Logothetis, D.E.; Zhang, J.-F. Selective phosphorylation modulates the PIP2 sensitivity of the CaM–SK channel complex. Nat. Chem. Biol. 2014, 10, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Liepiņa, I.; Czaplewski, C.; Janmey, P.; Liwo, A. Molecular dynamics study of a gelsolin-derived peptide binding to a lipid bilayer containing phosphatidylinositol 4,5-bisphosphate. Biopolymers 2003, 71, 49–70. [Google Scholar] [CrossRef] [PubMed]
- McLean, M.A.; Stephen, A.G.; Sligar, S.G. PIP2 Influences the Conformational Dynamics of Membrane-Bound KRAS4b. Biochemistry 2019, 58, 3537–3545. [Google Scholar] [CrossRef] [PubMed]
- Amano, T.; Tanabe, K.; Eto, T.; Narumiya, S.; Mizuno, K. LIM-kinase 2 induces formation of stress fibres, focal adhesions and membrane blebs, dependent on its activation by Rho-associated kinase-catalysed phosphorylation at threonine-505. Biochem. J. 2001, 354, 149–159. [Google Scholar] [CrossRef]
- Picas, L.; Viaud, J.; Schauer, K.; Vanni, S.; Hnia, K.; Fraisier, V.; Roux, A.; Bassereau, P.; Gaits-Iacovoni, F.; Payrastre, B.; et al. BIN1/M-Amphiphysin2 induces clustering of phosphoinositides to recruit its downstream partner dynamin. Nat. Commun. 2014, 5, 5647. [Google Scholar] [CrossRef]
- Mandal, K. Review of PIP2 in Cellular Signaling, Functions and Diseases. Int. J. Mol. Sci. 2020, 21, 8342. [Google Scholar] [CrossRef]
- Downes, C.P.; Gray, A.; Lucocq, J. Probing phosphoinositide functions in signaling and membrane trafficking. Trends Cell Biol. 2005, 15, 259–268. [Google Scholar] [CrossRef]
- Rameh, L.E.; Tolias, K.; Duckworth, B.C.; Cantley, L. A new pathway for synthesis of phosphatidylinositol-4,5-bisphosphate. Nature 1997, 390, 192–196. [Google Scholar] [CrossRef]
- Viaud, J.; Mansour, R.; Antkowiak, A.; Mujalli, A.; Valet, C.; Chicanne, G.; Xuereb, J.M.; Terrisse, A.D.; Severin, S.; Gratacap, M.-P.; et al. Phosphoinositides: Important lipids in the coordination of cell dynamics. Biochimie 2016, 125, 250–258. [Google Scholar] [CrossRef]
- Picas, L.; Gaits-Iacovoni, F.; Goud, B. The emerging role of phosphoinositide clustering in intracellular trafficking and signal transduction. F1000Research 2016, 5, 422. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M. Features of the Phosphatidylinositol Cycle and its Role in Signal Transduction. J. Membr. Biol. 2016, 250, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Stefan, C.J. Endoplasmic reticulum–plasma membrane contacts: Principals of phosphoinositide and calcium signaling. Curr. Opin. Cell Biol. 2020, 63, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Tuosto, L.; Capuano, C.; Muscolini, M.; Santoni, A.; Galandrini, R. The multifaceted role of PIP2 in leukocyte biology. Cell Mol. Life Sci. 2015, 72, 4461–4474. [Google Scholar] [CrossRef]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.-H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef]
- Hoth, M.; Niemeyer, B.A. The Neglected CRAC Proteins: Orai2, Orai3, and STIM2. Curr. Top. Membr. 2013, 71, 237–271. [Google Scholar] [CrossRef]
- Zhang, X.; Xin, P.; Yoast, R.E.; Emrich, S.M.; Johnson, M.T.; Pathak, T.; Benson, J.C.; Azimi, I.; Gill, D.L.; Monteith, G.R.; et al. Distinct pharmacological profiles of ORAI1, ORAI2, and ORAI3 channels. Cell Calcium 2020, 91, 102281. [Google Scholar] [CrossRef]
- Yoast, R.E.; Emrich, S.M.; Trebak, M. The anatomy of native CRAC channel(s). Curr. Opin. Physiol. 2020, 17, 89–95. [Google Scholar] [CrossRef]
- Ong, H.L.; Subedi, K.P.; Son, G.-Y.; Liu, X.; Ambudkar, I.S. Tuning store-operated calcium entry to modulate Ca2+-dependent physiological processes. Biochim. Biophys. Acta 2018, 1866, 1037–1045. [Google Scholar] [CrossRef]
- Subedi, K.P.; Ong, H.L.; Son, G.-Y.; Liu, X.; Ambudkar, I.S. STIM2 Induces Activated Conformation of STIM1 to Control Orai1 Function in ER-PM Junctions. Cell Rep. 2018, 23, 522–534. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.; Kim, M.L.; Do Heo, W.; Jones, J.T.; Myers, J.W.; Ferrell, J.E., Jr.; Meyer, T. STIM Is a Ca2+ Sensor Essential for Ca2+-Store-Depletion-Triggered Ca2+ Influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.M.; Buchanan, J.; Luik, R.M.; Lewis, R.S. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J. Cell Biol. 2006, 174, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Gudlur, A.; Zeraik, A.E.; Hirve, N.; Rajanikanth, V.; Bobkov, A.A.; Ma, G.; Zheng, S.; Wang, Y.; Zhou, Y.; Komives, E.A.; et al. Calcium sensing by the STIM1 ER-luminal domain. Nat. Commun. 2018, 9, 4536. [Google Scholar] [CrossRef] [PubMed]
- Schober, R.; Bonhenry, D.; Lunz, V.; Zhu, J.; Krizova, A.; Frischauf, I.; Fahrner, M.; Zhang, M.; Waldherr, L.; Schmidt, T.; et al. Sequential activation of STIM1 links Ca 2+ with luminal domain unfolding. Sci. Signal. 2019, 12, aax3194. [Google Scholar] [CrossRef] [PubMed]
- Sallinger, M.; Tiffner, A.; Schmidt, T.; Bonhenry, D.; Waldherr, L.; Frischauf, I.; Lunz, V.; Derler, I.; Schober, R.; Schindl, R. Luminal STIM1 Mutants that Cause Tubular Aggregate Myopathy Promote Autophagic Processes. Int. J. Mol. Sci. 2020, 21, 4410. [Google Scholar] [CrossRef]
- Yang, X.; Jin, H.; Cai, X.; Li, S.; Shen, Y. Structural and mechanistic insights into the activation of Stromal interaction molecule 1 (STIM1). Proc. Natl. Acad. Sci. USA 2012, 109, 5657–5662. [Google Scholar] [CrossRef]
- Fahrner, M.; Muik, M.; Schindl, R.; Butorac, C.; Stathopulos, P.; Zheng, L.; Jardin, I.; Ikura, M.; Romanin, C. A Coiled-coil Clamp Controls Both Conformation and Clustering of Stromal Interaction Molecule 1 (STIM1). J. Biol. Chem. 2014, 289, 33231–33244. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Stathopulos, P.B.; Schindl, R.; Li, G.-Y.; Romanin, C.; Ikura, M. Auto-inhibitory role of the EF-SAM domain of STIM proteins in store-operated calcium entry. Proc. Natl. Acad. Sci USA. 2011, 108, 1337–1342. [Google Scholar] [CrossRef]
- Ma, G.; Wei, M.; He, L.; Liu, C.; Wu, B.; Zhang, S.L.; Jing, J.; Liang, X.; Senes, A.; Tan, P.; et al. Inside-out Ca2+ signalling prompted by STIM1 conformational switch. Nat. Commun. 2015, 6, 7826. [Google Scholar] [CrossRef]
- Hirve, N.; Rajanikanth, V.; Hogan, P.G.; Gudlur, A. Coiled-Coil Formation Conveys a STIM1 Signal from ER Lumen to Cytoplasm. Cell Rep. 2018, 22, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Höglinger, C.; Grabmayr, H.; Maltan, L.; Horvath, F.; Krobath, H.; Muik, M.; Tiffner, A.; Renger, T.; Romanin, C.; Fahrner, M.; et al. Defects in the STIM1 SOARα2 domain affect multiple steps in the CRAC channel activation cascade. Experientia 2021, 78, 6645–6667. [Google Scholar] [CrossRef] [PubMed]
- van Dorp, S.; Qiu, R.; Choi, U.B.; Wu, M.M.; Yen, M.; Kirmiz, M.; Brunger, A.T.; Lewis, R.S. Conformational dynamics of auto-inhibition in the ER calcium sensor STIM1. eLife 2021, 10, e66194. [Google Scholar] [CrossRef] [PubMed]
- Frischauf, I.; Muik, M.; Derler, I.; Bergsmann, J.; Fahrner, M.; Schindl, R.; Groschner, K.; Romanin, C. Molecular Determinants of the Coupling between STIM1 and Orai Channels: Differential Activation of Orai1–3 Channels by a Stim1 Coiled-Coil Mutant. J. Biol. Chem. 2009, 284, 21696–21706. [Google Scholar] [CrossRef]
- Muik, M.; Fahrner, M.; Derler, I.; Schindl, R.; Bergsmann, J.; Frischauf, I.; Groschner, K.; Romanin, C. A Cytosolic Homomerization and a Modulatory Domain within STIM1 C Terminus Determine Coupling to ORAI1 Channels. J. Biol. Chem. 2009, 284, 8421–8426. [Google Scholar] [CrossRef]
- Yuan, J.P.; Zeng, W.; Dorwart, M.R.; Choi, Y.-J.; Worley, P.F.; Muallem, S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 2009, 11, 337–343. [Google Scholar] [CrossRef]
- Park, C.Y.; Hoover, P.J.; Mullins, F.M.; Bachhawat, P.; Covington, E.D.; Raunser, S.; Walz, T.; Garcia, K.C.; Dolmetsch, R.E.; Lewis, R.S. STIM1 Clusters and Activates CRAC Channels via Direct Binding of a Cytosolic Domain to Orai1. Cell 2009, 136, 876–890. [Google Scholar] [CrossRef]
- Kawasaki, T.; Ueyama, T.; Lange, I.; Feske, S.; Saito, N. Protein Kinase C-induced Phosphorylation of Orai1 Regulates the Intracellular Ca2+ Level via the Store-operated Ca2+ Channel. J. Biol. Chem. 2010, 285, 25720–25730. [Google Scholar] [CrossRef]
- Stathopulos, P.; Schindl, R.; Fahrner, M.; Zheng, L.; Gasmi-Seabrook, G.M.; Muik, M.; Romanin, C.; Ikura, M. STIM1/Orai1 coiled-coil interplay in the regulation of store-operated calcium entry. Nat. Commun. 2013, 4, 2963. [Google Scholar] [CrossRef]
- Muik, M.; Frischauf, I.; Derler, I.; Fahrner, M.; Bergsmann, J.; Eder, P.; Schindl, R.; Hesch, C.; Polzinger, B.; Fritsch, R.; et al. Dynamic Coupling of the Putative Coiled-coil Domain of ORAI1 with STIM1 Mediates ORAI1 Channel Activation. J. Biol. Chem. 2008, 283, 8014–8022. [Google Scholar] [CrossRef]
- Li, Z.; Lu, J.; Xu, P.; Xie, X.; Chen, L.; Xu, T. Mapping the Interacting Domains of STIM1 and Orai1 in Ca2+ Release-activated Ca2+ Channel Activation. J. Biol. Chem. 2007, 282, 29448–29456. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Zhou, Y.; Hendron, E.; Mancarella, S.; Andrake, M.D.; Rothberg, B.; Soboloff, J.; Gill, D.L. Distinct Orai-coupling domains in STIM1 and STIM2 define the Orai-activating site. Nat. Commun. 2014, 5, 3183. [Google Scholar] [CrossRef]
- Butorac, C.; Muik, M.; Derler, I.; Stadlbauer, M.; Lunz, V.; Krizova, A.; Lindinger, S.; Schober, R.; Frischauf, I.; Bhardwaj, R.; et al. A novel STIM1-Orai1 gating interface essential for CRAC channel activation. Cell Calcium 2019, 79, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Outhwaite, I.R.; Pedi, L.; Long, S.B. Cryo-EM structure of the calcium release-activated calcium channel Orai in an open conformation. eLife 2020, 9, e62772. [Google Scholar] [CrossRef] [PubMed]
- Derler, I.; Plenk, P.; Fahrner, M.; Muik, M.; Jardin, I.; Schindl, R.; Gruber, H.J.; Groschner, K.; Romanin, C. The Extended Transmembrane Orai1 N-terminal (ETON) Region Combines Binding Interface and Gate for Orai1 Activation by STIM1*. J. Biol. Chem. 2013, 288, 29025–29034. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Cai, X.; Loktionova, N.A.; Wang, X.; Nwokonko, R.; Wang, X.; Wang, Y.; Rothberg, B.S.; Trebak, M.; Gill, D.L. The STIM1-binding site nexus remotely controls Orai1 channel gating. Nat. Commun. 2016, 7, 13725. [Google Scholar] [CrossRef] [PubMed]
- Frischauf, I.; Zayats, V.; Deix, M.; Hochreiter, A.; Jardin, I.; Muik, M.; Lackner, B.; Svobodová, B.; Pammer, T.; Litviňuková, M.; et al. A calcium-accumulating region, CAR, in the channel Orai1 enhances Ca 2+ permeation and SOCE-induced gene transcription. Sci. Signal. 2015, 8, ra131. [Google Scholar] [CrossRef]
- Yamashita, M.; Ing, C.E.; Yeung, P.S.-W.; Maneshi, M.M.; Pomès, R.; Prakriya, M. The basic residues in the Orai1 channel inner pore promote opening of the outer hydrophobic gate. J. Gen. Physiol. 2019, 152. [Google Scholar] [CrossRef]
- Liu, X.; Wu, G.; Yu, Y.; Chen, X.; Ji, R.; Lu, J.; Li, X.; Zhang, X.; Yang, X.; Shen, Y. Molecular understanding of calcium permeation through the open Orai channel. PLoS Biol. 2019, 17, e3000096. [Google Scholar] [CrossRef]
- Vig, M.; Beck, A.; Billingsley, J.M.; Lis, A.; Parvez, S.; Peinelt, C.; Koomoa, D.L.; Soboloff, J.; Gill, D.L.; Fleig, A.; et al. CRACM1 Multimers Form the Ion-Selective Pore of the CRAC Channel. Curr. Biol. 2006, 16, 2073–2079. [Google Scholar] [CrossRef]
- McNally, B.A.; Somasundaram, A.; Yamashita, M.; Prakriya, M. Gated regulation of CRAC channel ion selectivity by STIM1. Nature 2012, 482, 241–245. [Google Scholar] [CrossRef]
- Derler, I.; Fahrner, M.; Carugo, O.; Muik, M.; Bergsmann, J.; Schindl, R.; Frischauf, I.; Eshaghi, S.; Romanin, C. Increased Hydrophobicity at the N Terminus/Membrane Interface Impairs Gating of the Severe Combined Immunodeficiency-related ORAI1 Mutant. J. Biol. Chem. 2009, 284, 15903–15915. [Google Scholar] [CrossRef]
- Yeung, P.S.-W.; Yamashita, M.; Ing, C.E.; Pomès, R.; Freymann, D.; Prakriya, M. Mapping the functional anatomy of Orai1 transmembrane domains for CRAC channel gating. Proc. Natl. Acad. Sci USA 2018, 115, E5193–E5202. [Google Scholar] [CrossRef]
- Lacruz, R.S.; Feske, S. Diseases caused by mutations in ORAI1 and STIM1. Ann. N. Y. Acad. Sci. 2015, 1356, 45–79. [Google Scholar] [CrossRef]
- Böhm, J.; Bulla, M.; Urquhart, J.E.; Malfatti, E.; Williams, S.G.; O’Sullivan, J.; Szlauer, A.; Koch, C.; Baranello, G.; Mora, M.; et al. ORAI1 Mutations with Distinct Channel Gating Defects in Tubular Aggregate Myopathy. Hum. Mutat. 2017, 38, 426–438. [Google Scholar] [CrossRef]
- Bulla, M.; Gyimesi, G.; Kim, J.; Bhardwaj, R.; Hediger, M.; Frieden, M.; Demaurex, N. ORAI1 channel gating and selectivity is differentially altered by natural mutations in the first or third transmembrane domain. J. Physiol. 2018, 597, 561–582. [Google Scholar] [CrossRef]
- Tiffner, A.; Schober, R.; Höglinger, C.; Bonhenry, D.; Pandey, S.; Lunz, V.; Sallinger, M.; Frischauf, I.; Fahrner, M.; Lindinger, S.; et al. CRAC channel opening is determined by a series of Orai1 gating checkpoints in the transmembrane and cytosolic regions. J. Biol. Chem. 2021, 296, 100224. [Google Scholar] [CrossRef]
- Palty, R.; Stanley, C.; Isacoff, E.Y. Critical role for Orai1 C-terminal domain and TM4 in CRAC channel gating. Cell Res. 2015, 25, 963–980. [Google Scholar] [CrossRef]
- Frischauf, I.; Litviňuková, M.; Schober, R.; Zayats, V.; Svobodová, B.; Bonhenry, D.; Lunz, V.; Cappello, S.; Tociu, L.; Reha, D.; et al. Transmembrane helix connectivity in Orai1 controls two gates for calcium-dependent transcription. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef]
- Derler, I.; Butorac, C.; Krizova, A.; Stadlbauer, M.; Muik, M.; Fahrner, M.; Frischauf, I.; Romanin, C. Authentic CRAC channel activity requires STIM1 and the conserved portion of the Orai N terminus. J. Biol. Chem. 2018, 293, 1259–1270. [Google Scholar] [CrossRef]
- Tiffner, A.; Maltan, L.; Weiß, S.; Derler, I. The Orai Pore Opening Mechanism. Int. J. Mol. Sci. 2021, 22, 533. [Google Scholar] [CrossRef]
- Tiffner, A.; Maltan, L.; Fahrner, M.; Sallinger, M.; Weiß, S.; Grabmayr, H.; Höglinger, C.; Derler, I. Transmembrane Domain 3 (TM3) Governs Orai1 and Orai3 Pore Opening in an Isoform-Specific Manner. Front. Cell Dev. Biol. 2021, 9, 635705. [Google Scholar] [CrossRef]
- Tiffner, A.; Derler, I. Isoform-Specific Properties of Orai Homologues in Activation, Downstream Signaling, Physiology and Pathophysiology. Int. J. Mol. Sci. 2021, 22, 8020. [Google Scholar] [CrossRef]
- Zhou, Y.; Ramachandran, S.; Oh-Hora, M.; Rao, A.; Hogan, P.G. Pore architecture of the ORAI1 store-operated calcium channel. Proc. Natl. Acad. Sci. USA 2010, 107, 4896–4901. [Google Scholar] [CrossRef]
- Hogan, P.G. The STIM1–ORAI1 microdomain. Cell Calcium 2015, 58, 357–367. [Google Scholar] [CrossRef]
- Cao, X.; Choi, S.; Maléth, J.J.; Park, S.; Ahuja, M.; Muallem, S. The ER/PM microdomain, PI(4,5)P2 and the regulation of STIM1–Orai1 channel function. Cell Calcium 2015, 58, 342–348. [Google Scholar] [CrossRef]
- Balla, T. Ca2+ and lipid signals hold hands at endoplasmic reticulum-plasma membrane contact sites. J. Physiol. 2018, 596, 2709–2716. [Google Scholar] [CrossRef]
- Gwozdz, T.; Dutko-Gwozdz, J.; Schafer, C.; Bolotina, V.M. Overexpression of Orai1 and STIM1 Proteins Alters Regulation of Store-operated Ca2+ Entry by Endogenous Mediators. J. Biol. Chem. 2012, 287, 22865–22872. [Google Scholar] [CrossRef]
- Derler, I.; Jardin, I.; Stathopulos, P.B.; Muik, M.; Fahrner, M.; Zayats, V.; Pandey, S.K.; Poteser, M.; Lackner, B.; Absolonova, M.; et al. Cholesterol modulates Orai1 channel function. Sci. Signal. 2016, 9, ra10. [Google Scholar] [CrossRef]
- Pacheco, J.; Dominguez, L.; Hernandez, A.B.; Asanov, A.; Vaca, L. A cholesterol-binding domain in STIM1 modulates STIM1-Orai1 physical and functional interactions. Sci. Rep. 2016, 6, 29634. [Google Scholar] [CrossRef]
- Bohórquez-Hernández, A.; Gratton, E.; Pacheco, J.; Asanov, A.; Vaca, L. Cholesterol modulates the cellular localization of Orai1 channels and its disposition among membrane domains. Biochim. Biophys. Acta 2017, 1862, 1481–1490. [Google Scholar] [CrossRef]
- Carreras-Sureda, A.; Abrami, L.; Ji-Hee, K.; Wang, W.-A.; Henry, C.; Frieden, M.; Didier, M.; van der Goot, F.G.; Demaurex, N. S-acylation by ZDHHC20 targets ORAI1 channels to lipid rafts for efficient Ca2+ signaling by Jurkat T cell receptors at the immune synapse. eLife 2021, 10, e72051. [Google Scholar] [CrossRef]
- Son, A.; Park, S.; Shin, D.M.; Muallem, S. Orai1 and STIM1 in ER/PM junctions: Roles in pancreatic cell function and dysfunction. Am. J. Physiol. Physiol. 2016, 310, C414–C422. [Google Scholar] [CrossRef]
- Gamper, N.; Rohacs, T. Phosphoinositide Sensitivity of Ion Channels, a Functional Perspective. Phosphoinositides II Divers. Biol. Funct. 2012, 59, 289–333. [Google Scholar] [CrossRef]
- Hille, B.; Dickson, E.J.; Kruse, M.; Vivas, O.; Suh, B.-C. Phosphoinositides regulate ion channels. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2015, 1851, 844–856. [Google Scholar] [CrossRef]
- Morales-Lázaro, S.L.; Rosenbaum, T. Multiple Mechanisms of Regulation of Transient Receptor Potential Ion Channels by Cholesterol. Curr. Top. Membr. 2017, 80, 139–161. [Google Scholar] [CrossRef]
- Combs, D.J.; Lu, Z. Sphingomyelinase D inhibits store-operated Ca2+ entry in T lymphocytes by suppressing ORAI current. J. Gen. Physiol. 2015, 146, 161–172. [Google Scholar] [CrossRef]
- Calloway, N.; Owens, T.; Corwith, K.; Rodgers, W.; Holowka, D.; Baird, B. Stimulated association of STIM1 and Orai1 is regulated by the balance of PtdIns(4,5)P2 between distinct membrane pools. J. Cell Sci. 2011, 124, 2602–2610. [Google Scholar] [CrossRef]
- Korzeniowski, M.K.; Popovic, M.A.; Szentpetery, Z.; Varnai, P.; Stojilkovic, S.S.; Balla, T. Dependence of STIM1/Orai1-mediated calcium entry on plasma membrane phosphoinositides. J. Biol. Chem. 2009, 284, 21027–21035. [Google Scholar] [CrossRef]
- Rosado, J.; Sage, S. Phosphoinositides Are Required for Store-mediated Calcium Entry in Human Platelets. J. Biol. Chem. 2000, 275, 9110–9113. [Google Scholar] [CrossRef]
- Broad, L.M.; Braun, F.J.; Lievremont, J.P.; Bird, G.S.J.; Kurosaki, T.; Putney, J.W. Role of the Phospholipase C-Inositol 1,4,5-Trisphosphate Pathway in Calcium Release-activated Calcium Current and Capacitative Calcium Entry. J. Biol. Chem. 2001, 276, 15945–15952. [Google Scholar] [CrossRef]
- Bojjireddy, N.; Botyanszki, J.; Hammond, G.; Creech, D.; Peterson, R.; Kemp, D.C.; Snead, M.; Brown, R.; Morrison, A.; Wilson, S.; et al. Pharmacological and Genetic Targeting of the PI4KA Enzyme Reveals Its Important Role in Maintaining Plasma Membrane Phosphatidylinositol 4-Phosphate and Phosphatidylinositol 4,5-Bisphosphate Levels. J. Biol. Chem. 2014, 289, 6120–6132. [Google Scholar] [CrossRef]
- Galan, C.; Woodard, G.E.; Dionisio, N.; Salido, G.M.; Rosado, J.A. Lipid rafts modulate the activation but not the maintenance of store-operated Ca2+ entry. Biochim. Biophys. Acta 2010, 1803, 1083–1093. [Google Scholar] [CrossRef]
- Pani, B.; Ong, H.L.; Liu, X.; Rauser, K.; Ambudkar, I.S.; Singh, B.B. Lipid Rafts Determine Clustering of STIM1 in Endoplasmic Reticulum-Plasma Membrane Junctions and Regulation of Store-operated Ca2+ Entry (SOCE). J. Biol. Chem. 2008, 283, 17333–17340. [Google Scholar] [CrossRef]
- Giordano, F.; Saheki, Y.; Idevall-Hagren, O.; Colombo, S.F.; Pirruccello-Straub, M.; Milosevic, I.; O Gracheva, E.; Bagriantsev, S.; Borgese, N.; De Camilli, P. PI(4,5)P2-Dependent and Ca2+-Regulated ER-PM Interactions Mediated by the Extended Synaptotagmins. Cell 2013, 153, 1494–1509. [Google Scholar] [CrossRef]
- Schauder, C.M.; Wu, X.; Saheki, Y.; Narayanaswamy, P.; Torta, F.; Wenk, M.R.; De Camilli, P.; Reinisch, K.M. Structure of a lipid-bound extended synaptotagmin indicates a role in lipid transfer. Nature 2014, 510, 552–555. [Google Scholar] [CrossRef]
- Yu, H.; Liu, Y.; Gulbranson, D.R.; Paine, A.; Rathore, S.S.; Shen, J. Extended synaptotagmins are Ca2+-dependent lipid transfer proteins at membrane contact sites. Proc. Natl. Acad. Sci. USA 2016, 113, 4362–4367. [Google Scholar] [CrossRef]
- Idevall-Hagren, O.; Lü, A.; Xie, B.; De Camilli, P. Triggered Ca 2+ influx is required for extended synaptotagmin 1-induced ER -plasma membrane tethering. EMBO J. 2015, 34, 2291–2305. [Google Scholar] [CrossRef]
- Chang, C.-L.; Hsieh, T.-S.; Yang, T.T.; Rothberg, K.G.; Azizoglu, D.B.; Volk, E.; Liao, J.-C.; Liou, J. Feedback Regulation of Receptor-Induced Ca2+ Signaling Mediated by E-Syt1 and Nir2 at Endoplasmic Reticulum-Plasma Membrane Junctions. Cell Rep. 2013, 5, 813–825. [Google Scholar] [CrossRef]
- Maléth, J.; Choi, S.; Muallem, S.; Ahuja, M. Translocation between PI(4,5)P2-poor and PI(4,5)P2-rich microdomains during store depletion determines STIM1 conformation and Orai1 gating. Nat. Commun. 2014, 5, 5843. [Google Scholar] [CrossRef]
- Kang, F.; Zhou, M.; Huang, X.; Fan, J.; Wei, L.; Boulanger, J.; Liu, Z.; Salamero, J.; Liu, Y.; Chen, L. E-syt1 Re-arranges STIM1 Clusters to Stabilize Ring-shaped ER-PM Contact Sites and Accelerate Ca2+ Store Replenishment. Sci. Rep. 2019, 9, 3975. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.S.; Sun, Z.; Srikanth, S.; Gwack, Y. The short isoform of extended synaptotagmin-2 controls Ca2+ dynamics in T cells via interaction with STIM1. Sci. Rep. 2020, 10, 14433. [Google Scholar] [CrossRef] [PubMed]
- Saheki, Y.; Bian, X.; Schauder, C.M.; Sawaki, Y.; Surma, M.; Klose, C.; Pincet, F.; Reinisch, K.M.; De Camilli, P. Control of plasma membrane lipid homeostasis by the extended synaptotagmins. Nat. Cell Biol. 2016, 18, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.-S.; Chen, Y.-J.; Chang, C.-L.; Lee, W.-R.; Liou, J. Cortical actin contributes to spatial organization of ER–PM junctions. Mol. Biol. Cell 2017, 28, 3171–3180. [Google Scholar] [CrossRef]
- Chang, C.-L.; Liou, J. Homeostatic regulation of the PI(4,5)P 2 –Ca2+ signaling system at ER–PM junctions. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2016, 1861, 862–873. [Google Scholar] [CrossRef]
- Loewen, C.J.R.; Levine, T. A Highly Conserved Binding Site in Vesicle-associated Membrane Protein-associated Protein (VAP) for the FFAT Motif of Lipid-binding Proteins. J. Biol. Chem. 2005, 280, 14097–14104. [Google Scholar] [CrossRef]
- Loewen, C.J.; Roy, A.; Levine, T.P. A conserved ER targeting motif in three families of lipid binding proteins and in Opi1p binds VAP. EMBO J. 2003, 22, 2025–2035. [Google Scholar] [CrossRef]
- Murphy, S.E.; Levine, T.P. VAP, a Versatile Access Point for the Endoplasmic Reticulum: Review and analysis of FFAT-like motifs in the VAPome. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2016, 1861, 952–961. [Google Scholar] [CrossRef]
- Weber-Boyvat, M.; Kentala, H.; Lilja, J.; Vihervaara, T.; Hanninen, R.; Zhou, Y.; Peränen, J.; Nyman, T.; Ivaska, J.; Olkkonen, V.M. OSBP-related protein 3 (ORP3) coupling with VAMP-associated protein A regulates R-Ras activity. Exp. Cell Res. 2015, 331, 278–291. [Google Scholar] [CrossRef]
- Gulyás, G.; Sohn, M.; Kim, Y.J.; Várnai, P.; Balla, T. ORP3 phosphorylation regulates phosphatidylinositol 4-phosphate and Ca2+ dynamics at PM-ER contact sites. J. Cell Sci. 2020, 133, jcs237388. [Google Scholar] [CrossRef]
- Nakatsu, F.; Kawasaki, A. Functions of Oxysterol-Binding Proteins at Membrane Contact Sites and Their Control by Phosphoinositide Metabolism. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Stefan, C.J.; Manford, A.G.; Baird, D.; Yamada-Hanff, J.; Mao, Y.; Emr, S.D. Osh Proteins Regulate Phosphoinositide Metabolism at ER-Plasma Membrane Contact Sites. Cell 2011, 144, 389–401. [Google Scholar] [CrossRef]
- Pulli, I.; Lassila, T.; Pan, G.; Yan, D.; Olkkonen, V.M.; Törnquist, K. Oxysterol-binding protein related-proteins (ORPs) 5 and 8 regulate calcium signaling at specific cell compartments. Cell Calcium 2018, 72, 62–69. [Google Scholar] [CrossRef]
- Begley, M.J.; Taylor, G.S.; Kim, S.-A.; Veine, D.M.; E Dixon, J.; A Stuckey, J. Crystal Structure of a Phosphoinositide Phosphatase, MTMR2: Insights into Myotubular Myopathy and Charcot-Marie-Tooth Syndrome. Mol. Cell 2003, 12, 1391–1402. [Google Scholar] [CrossRef]
- Berger, P.; Schaffitzel, C.; Berger, I.; Ban, N.; Suter, U. Membrane association of myotubularin-related protein 2 is mediated by a pleckstrin homology-GRAM domain and a coiled-coil dimerization module. Proc. Natl. Acad. Sci. USA 2003, 100, 12177–12182. [Google Scholar] [CrossRef]
- Gatta, A.T.; Wong, L.; Sere, Y.Y.; Calderón-Noreña, D.M.; Cockcroft, S.; Menon, A.K.; Levine, T.P. A new family of StART domain proteins at membrane contact sites has a role in ER-PM sterol transport. eLife 2015, 4, e07253. [Google Scholar] [CrossRef]
- Murley, A.; Sarsam, R.D.; Toulmay, A.; Yamada, J.; Prinz, W.A.; Nunnari, J. Ltc1 is an ER-localized sterol transporter and a component of ER–mitochondria and ER–vacuole contacts. J. Cell Biol. 2015, 209, 539–548. [Google Scholar] [CrossRef]
- Besprozvannaya, M.; Dickson, E.; Li, H.; Ginburg, K.S.; Bers, D.M.; Auwerx, J.; Nunnari, J. GRAM domain proteins specialize functionally distinct ER-PM contact sites in human cells. eLife 2018, 7, e31019. [Google Scholar] [CrossRef]
- Jha, A.; Chung, W.Y.; Vachel, L.; Maleth, J.; Lake, S.; Zhang, G.; Ahuja, M.; Muallem, S. Anoctamin 8 tethers endoplasmic reticulum and plasma membrane for assembly of Ca2+ signaling complexes at the ER/PM compartment. EMBO J. 2019, 38, e101452. [Google Scholar] [CrossRef]
- Yu, F.; Sun, L.; Machaca, K. Constitutive recycling of the store-operated Ca2+ channel Orai1 and its internalization during meiosis. J. Cell Biol. 2010, 191, 523–535. [Google Scholar] [CrossRef]
- Jiao, H.-X.; Mu, Y.-P.; Gui, L.-X.; Yan, F.-R.; Lin, D.-C.; Sham, J.S.; Lin, M.-J. Increase in caveolae and caveolin-1 expression modulates agonist-induced contraction and store- and receptor-operated Ca2+ entry in pulmonary arteries of pulmonary hypertensive rats. Vasc. Pharmacol. 2016, 84, 55–66. [Google Scholar] [CrossRef]
- Zhu, H.; Weisleder, N.; Wu, P.; Cai, C.; Chen, J.-W. Caveolae/caveolin-1 are important modulators of store-operated calcium entry in Hs578/T breast cancer cells. J. Pharmacol. Sci. 2008, 106, 287–294. [Google Scholar] [CrossRef]
- Yeh, Y.-C.; Parekh, A.B. Distinct Structural Domains of Caveolin-1 Independently Regulate Ca 2+ Release-Activated Ca2+ Channels and Ca2+ Microdomain-Dependent Gene Expression. Mol. Cell. Biol. 2015, 35, 1341–1349. [Google Scholar] [CrossRef]
- Sathish, V.; Abcejo, A.J.; Thompson, M.A.; Sieck, G.C.; Prakash, Y.S.; Pabelick, C.M. Caveolin-1 regulation of store-operated Ca2+ influx in human airway smooth muscle. Eur. Respir. J. 2012, 40, 470–478. [Google Scholar] [CrossRef]
- Srikanth, S.; Jew, M.; Kim, K.-D.; Yee, M.-K.; Abramson, J.; Gwack, Y. Junctate is a Ca2+-sensing structural component of Orai1 and stromal interaction molecule 1 (STIM1). Proc. Natl. Acad. Sci. USA 2012, 109, 8682–8687. [Google Scholar] [CrossRef]
- Woo, J.S.; Srikanth, S.; Nishi, M.; Ping, P.; Takeshima, H.; Gwack, Y. Junctophilin-4, a component of the endoplasmic reticulum–plasma membrane junctions, regulates Ca2+ dynamics in T cells. Proc. Natl. Acad. Sci. USA 2016, 113, 2762–2767. [Google Scholar] [CrossRef]
- Jha, A.; Ahuja, M.; Maléth, J.; Moreno, C.M.; Yuan, J.P.; Kim, M.S.; Muallem, S. The STIM1 CTID domain determines access of SARAF to SOAR to regulate Orai1 channel function. J. Cell Biol. 2013, 202, 71–79. [Google Scholar] [CrossRef]
- Palty, R.; Raveh, A.; Kaminsky, I.; Meller, R.; Reuveny, E. SARAF Inactivates the Store Operated Calcium Entry Machinery to Prevent Excess Calcium Refilling. Cell 2012, 149, 425–438. [Google Scholar] [CrossRef]
- Sharma, S.; Quintana, A.; Findlay, G.; Mettlen, M.; Baust, B.; Jain, M.; Nilsson, R.; Rao, A.; Hogan, P.G. An siRNA screen for NFAT activation identifies septins as coordinators of store-operated Ca2+ entry. Nature 2013, 499, 238–242. [Google Scholar] [CrossRef]
- Deb, B.K.; Hasan, G. Regulation of Store-Operated Ca2+ Entry by Septins. Front. Cell Dev. Biol. 2016, 4. [Google Scholar] [CrossRef]
- Katz, Z.; Zhang, C.; Quintana, A.; Lillemeier, B.F.; Hogan, P.G. Septins organize endoplasmic reticulum-plasma membrane junctions for STIM1-ORAI1 calcium signalling. Sci. Rep. 2019, 9, 10839. [Google Scholar] [CrossRef] [PubMed]
- Deb, B.K.; Pathak, T.; Hasan, G. Store-independent modulation of Ca2+ entry through Orai by Septin 7. Nat. Commun. 2016, 7, 11751. [Google Scholar] [CrossRef] [PubMed]
- Deb, B.K.; Chakraborty, P.; Gopurappilly, R.; Hasan, G. SEPT7 regulates Ca2+ entry through Orai channels in human neural progenitor cells and neurons. Cell Calcium 2020, 90, 102252. [Google Scholar] [CrossRef] [PubMed]
- Jing, J.; He, L.; Sun, A.; Quintana, A.; Ding, Y.; Ma, G.; Tan, P.; Liang, X.; Zheng, X.; Chen, L.; et al. Proteomic mapping of ER–PM junctions identifies STIMATE as a regulator of Ca2+ influx. Nat. Cell Biol. 2015, 17, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Chang, C.-L.; Lee, W.-R.; Liou, J. RASSF4 controls SOCE and ER–PM junctions through regulation of PI(4,5)P2. J. Cell Biol. 2017, 216, 2011–2025. [Google Scholar] [CrossRef]
- Delint-Ramirez, I.; Willoughby, D.; Hammond, G.V.R.; Ayling, L.J.; Cooper, D.M.F. Palmitoylation Targets AKAP79 Protein to Lipid Rafts and Promotes Its Regulation of Calcium-sensitive Adenylyl Cyclase Type 8. J. Biol. Chem. 2011, 286, 32962–32975. [Google Scholar] [CrossRef]
- Dell’Acqua, M.L.; Faux, M.C.; Thorburn, J.; Thorburn, A.; Scott, J.D. Membrane-targeting sequences on AKAP79 bind phosphatidylinositol-4, 5-bisphosphate. EMBO J. 1998, 17, 2246–2260. [Google Scholar] [CrossRef]
- Liou, J.; Fivaz, M.; Inoue, T.; Meyer, T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc. Natl. Acad. Sci. USA 2007, 104, 9301–9306. [Google Scholar] [CrossRef]
- Ercan, E.; Momburg, F.; Engel, U.; Temmerman, K.; Nickel, W.; Seedorf, M. A Conserved, Lipid-Mediated Sorting Mechanism of Yeast Ist2 and Mammalian STIM Proteins to the Peripheral ER. Traffic 2009, 10, 1802–1818. [Google Scholar] [CrossRef]
- Collins, S.; Meyer, T. Evolutionary origins of STIM1 and STIM2 within ancient Ca2+ signaling systems. Trends Cell Biol. 2011, 21, 202–211. [Google Scholar] [CrossRef]
- Heo, W.D.; Inoue, T.; Park, W.S.; Kim, M.L.; Park, B.O.; Wandless, T.J.; Meyer, T. PI(3,4,5)P 3 and PI(4,5)P 2 Lipids Target Proteins with Polybasic Clusters to the Plasma Membrane. Science 2006, 314, 1458–1461. [Google Scholar] [CrossRef] [PubMed]
- Chvanov, M.; Walsh, C.M.; Haynes, L.P.; Voronina, S.G.; Lur, G.; Gerasimenko, O.V.; Barraclough, R.; Rudland, P.S.; Petersen, O.H.; Burgoyne, R.D.; et al. ATP depletion induces translocation of STIM1 to puncta and formation of STIM1-ORAI1 clusters: Translocation and re-translocation of STIM1 does not require ATP. Pflügers Arch. Eur. J. Physiol. 2008, 457, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.M.; Doherty, M.K.; Tepikin, A.V.; Burgoyne, R.D. Evidence for an interaction between Golli and STIM1 in store-operated calcium entry. Biochem. J. 2010, 430, 453–460. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bhardwaj, R.; Müller, H.-M.; Nickel, W.; Seedorf, M. Oligomerization and Ca2+/calmodulin control binding of the ER Ca2+-sensors STIM1 and STIM2 to plasma membrane lipids. Biosci. Rep. 2013, 33, 833–845. [Google Scholar] [CrossRef]
- Wu, M.M.; Covington, E.D.; Lewis, R.S. Single-molecule analysis of diffusion and trapping of STIM1 and Orai1 at endoplasmic reticulum-plasma membrane junctions. Mol. Biol. Cell 2014, 25, 3672–3685. [Google Scholar] [CrossRef]
- Li, H.; Papadopoulos, V. Peripheral-Type Benzodiazepine Receptor Function in Cholesterol Transport. Identification of a Putative Cholesterol Recognition/Interaction Amino Acid Sequence and Consensus Pattern1. Endocrinology 1998, 139, 4991–4997. [Google Scholar] [CrossRef]
- Epand, R.M. Cholesterol and the interaction of proteins with membrane domains. Prog. Lipid Res. 2006, 45, 279–294. [Google Scholar] [CrossRef]
- Epand, R.M. Proteins and cholesterol-rich domains. Biochim. Biophys. Acta Biomembr. 2008, 1778, 1576–1582. [Google Scholar] [CrossRef]
- Vieira-Pires, R.; Morais-Cabral, J.H. 310 helices in channels and other membrane proteins. J. Gen. Physiol. 2010, 136, 585–592. [Google Scholar] [CrossRef]
- Kovarova, M.; Wassif, C.; Odom, S.; Liao, K.; Porter, F.D.; Rivera, J. Cholesterol deficiency in a mouse model of Smith-Lemli-Opitz syndrome reveals increased mast cell responsiveness. J. Exp. Med. 2006, 203, 1161–1171. [Google Scholar] [CrossRef]
- Butorac, C.; Krizova, A.; Derler, I. Review: Structure and Activation Mechanisms of CRAC Channels. In Calcium Signaling; Springer: Cham, Switzerland, 2019; Volume 1131, pp. 547–604. [Google Scholar] [CrossRef]
- Krizova, A.; Maltan, L.; Derler, I. Critical parameters maintaining authentic CRAC channel hallmarks. Eur. Biophys. J. 2019, 48, 425–445. [Google Scholar] [CrossRef] [PubMed]
- Fahrner, M.; Pandey, S.K.; Muik, M.; Traxler, L.; Butorac, C.; Stadlbauer, M.; Zayats, V.; Krizova, A.; Plenk, P.; Frischauf, I.; et al. Communication between N terminus and loop2 tunes Orai activation. J. Biol. Chem. 2018, 293, 1271–1285. [Google Scholar] [CrossRef] [PubMed]
- Derler, I.; Jardin, I.; Romanin, C. Molecular mechanisms of STIM/Orai communication. Am. J. Physiol. Physiol. 2016, 310, C643–C662. [Google Scholar] [CrossRef] [PubMed]
- Hogan, P.G. Sphingomyelin, ORAI1 channels, and cellular Ca2+ signaling. J. Gen. Physiol. 2015, 146, 195–200. [Google Scholar] [CrossRef][Green Version]
- Zachowski, A. Phospholipids in animal eukaryotic membranes: Transverse asymmetry and movement. Biochem. J. 1993, 294 Pt 1, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.P.; Vaidyanathan, N.; Rengasamy, M.; Oommen, A.M.; Somaiya, N.; Jagannath, M.R. Sphingolipid Metabolic Pathway: An Overview of Major Roles Played in Human Diseases. J. Lipids 2013, 2013, 178910. [Google Scholar] [CrossRef]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An Overview of Sphingolipid Metabolism: From Synthesis to Breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar] [CrossRef]
- Rohacs, T. Phosphoinositide regulation of TRPV1 revisited. Pflügers Arch. -Eur. J. Physiol. 2015, 467, 1851–1869. [Google Scholar] [CrossRef]
- Hansen, S.B. Lipid agonism: The PIP2 paradigm of ligand-gated ion channels. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2015, 1851, 620–628. [Google Scholar] [CrossRef]
- Suh, B.-C.; Hille, B. PIP2 Is a Necessary Cofactor for Ion Channel Function: How and Why? Annu. Rev. Biophys. 2008, 37, 175–195. [Google Scholar] [CrossRef]
- Chen, J.J.; Fan, Y.; Boehning, D. Regulation of Dynamic Protein S-Acylation. Front. Mol. Biosci. 2021, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- Kordyukova, L.; Krabben, L.; Serebryakova, M.; Veit, M. S-Acylation Proteins. In Post-Translational Modification of Proteins; Humana: New York, NY, USA, 2019; Volume 1934, pp. 265–291. [Google Scholar] [CrossRef]
- Shipston, M.J. Ion Channel Regulation by Protein Palmitoylation. J. Biol. Chem. 2011, 286, 8709–8716. [Google Scholar] [CrossRef] [PubMed]
- Ohno, Y.; Kihara, A.; Sano, T.; Igarashi, Y. Intracellular localization and tissue-specific distribution of human and yeast DHHC cysteine-rich domain-containing proteins. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2006, 1761, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, N.; Fukata, Y.; Sekiya, A.; Murakami, T.; Kobayashi, K.; Fukata, M. Identification of PSD-95 Depalmitoylating Enzymes. J. Neurosci. 2016, 36, 6431–6444. [Google Scholar] [CrossRef]
- Tabaczar, S.; Czogalla, A.; Podkalicka, J.; Biernatowska, A.; Sikorski, A.F. Protein palmitoylation: Palmitoyltransferases and their specificity. Exp. Biol. Med. 2017, 242, 1150–1157. [Google Scholar] [CrossRef]
- Rocks, O.; Gerauer, M.; Vartak, N.; Koch, S.; Huang, Z.-P.; Pechlivanis, M.; Kuhlmann, J.; Brunsveld, L.; Chandra, A.; Ellinger, B.; et al. The Palmitoylation Machinery Is a Spatially Organizing System for Peripheral Membrane Proteins. Cell 2010, 141, 458–471. [Google Scholar] [CrossRef]
- Elbaz, Y.; Schuldiner, M. Staying in touch: The molecular era of organelle contact sites. Trends Biochem. Sci. 2011, 36, 616–623. [Google Scholar] [CrossRef]
- Helle, S.C.; Kanfer, G.; Kolar, K.; Lang, A.; Michel, A.H.; Kornmann, B. Organization and function of membrane contact sites. Biochim. Biophys. Acta 2013, 1833, 2526–2541. [Google Scholar] [CrossRef]
- Prinz, W.A. Bridging the gap: Membrane contact sites in signaling, metabolism, and organelle dynamics. J. Cell Biol. 2014, 205, 759–769. [Google Scholar] [CrossRef]
- Burgoyne, T.; Patel, S.; Eden, E.R. Calcium signaling at ER membrane contact sites. Biochim. Biophys. Acta 2015, 1853, 2012–2017. [Google Scholar] [CrossRef]
- Carrasco, S.; Meyer, T. STIM Proteins and the Endoplasmic Reticulum-Plasma Membrane Junctions. Annu. Rev. Biochem. 2011, 80, 973–1000. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum–mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.J.; Voeltz, G.K. Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol. 2015, 17, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L.; De Matteis, M.A.; Emr, S.; Giordano, F.; Hajnóczky, G.; Kornmann, B.; Lackner, L.L.; Levine, T.P.; Pellegrini, L.; Reinisch, K.; et al. Coming together to define membrane contact sites. Nat. Commun. 2019, 10, 1287. [Google Scholar] [CrossRef]
- Jing, J.; Liu, G.; Huang, Y.; Zhou, Y. A molecular toolbox for interrogation of membrane contact sites. J. Physiol. 2019, 598, 1725–1739. [Google Scholar] [CrossRef]
- Schulz, O.; Pieper, C.; Clever, M.; Pfaff, J.; Ruhlandt, A.; Kehlenbach, R.; Wouters, F.S.; Grosshans, J.; Bunt, G.; Enderlein, J. Resolution doubling in fluorescence microscopy with confocal spinning-disk image scanning microscopy. Proc. Natl. Acad. Sci. USA 2013, 110, 21000–21005. [Google Scholar] [CrossRef]
- MacDonald, L.; Baldini, G.; Storrie, B. Does Super-Resolution Fluorescence Microscopy Obsolete Previous Microscopic Approaches to Protein Co-localization? Membr. Traffick. 2014, 1270, 255–275. [Google Scholar] [CrossRef]
- Sengupta, P.; van Engelenburg, S.B.; Lippincott-Schwartz, J. Superresolution Imaging of Biological Systems Using Photoactivated Localization Microscopy. Chem. Rev. 2014, 114, 3189–3202. [Google Scholar] [CrossRef]
- Shim, S.-H.; Xia, C.; Zhong, G.; Babcock, H.; Vaughan, J.C.; Huang, B.; Wang, X.; Xu, C.; Bi, G.-Q.; Zhuang, X. Super-resolution fluorescence imaging of organelles in live cells with photoswitchable membrane probes. Proc. Natl. Acad. Sci. USA 2012, 109, 13978–13983. [Google Scholar] [CrossRef]
- Shai, N.; Yifrach, E.; Van Roermund, C.W.T.; Cohen, N.; Bibi, C.; Ijlst, L.; Cavellini, L.; Meurisse, J.; Schuster, R.; Zada, L.; et al. Systematic mapping of contact sites reveals tethers and a function for the peroxisome-mitochondria contact. Nat. Commun. 2018, 9, 1761. [Google Scholar] [CrossRef]
- Chang, C.-L.; Liou, J. Phosphatidylinositol 4,5-Bisphosphate Homeostasis Regulated by Nir2 and Nir3 Proteins at Endoplasmic Reticulum-Plasma Membrane Junctions. J. Biol. Chem. 2015, 290, 14289–14301. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Jing, J.; Zhu, L.; Tan, P.; Ma, G.; Zhang, Q.; Nguyen, N.T.; Wang, J.; Zhou, Y.; Huang, Y. Optical control of membrane tethering and interorganellar communication at nanoscales. Chem. Sci. 2017, 8, 5275–5281. [Google Scholar] [CrossRef] [PubMed]
- Orci, L.; Ravazzola, M.; Le Coadic, M.; Shen, W.-W.; Demaurex, N.; Cosson, P. STIM1-induced precortical and cortical subdomains of the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2009, 106, 19358–19362. [Google Scholar] [CrossRef]
- Perni, S.; Dynes, J.L.; Yeromin, A.V.; Cahalan, M.D.; Franzini-Armstrong, C. Nanoscale patterning of STIM1 and Orai1 during store-operated Ca2+ entry. Proc. Natl. Acad. Sci. USA 2015, 112, E5533–E5542. [Google Scholar] [CrossRef] [PubMed]
- Várnai, P.; Tóth, B.; Tóth, D.; Hunyady, L.; Balla, T. Visualization and Manipulation of Plasma Membrane-Endoplasmic Reticulum Contact Sites Indicates the Presence of Additional Molecular Components within the STIM1-Orai1 Complex. J. Biol. Chem. 2007, 282, 29678–29690. [Google Scholar] [CrossRef] [PubMed]
- Gudlur, A.; Quintana, A.; Zhou, Y.; Hirve, N.; Mahapatra, S.; Hogan, P.G. STIM1 triggers a gating rearrangement at the extracellular mouth of the ORAI1 channel. Nat. Commun. 2014, 5, 5164. [Google Scholar] [CrossRef]
- Peretti, M.; Badaoui, M.; Girault, A.; Van Gulick, L.; Mabille, M.-P.; Tebbakha, R.; Sevestre, H.; Morjani, H.; Ouadid-Ahidouch, H. Original association of ion transporters mediates the ECM-induced breast cancer cell survival: Kv10.1-Orai1-SPCA2 partnership. Sci. Rep. 2019, 9, 1175. [Google Scholar] [CrossRef]
- Sclip, A.; Bacaj, T.; Giam, L.R.; Südhof, T.C. Extended Synaptotagmin (ESyt) Triple Knock-Out Mice Are Viable and Fertile without Obvious Endoplasmic Reticulum Dysfunction. PLoS ONE 2016, 11, e0158295. [Google Scholar] [CrossRef]
- Kim, Y.J.; Guzman, M.; Wisniewski, E.; Balla, T. Phosphatidylinositol-Phosphatidic Acid Exchange by Nir2 at ER-PM Contact Sites Maintains Phosphoinositide Signaling Competence. Dev. Cell 2015, 33, 549–561. [Google Scholar] [CrossRef]
- Kim, S.; Kedan, A.; Marom, M.; Gavert, N.; Keinan, O.; Selitrennik, M.; Laufman, O.; Lev, S. The phosphatidylinositol-transfer protein Nir2 binds phosphatidic acid and positively regulates phosphoinositide signalling. EMBO Rep. 2013, 14, 891–899. [Google Scholar] [CrossRef]
- Yadav, S.; Garner, K.; Georgiev, P.; Li, M.; Gomez-Espinosa, E.; Panda, A.; Mathre, S.; Okkenhaug, H.; Cockcroft, S.; Raghu, P. RDGBα, a PI-PA transfer protein regulates G-protein coupled PtdIns(4,5)P2 signalling during Drosophila phototransduction. J. Cell Sci. 2015, 128, 3330–3344. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-L.; Chen, Y.-J.; Liou, J. ER-plasma membrane junctions: Why and how do we study them? Biochim. Biophys. Acta 2017, 1864, 1494–1506. [Google Scholar] [CrossRef] [PubMed]
- Dickson, E.J. RASSF4: Regulator of plasma membrane PI(4,5)P2. J. Cell Biol. 2017, 216, 1879–1881. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, S.; Miki, H.; Zhou, R.; Kido, Y.; Nishimura, W.; Kikuchi, M.; Noda, Y. Oxysterol-binding protein-related protein (ORP) 6 localizes to the ER and ER-plasma membrane contact sites and is involved in the turnover of PI4P in cerebellar granule neurons. Exp. Cell Res. 2018, 370, 601–612. [Google Scholar] [CrossRef]
- Chung, J.; Torta, F.; Masai, K.; Lucast, L.; Czapla, H.; Tanner, L.B.; Narayanaswamy, P.; Wenk, M.R.; Nakatsu, F.; De Camilli, P. PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER–plasma membrane contacts. Science 2015, 349, 428–432. [Google Scholar] [CrossRef]
- von Filseck, J.M.; Čopič, A.; Delfosse, V.; Vanni, S.; Jackson, C.L.; Bourguet, W.; Drin, G. Phosphatidylserine transport by ORP/Osh proteins is driven by phosphatidylinositol 4-phosphate. Science 2015, 349, 432–436. [Google Scholar] [CrossRef]
- Von Filseck, J.M.; Vanni, S.; Mesmin, B.; Antonny, B.; Drin, G. A phosphatidylinositol-4-phosphate powered exchange mechanism to create a lipid gradient between membranes. Nat. Commun. 2015, 6, 6671. [Google Scholar] [CrossRef]
- Schulz, T.A.; Choi, M.-G.; Raychaudhuri, S.; Mears, J.A.; Ghirlando, R.; Hinshaw, J.E.; Prinz, W.A. Lipid-regulated sterol transfer between closely apposed membranes by oxysterol-binding protein homologues. J. Cell Biol. 2009, 187, 889–903. [Google Scholar] [CrossRef]
- Ghai, R.; Du, X.; Wang, H.; Dong, J.; Ferguson, C.; Brown, A.J.; Parton, R.G.; Wu, J.-W.; Yang, H. ORP5 and ORP8 bind phosphatidylinositol-4, 5-biphosphate (PtdIns(4,5)P 2) and regulate its level at the plasma membrane. Nat. Commun. 2017, 8, 757. [Google Scholar] [CrossRef]
- Sohn, M.; Korzeniowski, M.; Zewe, J.P.; Wills, R.C.; Hammond, G.R.; Humpolickova, J.; Vrzal, L.; Chalupska, D.; Veverka, V.; Fairn, G.D.; et al. PI(4,5)P2 controls plasma membrane PI4P and PS levels via ORP5/8 recruitment to ER–PM contact sites. J. Cell Biol. 2018, 217, 1797–1813. [Google Scholar] [CrossRef]
- Balla, T. Imaging and manipulating phosphoinositides in living cells. J. Physiol. 2007, 582, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, F.; Baskin, J.M.; Chung, J.; Tanner, L.B.; Shui, G.; Lee, S.Y.; Pirruccello, M.; Hao, M.; Ingolia, N.T.; Wenk, M.R.; et al. PtdIns4P synthesis by PI4KIIIα at the plasma membrane and its impact on plasma membrane identity. J. Cell Biol. 2012, 199, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Gecht, M.; Covino, R.; Hummer, G.; Surma, M.; Klose, C.; Arai, H.; Kono, N.; Stefan, C.J. Osh Proteins Control Nanoscale Lipid Organization Necessary for PI(4,5)P2 Synthesis. Mol. Cell 2019, 75, 1043–1057.e8. [Google Scholar] [CrossRef] [PubMed]
- Hammond, G.R.V.; Fischer, M.J.; Anderson, K.E.; Holdich, J.; Koteci, A.; Balla, T.; Irvine, R.F. PI4P and PI(4,5)P2 Are Essential But Independent Lipid Determinants of Membrane Identity. Science 2012, 337, 727–730. [Google Scholar] [CrossRef]
- Sohn, M.; Ivanova, P.; Brown, H.A.; Toth, D.J.; Varnai, P.; Kim, Y.J.; Balla, T. Lenz-Majewski mutations in PTDSS1 affect phosphatidylinositol 4-phosphate metabolism at ER-PM and ER-Golgi junctions. Proc. Natl. Acad. Sci. USA 2016, 113, 4314–4319. [Google Scholar] [CrossRef]
- Balla, M.S.A.T. Lenz-Majewski syndrome: How a single mutation leads to complex changes in lipid metabolism. J. Rare Dis. Res. Treat. 2017, 2, 47–51. [Google Scholar] [CrossRef]
- Wu, W.; Shi, X.; Xu, C. Regulation of T cell signalling by membrane lipids. Nat. Rev. Immunol. 2016, 16, 690–701. [Google Scholar] [CrossRef]
- Elbaz-Alon, Y.; Eisenberg-Bord, M.; Shinder, V.; Stiller, S.B.; Shimoni, E.; Wiedemann, N.; Geiger, T.; Schuldiner, M. Lam6 Regulates the Extent of Contacts between Organelles. Cell Rep. 2015, 12, 7–14. [Google Scholar] [CrossRef]
- Naito, T.; Ercan, B.; Krshnan, L.; Triebl, A.; Koh, D.; Wei, F.-Y.; Tomizawa, K.; Torta, F.T.; Wenk, M.R.; Saheki, Y. Movement of accessible plasma membrane cholesterol by the GRAMD1 lipid transfer protein complex. eLife 2019, 8, e51401. [Google Scholar] [CrossRef]
- Whitlock, J.M.; Hartzell, H.C. Anoctamins/TMEM16 Proteins: Chloride Channels Flirting with Lipids and Extracellular Vesicles. Annu. Rev. Physiol. 2017, 79, 119–143. [Google Scholar] [CrossRef]
- Maass, K.; Fischer, M.A.; Seiler, M.; Temmerman, K.; Nickel, W.; Seedorf, M. A signal comprising a basic cluster and an amphipathic α-helix interacts with lipids and is required for the transport of Ist2 to the yeast cortical ER. J. Cell Sci. 2009, 122, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Dang, S.; Feng, S.; Tien, J.; Peters, C.; Bulkley, D.; Lolicato, M.; Zhao, J.; Zuberbühler, K.; Ye, W.; Qi, L.; et al. Cryo-EM structures of the TMEM16A calcium-activated chloride channel. Nature 2017, 552, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Paulino, C.; Kalienkova, V.; Lam, A.; Neldner, Y.; Dutzler, R. Activation mechanism of the calcium-activated chloride channel TMEM16A revealed by cryo-EM. Nature 2017, 552, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Paulino, C.; Neldner, Y.; Lam, A.K.; Kalienkova, V.; Brunner, J.D.; Schenck, S.; Dutzler, R. Structural basis for anion conduction in the calcium-activated chloride channel TMEM16A. eLife 2017, 6, e26232. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Xiao, S.; Huang, F.; Harfe, B.D.; Jan, Y.N.; Jan, L.Y. Calcium-Activated Chloride Channels (CaCCs) Regulate Action Potential and Synaptic Response in Hippocampal Neurons. Neuron 2012, 74, 179–192. [Google Scholar] [CrossRef]
- Suzuki, J.; Umeda, M.; Sims, P.J.; Nagata, S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature 2010, 468, 834–838. [Google Scholar] [CrossRef]
- Brunner, J.D.; Lim, N.K.; Schenck, S.; Duerst, A.; Dutzler, R. X-ray structure of a calcium-activated TMEM16 lipid scramblase. Nature 2014, 516, 207–212. [Google Scholar] [CrossRef]
- Alvadia, C.; Lim, N.K.; Mosina, V.C.; Oostergetel, G.T.; Dutzler, R.; Paulino, C. Cryo-EM structures and functional characterization of the murine lipid scramblase TMEM16F. eLife 2019, 8, e44365. [Google Scholar] [CrossRef]
- Stöhr, H.; Heisig, J.B.; Benz, P.M.; Schöberl, S.; Milenkovic, V.M.; Strauss, O.; Aartsen, W.M.; Wijnholds, J.; Weber, B.H.F.; Schulz, H.L. TMEM16B, A Novel Protein with Calcium-Dependent Chloride Channel Activity, Associates with a Presynaptic Protein Complex in Photoreceptor Terminals. J. Neurosci. 2009, 29, 6809–6818. [Google Scholar] [CrossRef]
- Prakash, Y.S.; Thompson, M.A.; Vaa, B.; Matabdin, I.; Peterson, T.E.; He, T.; Pabelick, C.M. Caveolins and intracellular calcium regulation in human airway smooth muscle. Am. J. Physiol. Cell. Mol. Physiol. 2007, 293, L1118–L1126. [Google Scholar] [CrossRef]
- Pani, B.; Singh, B.B. Lipid rafts/caveolae as microdomains of calcium signaling. Cell Calcium 2009, 45, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Treves, S.; Franzini-Armstrong, C.; Moccagatta, L.; Arnoult, C.; Grasso, C.; Schrum, A.; Ducreux, S.; Zhu, M.X.; Mikoshiba, K.; Girard, T.; et al. Junctate is a key element in calcium entry induced by activation of InsP3 receptors and/or calcium store depletion. J. Cell Biol. 2004, 166, 537–548. [Google Scholar] [CrossRef] [PubMed]
- de Souza, L.B.; Ong, H.L.; Liu, X.; Ambudkar, I.S. PIP2 and septin control STIM1/Orai1 assembly by regulating cytoskeletal remodeling via a CDC42-WASP/WAVE-ARP2/3 protein complex. Cell Calcium 2021, 99, 102475. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.J.; Flatters, D.; Rodrigues-Lima, F.; Yan, J.; Thalassinos, K.; Katan, M. Comparative analysis of interactions of RASSF1-10. Adv. Biol. Regul. 2013, 53, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, V.; Recino, A.; Jeffries, A.; Ward, A.; Chalmers, A.D. The N-terminal RASSF family: A new group of Ras-association-domaincontaining proteins, with emerging links to cancer formation. Biochem. J. 2009, 425, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Scheel, H.; Hofmann, K. A novel inter action motif, SARAH, connects three classes of tumor suppressor. Curr. Biol. 2003, 13, R899–R900. [Google Scholar] [CrossRef]
- Dittfeld, C.; Richter, A.M.; Steinmann, K.; Klagge-Ulonska, A.; Dammann, R.H. The SARAH Domain of RASSF1A and Its Tumor Suppressor Function. Mol. Biol. Int. 2012, 2012, 196715. [Google Scholar] [CrossRef]
- Makbul, C.; Aruxandei, D.C.; Hofmann, E.; Schwarz, D.; Wolf, E.; Herrmann, C. Structural and Thermodynamic Characterization of Nore1-SARAH: A Small, Helical Module Important in Signal Transduction Networks. Biochemistry 2013, 52, 1045–1054. [Google Scholar] [CrossRef]
- Halder, G.; Johnson, R.L. Hippo signaling: Growth control and beyond. Development 2011, 138, 9–22. [Google Scholar] [CrossRef]
- Pan, D. The Hippo Signaling Pathway in Development and Cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef]
- Ando, H.; Mizutani, A.; Matsu-Ura, T.; Mikoshiba, K. IRBIT, a Novel Inositol 1,4,5-Trisphosphate (IP3) Receptor-binding Protein, Is Released from the IP3 Receptor upon IP3 Binding to the Receptor. J. Biol. Chem. 2003, 278, 10602–10612. [Google Scholar] [CrossRef] [PubMed]
- Stathopulos, P.; Li, G.-Y.; Plevin, M.; Ames, J.B.; Ikura, M. Stored Ca2+ Depletion-induced Oligomerization of Stromal Interaction Molecule 1 (STIM1) via the EF-SAM Region. J. Biol. Chem. 2006, 281, 35855–35862. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.; Clynes, M.A.; Masada, N.; Ciruela, A.; Ayling, L.-J.; Wachten, S.; Cooper, D.M.F. Insights into the residence in lipid rafts of adenylyl cyclase AC8 and its regulation by capacitative calcium entry. Am. J. Physiol. Physiol. 2009, 296, C607–C619. [Google Scholar] [CrossRef] [PubMed]
- Tabbasum, V.G.; Cooper, D.M.F. Structural and Functional Determinants of AC8 Trafficking, Targeting and Responsiveness in Lipid Raft Microdomains. J. Membr. Biol. 2019, 252, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Ayling, L.J.; Briddon, S.; Halls, M.; Hammond, G.; Vaca, L.; Pacheco, J.; Hill, S.; Cooper, D.M.F. Adenylyl cyclase AC8 directly controls its micro-environment by recruiting the actin cytoskeleton in a cholesterol-rich milieu. J. Cell Sci. 2012, 125, 869–886. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Pathak, T.; Yoast, R.; Emrich, S.; Xin, P.; Nwokonko, R.M.; Johnson, M.; Wu, S.; Delierneux, C.; Gueguinou, M.; et al. A calcium/cAMP signaling loop at the ORAI1 mouth drives channel inactivation to shape NFAT induction. Nat. Commun. 2019, 10, 1971. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, D.; Everett, K.L.; Halls, M.L.; Pacheco, J.; Skroblin, P.; Vaca, L.; Klussmann, E.; Cooper, D.M.F. Direct Binding between Orai1 and AC8 Mediates Dynamic Interplay Between Ca2+ and cAMP Signaling. Sci. Signal. 2012, 5, ra29. [Google Scholar] [CrossRef]
- Kar, P.; Barak, P.; Zerio, A.; Lin, Y.P.; Parekh, A.J.; Watts, V.J.; Cooper, D.M.F.; Zaccolo, M.; Kramer, H.; Parekh, A.B. AKAP79 Orchestrates a Cyclic AMP Signalosome Adjacent to Orai1 Ca(2+) Channels. Function 2021, 2, zqab036. [Google Scholar] [CrossRef]
- Kar, P.; Lin, Y.-P.; Bhardwaj, R.; Tucker, C.J.; Bird, G.S.; Hediger, M.A.; Monico, C.; Amin, N.; Parekh, A.B. The N terminus of Orai1 couples to the AKAP79 signaling complex to drive NFAT1 activation by local Ca2+ entry. Proc. Natl. Acad. Sci. USA 2021, 118, e2012908118. [Google Scholar] [CrossRef]
- Jardin, I.; Salido, G.M.; Rosado, J.A. Role of lipid rafts in the interaction between hTRPC1, Orai1 and STIM1. Channels 2008, 2, 401–403. [Google Scholar] [CrossRef]
- Lockwich, T.P.; Liu, X.; Singh, B.; Jadlowiec, J.; Weiland, S.; Ambudkar, I.S. Assembly of Trp1 in a Signaling Complex Associated with Caveolin-Scaffolding Lipid Raft Domains. J. Biol. Chem. 2000, 275, 11934–11942. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Lin, M.I.; Stan, R.; Bauer, P.M.; Yu, J.; Sessa, W. Genetic Evidence Supporting Caveolae Microdomain Regulation of Calcium Entry in Endothelial Cells. J. Biol. Chem. 2007, 282, 16631–16643. [Google Scholar] [CrossRef] [PubMed]
- Pani, B.; Liu, X.; Bollimuntha, S.; Cheng, K.T.; Niesman, I.R.; Zheng, C.; Achen, V.R.; Patel, H.H.; Ambudkar, I.S.; Singh, B.B. Impairment of TRPC1–STIM1 channel assembly and AQP5 translocation compromise agonist-stimulated fluid secretion in mice lacking caveolin1. J. Cell Sci. 2013, 126, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Rathor, N.; Chung, H.K.; Wang, S.R.; Wang, J.; Turner, U.J.; Rao, J.N. Caveolin-1 enhances rapid mucosal restitution by activating TRPC1-mediated Ca2+ signaling. Physiol. Rep. 2014, 2, e12193. [Google Scholar] [CrossRef] [PubMed]
- Alicia, S.; Angélica, Z.; Carlos, S.; Alfonso, S.; Vaca, L. STIM1 converts TRPC1 from a receptor-operated to a store-operated channel: Moving TRPC1 in and out of lipid rafts. Cell Calcium 2008, 44, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Pani, B.; Ong, H.L.; Brazer, S.-C.W.; Liu, X.; Rauser, K.; Singh, B.B.; Ambudkar, I.S. Activation of TRPC1 by STIM1 in ER-PM microdomains involves release of the channel from its scaffold caveolin-1. Proc. Natl. Acad. Sci. USA 2009, 106, 20087–20092. [Google Scholar] [CrossRef]
- Duffy, S.M.; Ashmole, I.; Smallwood, D.T.; Leyland, M.L.; Bradding, P. Orai/CRACM1 and KCa3.1 ion channels interact in the human lung mast cell plasma membrane. Cell Commun. Signal. 2015, 13, 32. [Google Scholar] [CrossRef]
- Ferreira, R.; Schlichter, L.C. Selective Activation of KCa3.1 and CRAC Channels by P2Y2 Receptors Promotes Ca2+ Signaling, Store Refilling and Migration of Rat Microglial Cells. PLoS ONE 2013, 8, e62345. [Google Scholar] [CrossRef]
- Guéguinou, M.; Harnois, T.; Crottes, D.; Uguen, A.; Deliot, N.; Gambade, A.; Chantôme, A.; Haelters, J.P.; Jaffrès, P.A.; Jourdan, M.L.; et al. SK3/TRPC1/Orai1 complex regulates SOCE-dependent colon cancer cell migration: A novel opportunity to modulate anti-EGFR mAb action by the alkyl-lipid Ohmline. Oncotarget 2016, 7, 36168–36184. [Google Scholar] [CrossRef]
- Chantôme, A.; Potier-Cartereau, M.; Clarysse, L.; Fromont, G.; Lambot, S.M.; Guéguinou, M.; Pages, J.-C.; Collin, C.; Oullier, T.; Girault, A.; et al. Pivotal Role of the Lipid Raft SK3–Orai1 Complex in Human Cancer Cell Migration and Bone Metastases. Cancer Res. 2013, 73, 4852–4861. [Google Scholar] [CrossRef]
- Gueguinou, M.; Crottès, D.; Chantôme, A.; Rapetti-Mauss, R.; Potier-Cartereau, M.; Clarysse, L.; Girault, A.; Fourbon, Y.; Jézéquel, P.; Guérin-Charbonnel, C.; et al. The SigmaR1 chaperone drives breast and colorectal cancer cell migration by tuning SK3-dependent Ca2+ homeostasis. Oncogene 2017, 36, 3640–3647. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Zhong, X.-G.; Xia, X.-M.; Huang, J.-H.; Fan, Y.-F.; Yuan, R.-X.; Xue, N.-R.; Du, J.; Han, W.-X.; Xu, A.-M.; et al. Orai1 forms a signal complex with SK3 channel in gallbladder smooth muscle. Biochem. Biophys. Res. Commun. 2015, 466, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Girault, A.; Haelters, J.-P.; Potier-Cartereau, M.; Chantome, A.; Pinault, M.; Lambot, S.M.; Oullier, T.; Simon, G.; Couthon-Gourves, H.; Jaffres, P.-A.; et al. New Alkyl-Lipid Blockers of SK3 Channels Reduce Cancer Cell Migration and Occurrence of Metastasis. Curr. Cancer Drug Targets 2011, 11, 1111–1125. [Google Scholar] [CrossRef]
- Klumpp, L.; Sezgin, E.C.; Skardelly, M.; Eckert, F.; Huber, S.M. KCa3.1 Channels and Glioblastoma: In Vitro Studies. Curr. Neuropharmacol. 2018, 16, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Potier, M.-C.; Tran, T.A.; Chantome, A.; Girault, A.; Joulin, V.; Bougnoux, P.; Vandier, C.; Pierre, F. Altered SK3/KCa2.3-mediated migration in adenomatous polyposis coli (Apc) mutated mouse colon epithelial cells. Biochem. Biophys. Res. Commun. 2010, 397, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Mignen, O.; Constantin, B.; Potier-Cartereau, M.; Penna, A.; Gautier, M.; Guéguinou, M.; Renaudineau, Y.; Shoji, K.F.; Félix, R.; Bayet, E.; et al. Constitutive calcium entry and cancer: Updated views and insights. Eur. Biophys. J. 2017, 46, 395–413. [Google Scholar] [CrossRef]
- Tiffner, A.; Hopl, V.; Schober, R.; Sallinger, M.; Grabmayr, H.; Höglinger, C.; Fahrner, M.; Lunz, V.; Maltan, L.; Frischauf, I.; et al. Orai1 Boosts SK3 Channel Activation. Cancers 2021, 13, 6357. [Google Scholar] [CrossRef]
- Tiffner, A.; Derler, I. Molecular Choreography and Structure of Ca2+ Release-Activated Ca2+ (CRAC) and KCa2+ Channels and Their Relevance in Disease with Special Focus on Cancer. Membranes 2020, 10, 425. [Google Scholar] [CrossRef]
- Clarysse, L.; Guéguinou, M.; Potier-Cartereau, M.; Vandecasteele, G.; Bougnoux, P.; Chevalier, S.; Chantôme, A.; Vandier, C. cAMP–PKA inhibition of SK3 channel reduced both Ca2+ entry and cancer cell migration by regulation of SK3–Orai1 complex. Pflügers Arch. -Eur. J. Physiol. 2014, 466, 1921–1932. [Google Scholar] [CrossRef]
- Herrera, F.E.; Sevrain, C.M.; Jaffrès, P.-A.; Couthon, H.; Grelard, A.; Dufourc, E.J.; Chantôme, A.; Potier-Cartereau, M.; Vandier, C.; Bouchet, A.M. Singular Interaction between an Antimetastatic Agent and the Lipid Bilayer: The Ohmline Case. ACS Omega 2017, 2, 6361–6370. [Google Scholar] [CrossRef]
- Kouba, S.; Braire, J.; Félix, R.; Chantôme, A.; Jaffrès, P.-A.; Lebreton, J.; Dubreuil, D.; Pipelier, M.; Zhang, X.X.; Trebak, M.; et al. Lipidic synthetic alkaloids as SK3 channel modulators. Synthesis and biological evaluation of 2-substituted tetrahydropyridine derivatives with potential anti-metastatic activity. Eur. J. Med. Chem. 2019, 186, 111854. [Google Scholar] [CrossRef] [PubMed]
- Srivats, S.; Balasuriya, D.; Pasche, M.; Vistal, G.; Edwardson, J.M.; Taylor, C.; Murrell-Lagnado, R.D. Sigma1 receptors inhibit store-operated Ca2+ entry by attenuating coupling of STIM1 to Orai1. J. Cell Biol. 2016, 213, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Koshy, C.; Ziegler, C. Structural insights into functional lipid–protein interactions in secondary transporters. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Stauderman, K.A. CRAC channels as targets for drug discovery and development. Cell Calcium 2018, 74, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Du, L.; Zhou, Y.; Li, M. Store-operated CRAC channel inhibitors: Opportunities and challenges. Futur. Med. Chem. 2016, 8, 817–832. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipid Type | Location | Effect on Endogenous CRAC | Effect on STIM1 | Effect on Orai1 | Direct Interaction with STIM1 or Orai1 | Ref. |
| PIP2 | PM | ↑ | ↑ STIM1 assembly at PIP2 rich region | ↑ | STIM1 K-rich region (aa 672–685) Orai1 N-terminus (aa 28–33) | [129,130] |
| PI4P | PM | ↑ | ↑ | n.d. | n.d. | [130,131,132,133] |
| cholesterol | PM | ↑ | ↓ | ↓ | STIM1 OASF (I364) Orai1 N-terminus (L74, Y80) | [119,120,121,122,129,134,135] |
| sphingo-myelin | PM | ↑ | n.d. | n.d. | n.d. | [128] |
| ER-phospho-lipids | ER | n.d. | ↓ | not applicable | STIM1 apex (F394K/D) | [83] |
| S-acylation (post-transla-tional tethering of fatty acids) | PM | ↑ | not applicable | ↑ | Orai1 C143 | [123] |
| Protein within ER-PM junction | Location | Effect on endogenous CRAC | Effect on STIM1 | Effect on Orai1 | Direct interaction with STIM1 or Orai1 | |
| E-Syt | ER-PM junction (E-Syt1 located there in a Ca2+ dependent manner) embedded in ER | no/minor effect (HeLa cells) | n.d. | n. d. | indirect | [136,137,138,139,140,141,142] |
| E-Syt1 | Rearrangement of ER-PM contact sites | no/minor effect | supports repletion of ER stores with Ca2+ (HEK293 cells) | n.d. | indirect | [142] |
| E-Syt2 | ER-PM junction | ↑ (E-Syt2S) | ↑ (E-Syt2S) ↓ (E-Syt2L) | n.d. | indirect | [143] |
| E-Syt3 | ER-PM junction | No effect/ minor (when knocked-out together with E-Syt1/2) | n.d. | n.d. | indirect | [136] |
| E-Syt1 + Nir2 | ER-PM junction | ↑ | ↑ Co-localization with STIM1 | n.d. | Indirect dependent on DAG levels in the membrane | [140,144,145,146] |
| VAP | n.d. | n.d. | n.d. | indirect | [147,148,149] | |
| VAP + ORP3/6 | ER-PM junction | ↓ | ↓ | n.d. | n.d. | [150,151,152] |
| ORP5/8 | ER-PM junction, ER-Mito | n.d. | n.d. | n.d. | n.d. | [148,150,152,153,154] |
| GRAMD2A | ER-PM junction | not required (more studies required) | ↑ Supports STIM1 translocation STIM1 puncta formation | n.d. | Indirect Co-localizes with E-Syt and STIM1 (PIP2 required) | [155,156,157,158,159] |
| ANO8 | ER-PM junction | ↑ | ↑ STIM1 oligomeri-zation/clustering | ↑ STIM1/Orai1 interaction; inactivation | Indirect (further studies necessary) | [160] |
| Caveolin-1 | PM | ↑ | independent of STIM1 | ↑ STIM1/Orai1 coupling | Direct with Orai1 (aa 52–60; aa 250–258) | [122,161,162,163,164,165] |
| Junctate | ER | ↑ | ↑ | ↑ | Direct with STIM1 (STIM1 N-terminus) | [166] |
| Juncto-philin 4 | ER | ↑ | ↑ supports recruitment of STIM1 to ER-PM | n.d. | Direct interaction with STIM1 | [167] |
| Junctate + juncto-philin 4 | ER-PM | ↑ | supports recruitment of STIM1 to ER-PM | n.d. | Direct interaction with STIM1 | [166,167] |
| SARAF | ER | ↓ | ↓ | ↓ | STIM1 CTID | [141,168,169] |
| Septin | PM | depending on the septin type | indirect | [170,171,172,173,174] | ||
| STIMATE | ER | ↑ | ↑ | ↑ | STIM1-CC1 | [175] |
| RASSF | PM | ↑ | ↑ | ↑ | indirect (modulates PIP2 levels) | [176] |
| AC8 | PM | ↑ (Regulated by its direct association with AKAP79) | ↑ | ↑ | Orai1 | [177,178] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maltan, L.; Andova, A.-M.; Derler, I. The Role of Lipids in CRAC Channel Function. Biomolecules 2022, 12, 352. https://doi.org/10.3390/biom12030352
Maltan L, Andova A-M, Derler I. The Role of Lipids in CRAC Channel Function. Biomolecules. 2022; 12(3):352. https://doi.org/10.3390/biom12030352
Chicago/Turabian StyleMaltan, Lena, Ana-Marija Andova, and Isabella Derler. 2022. "The Role of Lipids in CRAC Channel Function" Biomolecules 12, no. 3: 352. https://doi.org/10.3390/biom12030352
APA StyleMaltan, L., Andova, A.-M., & Derler, I. (2022). The Role of Lipids in CRAC Channel Function. Biomolecules, 12(3), 352. https://doi.org/10.3390/biom12030352