
RRM2 Alleviates Doxorubicin-Induced Cardiotoxicity through the AKT/mTOR Signaling Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Treatment
2.2. Cell Studies
2.3. Recombinant Adenovirus Was Delivered to Mice Ventricular and Primary Cardiomyocytes
2.4. Cell Proliferation Assay
2.5. Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick End Labeling (TUNEL)
2.6. Histological Analysis and Immunofluorescence Staining
2.7. Reverse-Transcription Quantitative PCR (qPCR)
| GAPDH (Mus musculus) | F | TGCACCACCAACTGCTTAG |
| GAPDH (Mus musculus) | R | GGATGCAGGGATGATGTTC |
| RRM2 (Mus musculus) | F | ACTGTGACTTTGCCTGCCTGATG |
| RRM2 (Mus musculus) | R | TCCGTGAGGAACTCCTGCTCTATC |
2.8. Western Blot
2.9. Data Analysis
3. Results
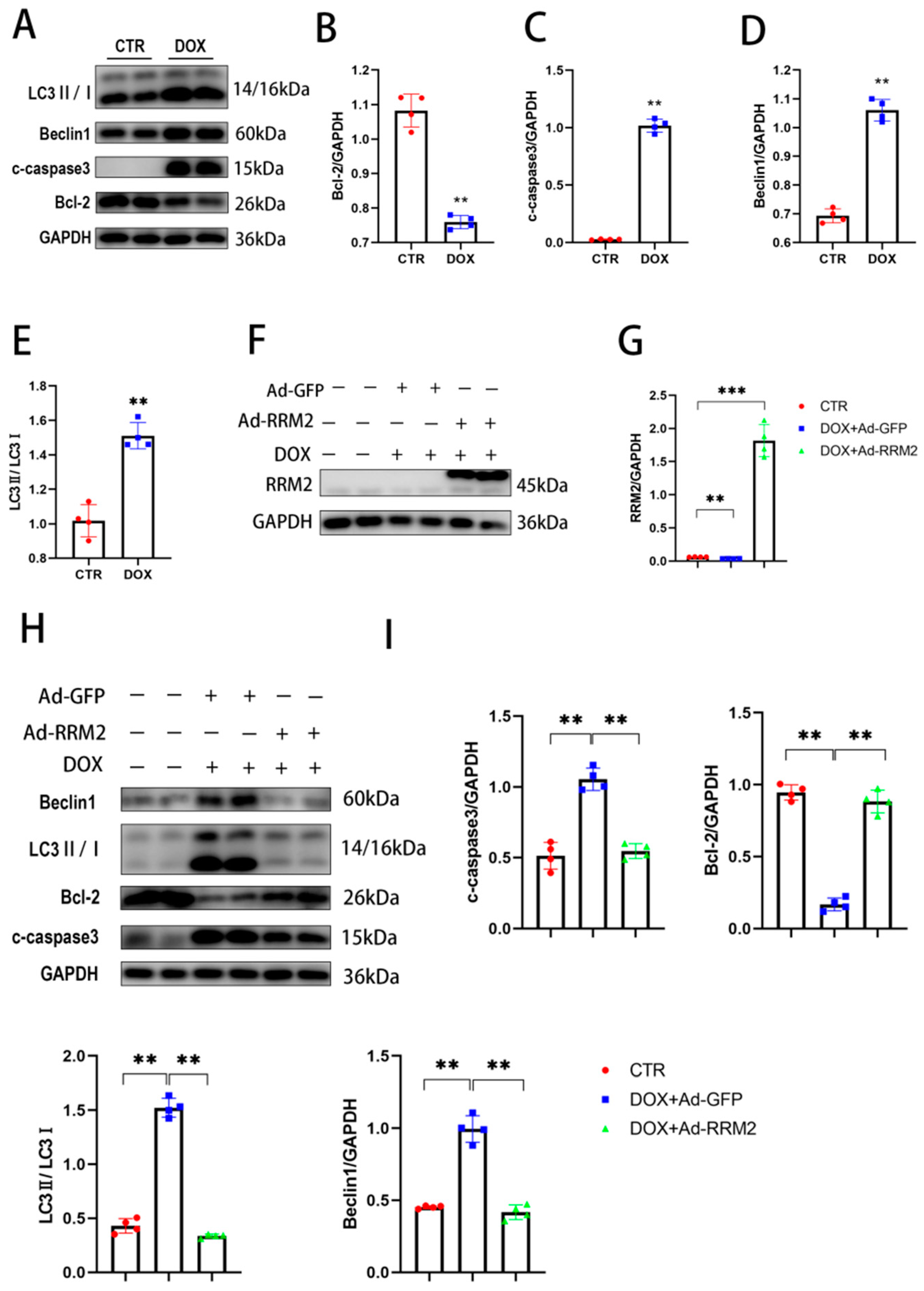
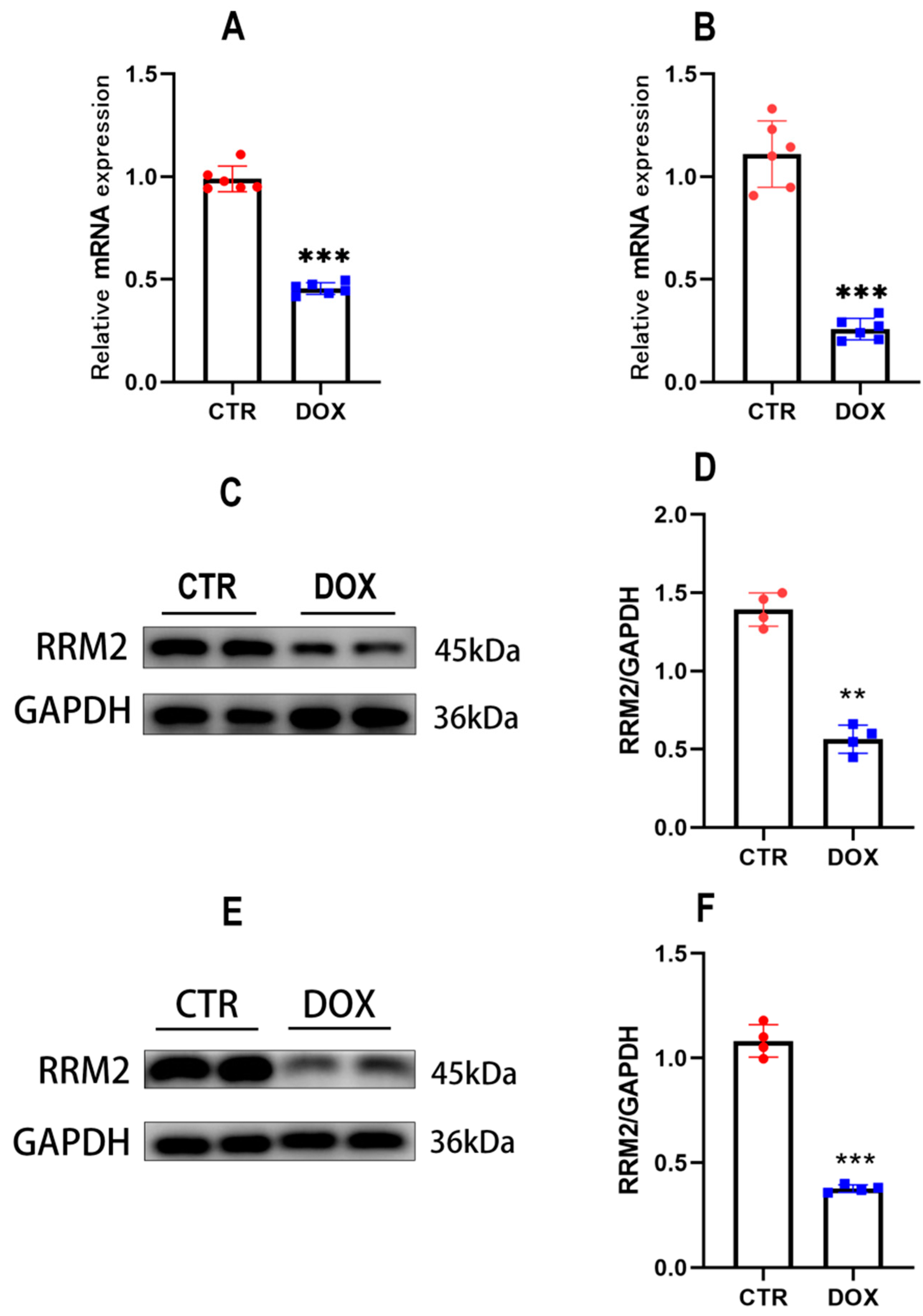
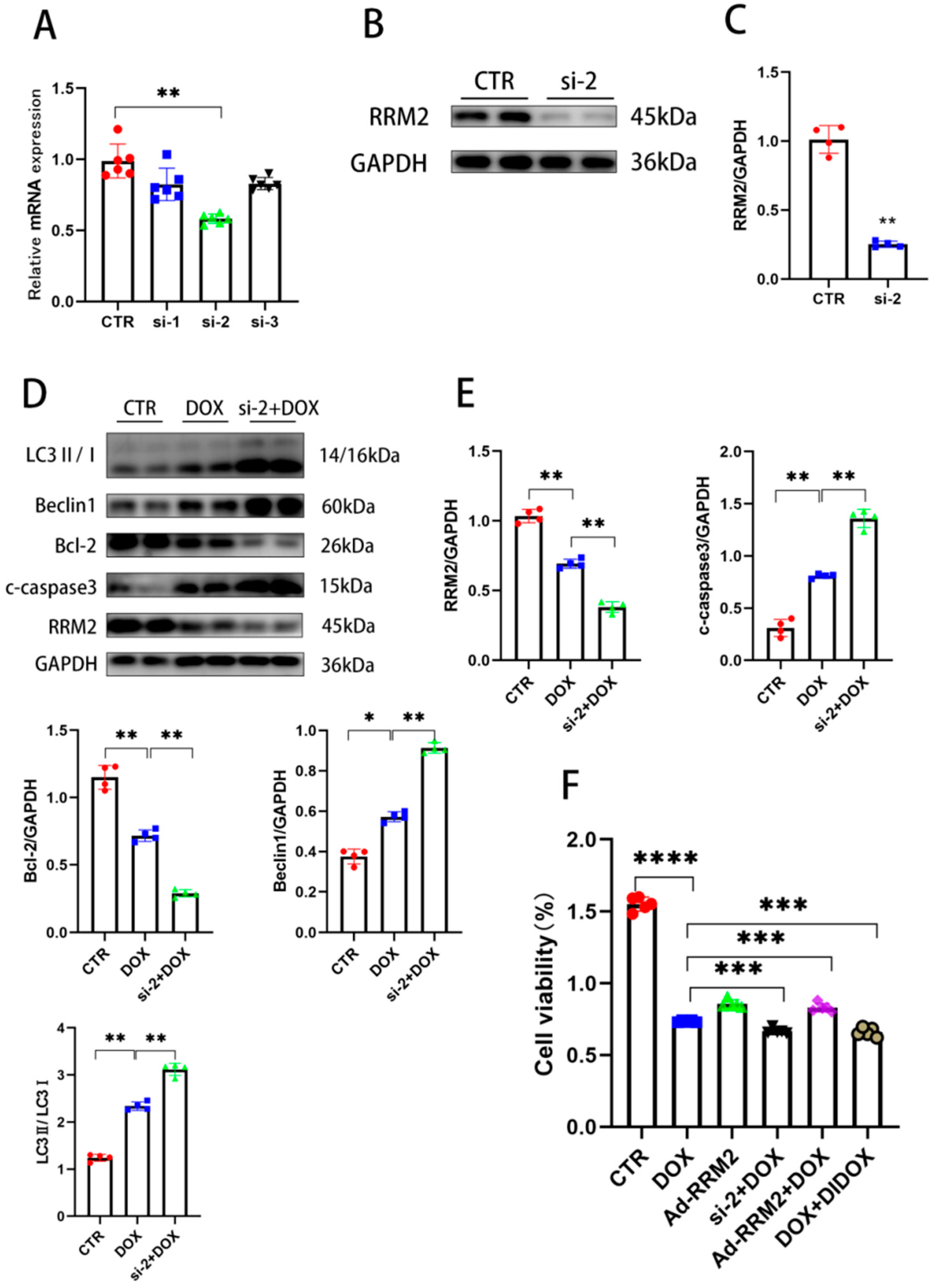
3.1. RRM2 Expression Is Decreased In Vitro and In Vivo after DOX Treatment
3.2. RRM2 Overexpression Abated DOX-Induced Apoptosis and Autophagy In Vitro
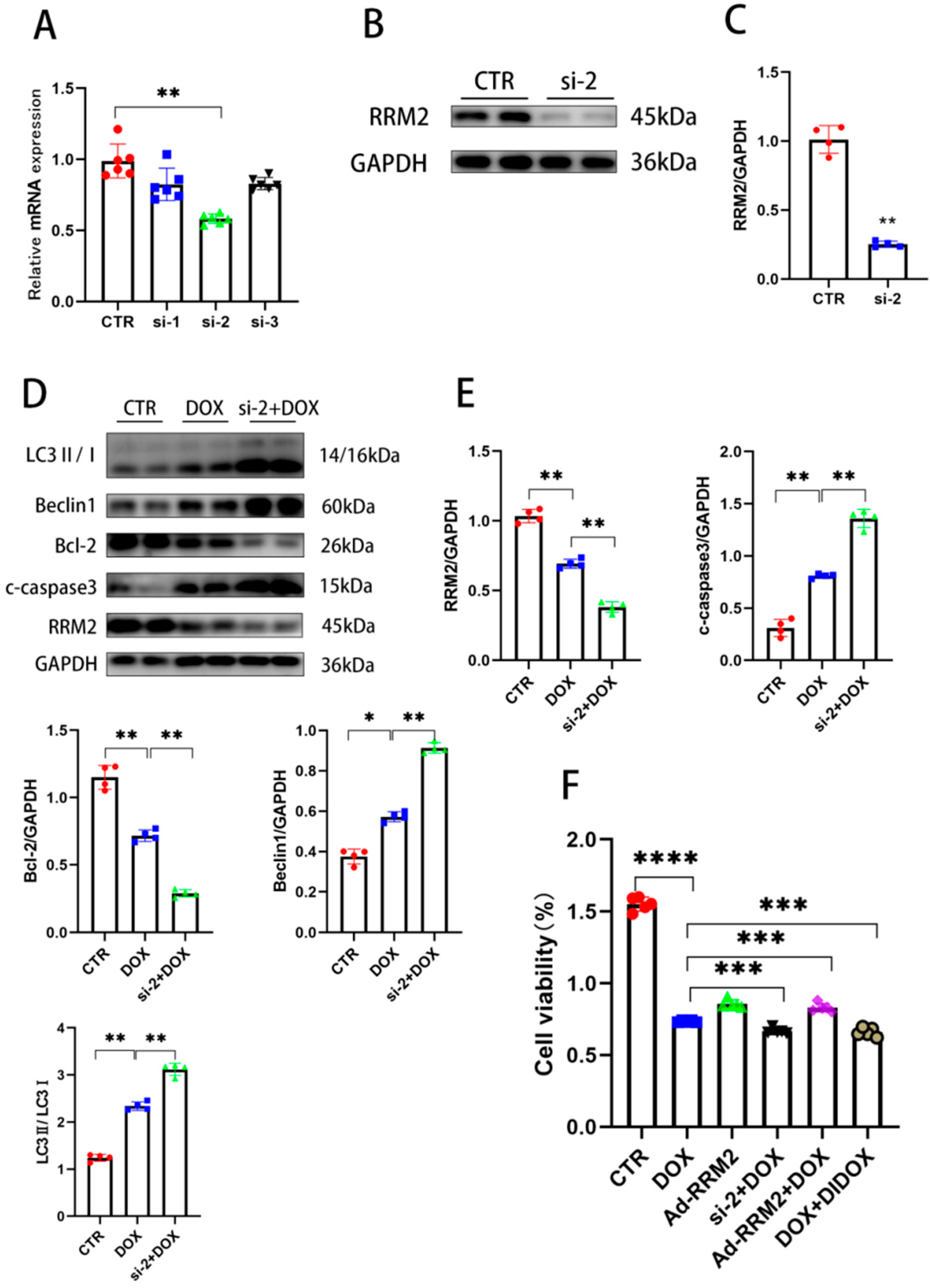
3.3. Knockdown of RRM2 Facilitates DOX-Induced Injury In Vitro
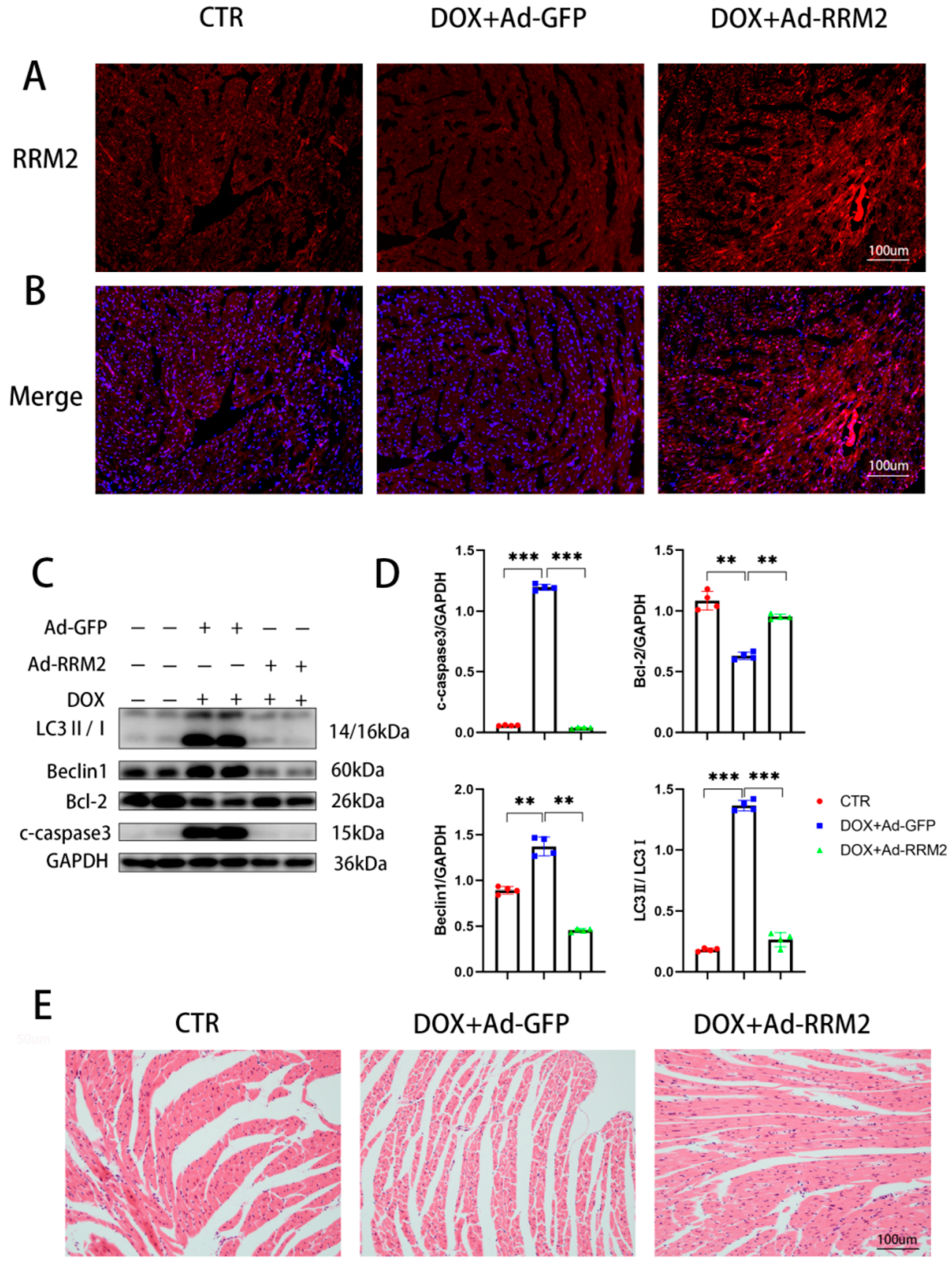
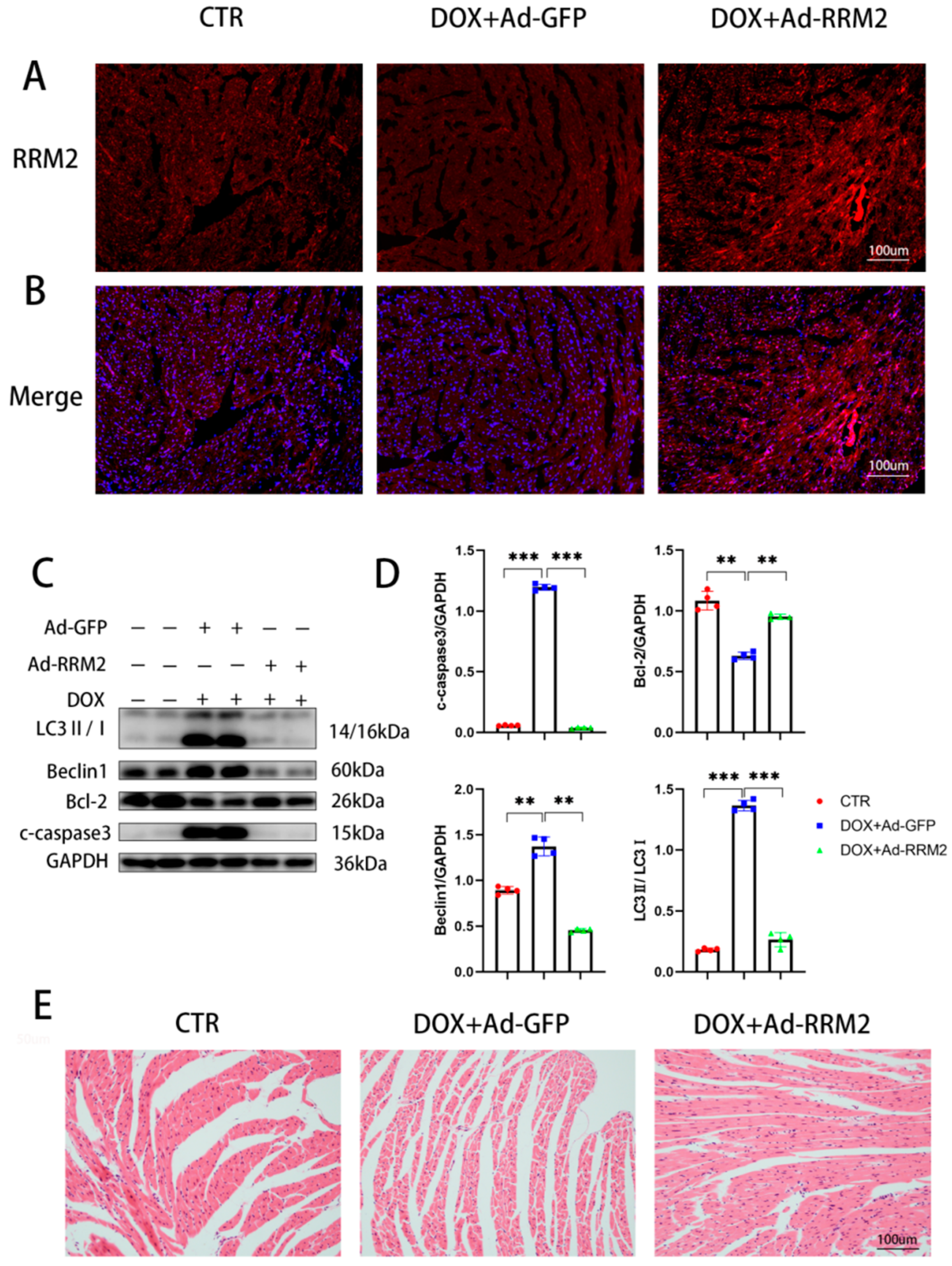
3.4. RRM2 Overexpression Alleviated DOX-Induced Cardiotoxicity In Vivo
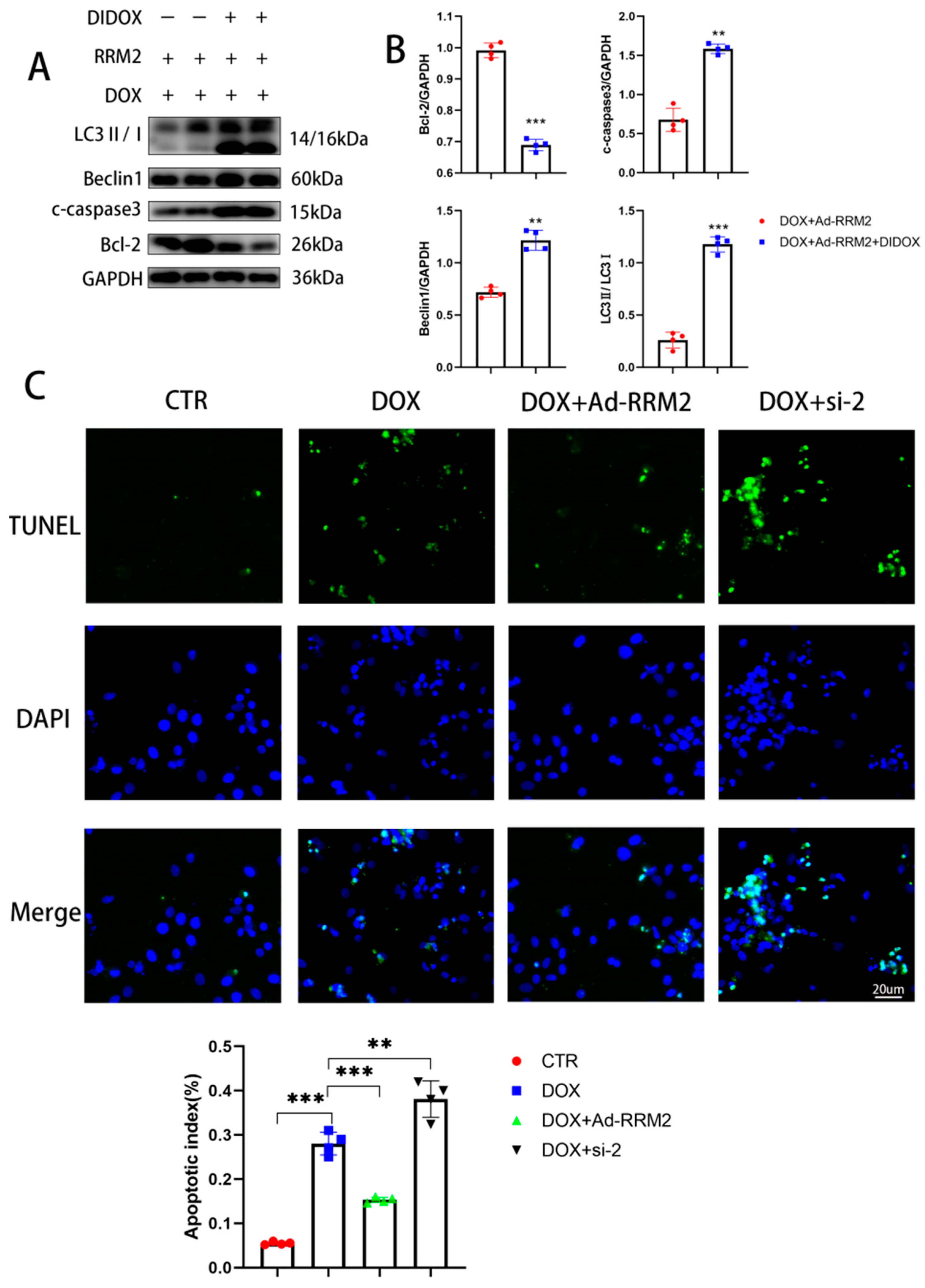
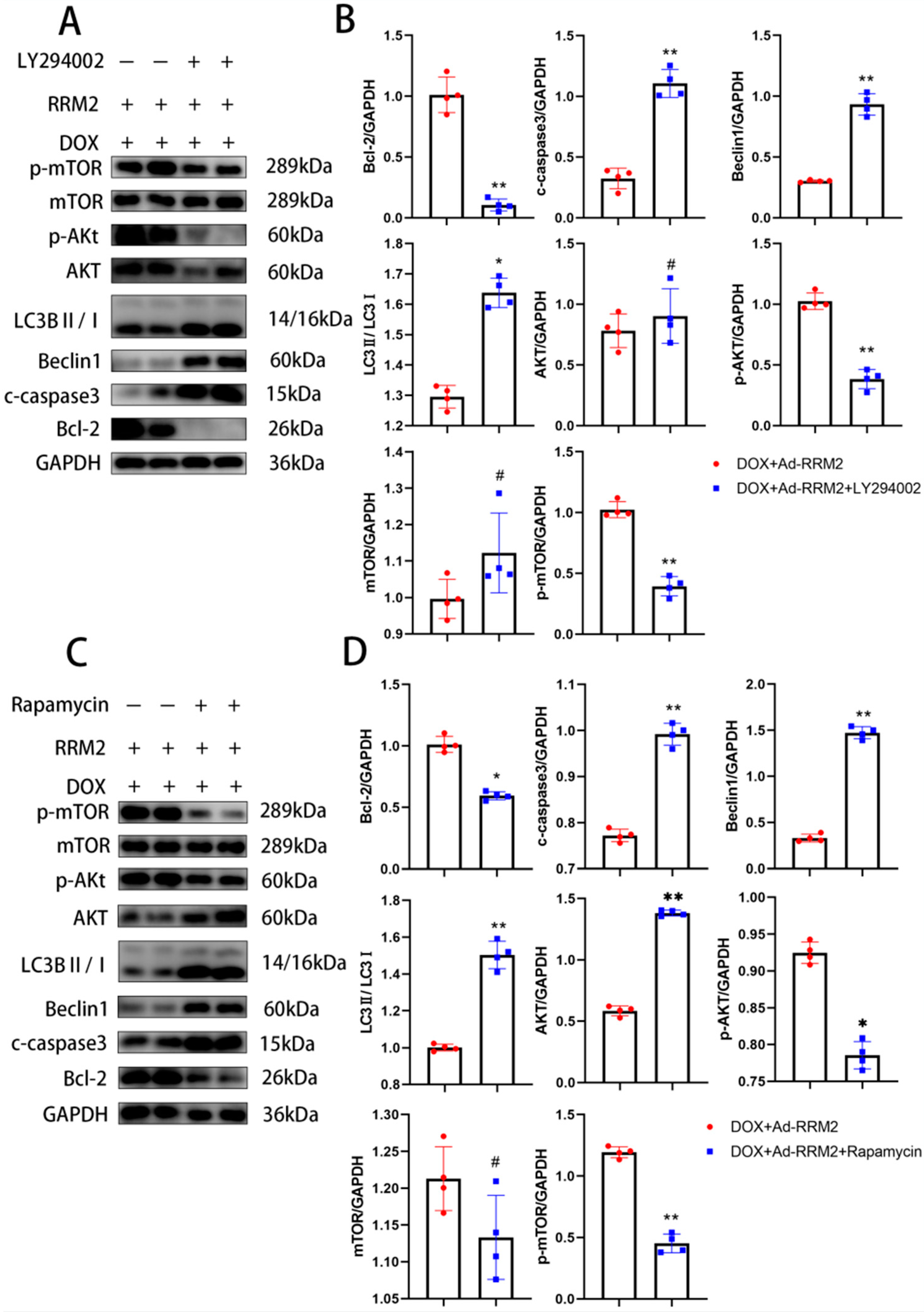
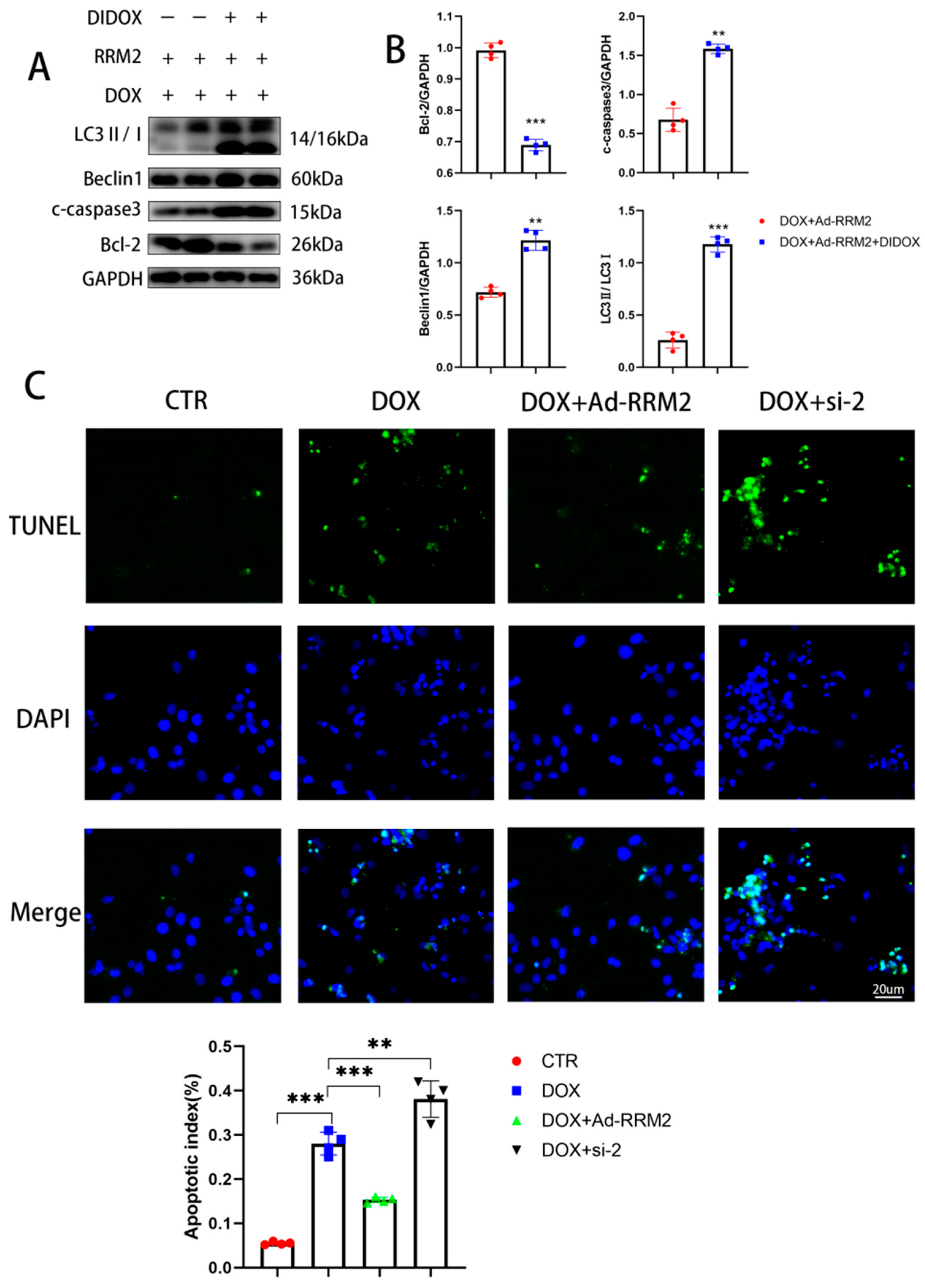
3.5. Blocking RRM2 Overexpression Can Reverse Its Protective Effect
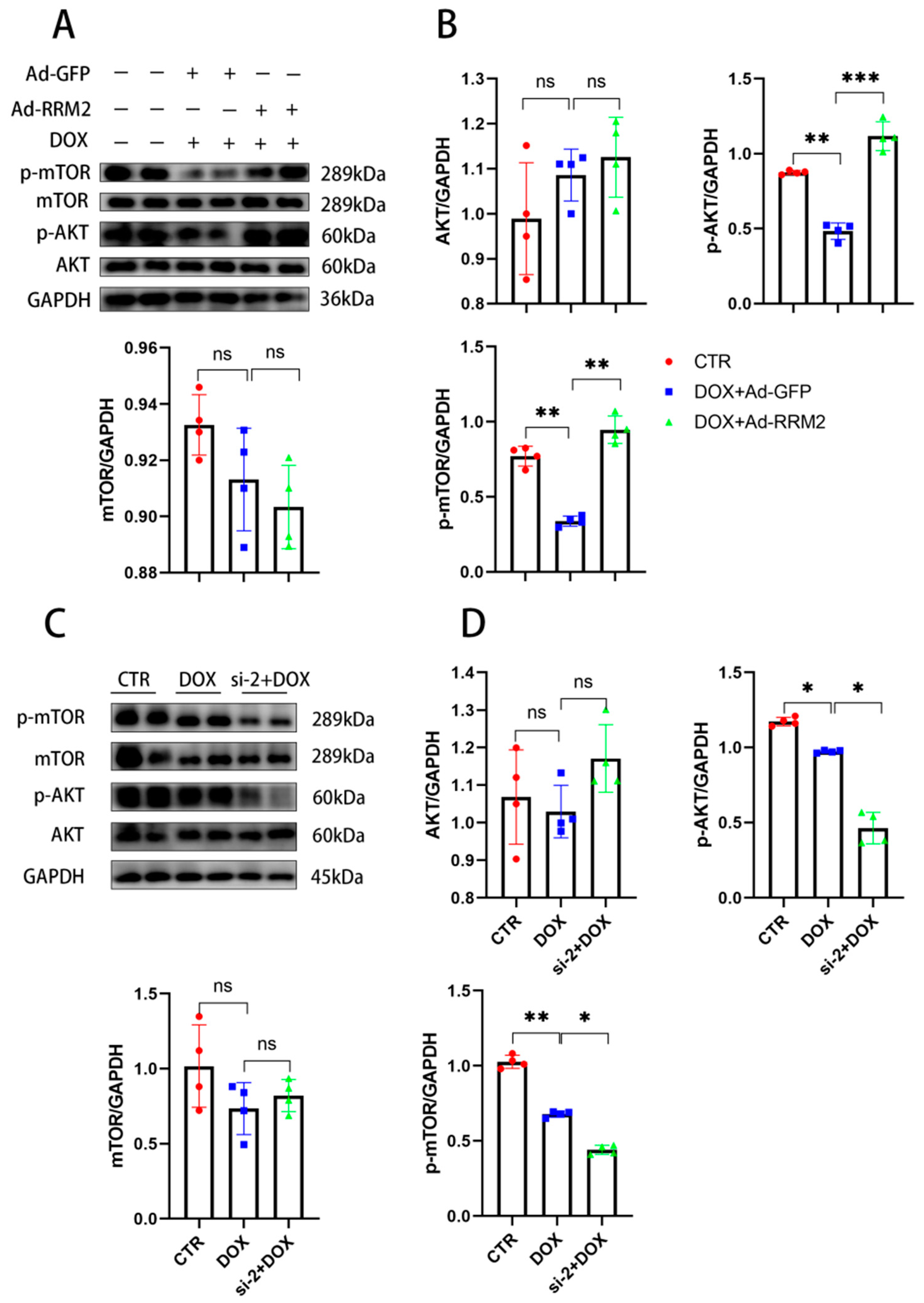
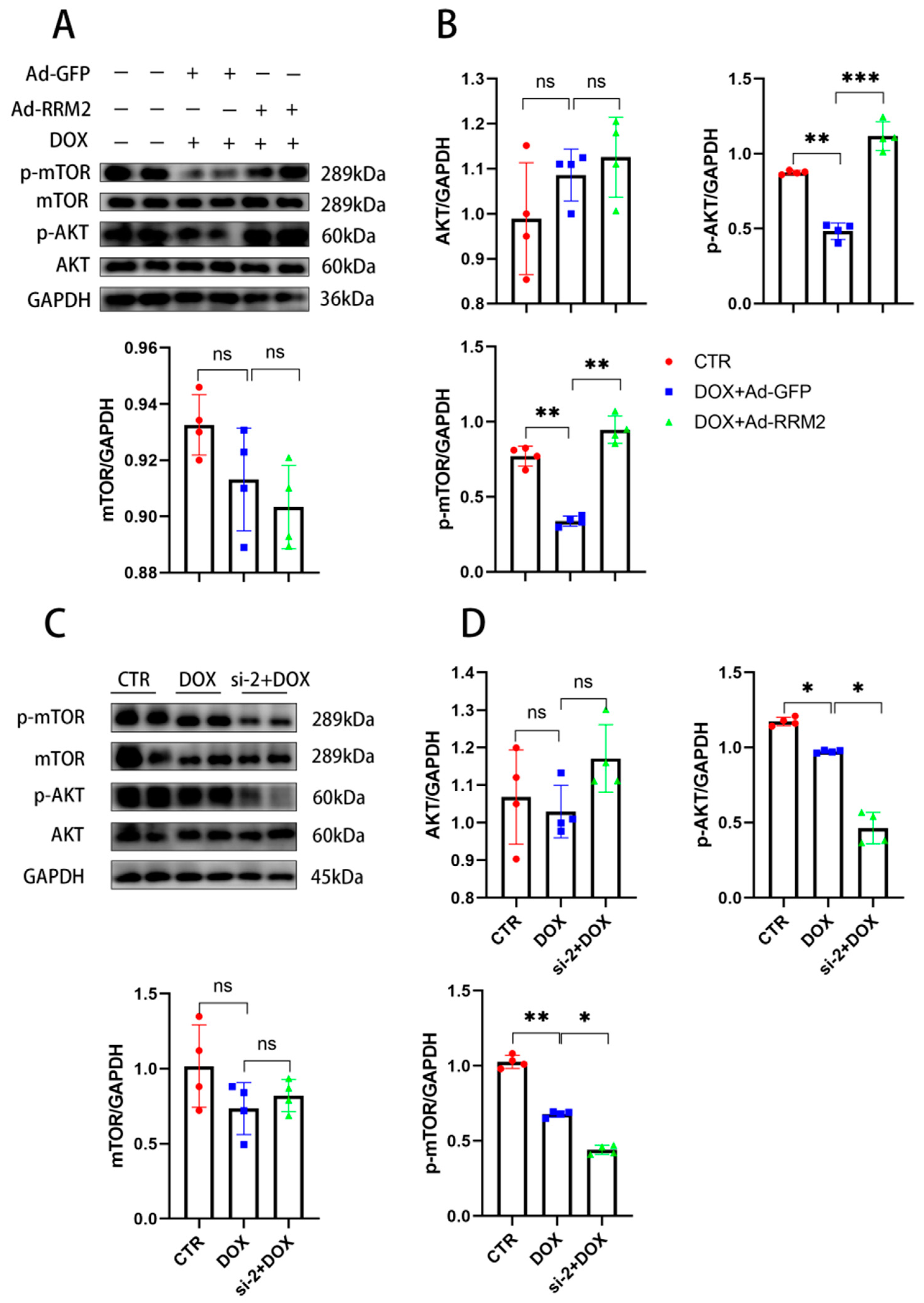
3.6. AKT/mTOR Signaling Is Involved in Regulating RRM2 in Cardiomyocytes
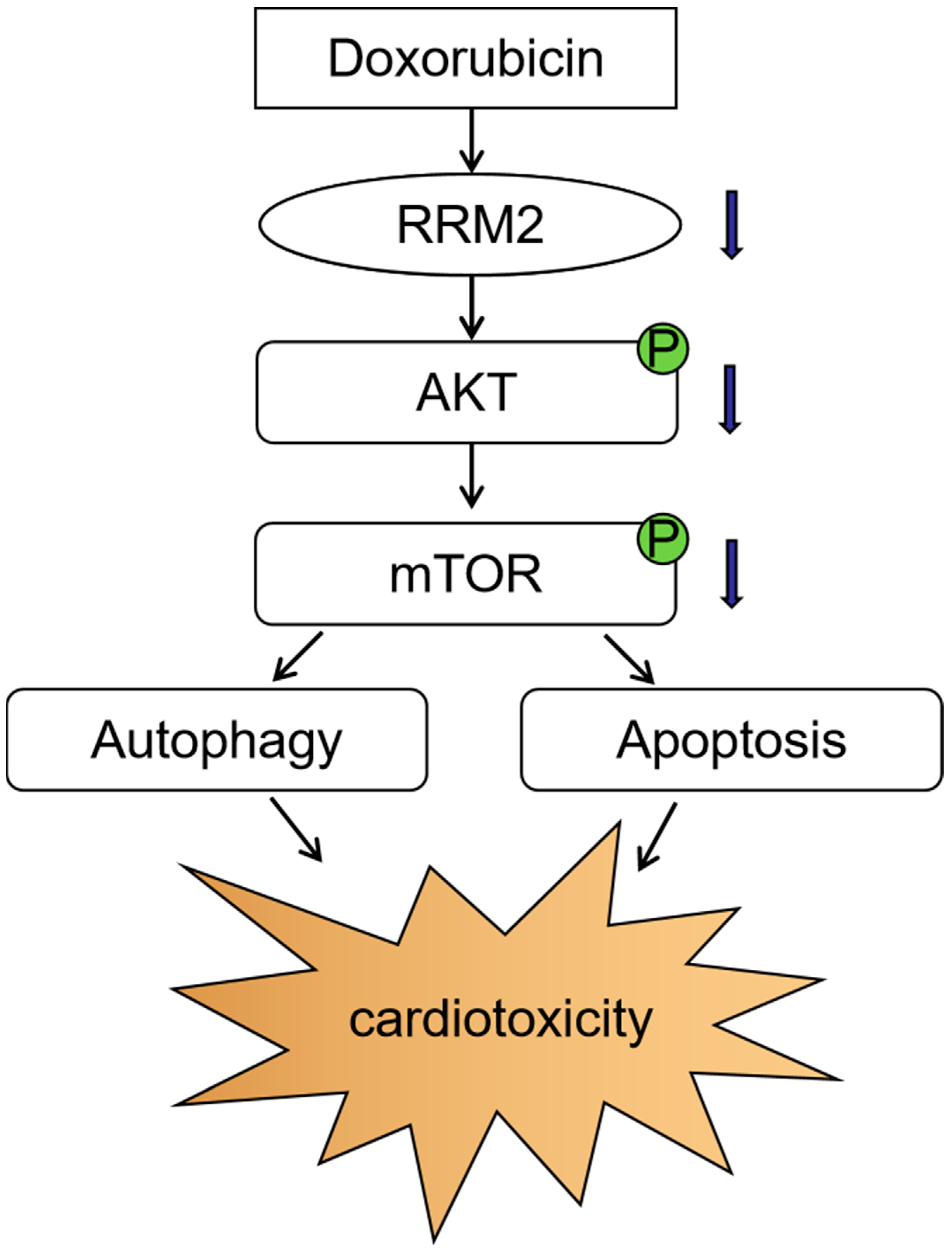
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Herrmann, J. Adverse cardiac effects of cancer therapies: Cardiotoxicity and arrhythmia. Nat. Rev. Cardiol. 2020, 17, 474–502. [Google Scholar] [CrossRef]
- Lu, J.; Li, J.; Hu, Y.; Guo, Z.; Sun, D.; Wang, P.; Guo, K.; Duan, D.D.; Gao, S.; Jiang, J.; et al. Chrysophanol protects against doxorubicin-induced cardiotoxicity by suppressing cellular PARylation. Acta Pharm. Sin. B 2019, 9, 782–793. [Google Scholar] [CrossRef] [PubMed]
- Gorini, S.; De Angelis, A.; Berrino, L.; Malara, N.; Rosano, G.; Ferraro, E. Chemotherapeutic Drugs and Mitochondrial Dysfunction: Focus on Doxorubicin, Trastuzumab, and Sunitinib. Oxidative Med. Cell. Longev. 2018, 2018, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Wang, Y.; Zheng, D.; Wei, M.; Xu, H.; Peng, T. Rac1 signalling mediates doxorubicin-induced cardiotoxicity through both reactive oxygen species-dependent and -independent pathways. Cardiovasc. Res. 2012, 97, 77–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, J.; Fan, Y.-Q.; Zhang, H.-L.; Pan, J.-A.; Yu, J.-Y.; Zhang, J.-F.; Wang, C.-Q. Resveratrol suppresses doxorubicin-induced cardiotoxicity by disrupting E2F1 mediated autophagy inhibition and apoptosis promotion. Biochem. Pharmacol. 2018, 150, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Hu, W.; Song, Z.-P.; Chen, Y.-G.; Zhang, D.-D.; Wang, C.-Q. Resveratrol-induced autophagy promotes survival and attenuates doxorubicin-induced cardiotoxicity. Int. Immunopharmacol. 2016, 32, 1–7. [Google Scholar] [CrossRef]
- Russo, M.; Guida, F.; Paparo, L.; Trinchese, G.; Aitoro, R.; Avagliano, C.; Fiordelisi, A.; Napolitano, F.; Mercurio, V.; Sala, V.; et al. The novel butyrate derivative phenylalanine-butyramide protects from doxorubicin-induced cardiotoxicity. Eur. J. Heart Fail. 2019, 21, 519–528. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, C.; Kong, C.-Y.; Song, P.; Wu, H.-M.; Xu, S.-C.; Yuan, Y.-P.; Deng, W.; Ma, Z.-G.; Tang, Q.-Z. FNDC5 alleviates oxidative stress and cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via activating AKT. Cell Death Differ. 2020, 27, 540–555. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Qi, Y.; Xu, L.; Tao, X.; Han, X.; Yin, L.; Peng, J. MicroRNA-140-5p aggravates doxorubicin-induced cardiotoxicity by promoting myocardial oxidative stress via targeting Nrf2 and Sirt2. Redox Biol. 2018, 15, 284–296. [Google Scholar] [CrossRef]
- Dong, Q.; Chen, L.; Lu, Q.; Sharma, S.; Li, L.; Morimoto, S.; Wang, G. Quercetin attenuates doxorubicin cardiotoxicity by modulating Bmi-1 expression. J. Cereb. Blood Flow Metab. 2014, 171, 4440–4454. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.-P.; Ma, Z.-G.; Zhang, X.; Xu, S.-C.; Zeng, X.-F.; Yang, Z.; Deng, W.; Tang, Q.-Z. CTRP3 protected against doxorubicin-induced cardiac dysfunction, inflammation and cell death via activation of Sirt1. J. Mol. Cell. Cardiol. 2018, 114, 38–47. [Google Scholar] [CrossRef]
- Rasmussen, R.D.; Gajjar, M.K.; Tuckova, L.; Jensen, K.E.; Maya-Mendoza, A.; Holst, C.B.; Møllgaard, K.; Rasmussen, J.S.; Brennum, J.; Bartek, J.; et al. BRCA1-regulated RRM2 expression protects glioblastoma cells from endogenous replication stress and promotes tumorigenicity. Nat. Commun. 2016, 7, 13398. [Google Scholar] [CrossRef]
- Wang, N.; Zhan, T.; Ke, T.; Huang, X.; Ke, D.; Wang, Q.; Li, H. Increased expression of RRM2 by human papillomavirus E7 oncoprotein promotes angiogenesis in cervical cancer. Br. J. Cancer 2014, 110, 1034–1044. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Zheng, J.; Chen, S.; Huang, B.; Li, G.; Feng, Z.; Wang, J.; Xu, S. RRM2 promotes the progression of human glioblastoma. J. Cell. Physiol. 2018, 233, 6759–6767. [Google Scholar] [CrossRef]
- Li, J.; Pang, J.; Liu, Y.; Zhang, J.; Zhang, C.; Shen, G.; Song, L. Suppression of RRM2 inhibits cell proliferation, causes cell cycle arrest and promotes the apoptosis of human neuroblastoma cells and in human neuroblastoma RRM2 is suppressed following chemotherapy. Oncol. Rep. 2018, 40, 355–360. [Google Scholar] [CrossRef] [Green Version]
- Jin, C.-Y.; Du, L.; Nuerlan, A.-H.; Wang, X.-L.; Yang, Y.-W.; Guo, R. High expression of RRM2 as an independent predictive factor of poor prognosis in patients with lung adenocarcinoma. Aging 2021, 13, 3518–3535. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Luo, H.; Cao, J.; Gao, C.; Fa, X.; Wang, G. Independent prognostic implications of RRM2 in lung adenocarcinoma. J. Cancer 2020, 11, 7009–7022. [Google Scholar] [CrossRef] [PubMed]
- Kolwicz, S.C.; Odom, G.L.; Nowakowski, S.G.; Moussavi-Harami, F.; Chen, X.; Reinecke, H.; Hauschka, S.D.; Murry, C.E.; Mahairas, G.G.; Regnier, M. AAV6-mediated Cardiac-specific Overexpression of Ribonucleotide Reductase Enhances Myocardial Contractility. Mol. Ther. 2016, 24, 240–250. [Google Scholar] [CrossRef] [Green Version]
- Louch, W.E.; Sheehan, K.A.; Wolska, B.M. Methods in cardiomyocyte isolation, culture, and gene transfer. J. Mol. Cell. Cardiol. 2011, 51, 288–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; Zhang, X.; Zhang, N.; Wei, W.; Li, L.; Ma, Z.; Tang, Q. Osteocrin attenuates inflammation, oxidative stress, apoptosis, and cardiac dysfunction in doxorubicin-induced cardiotoxicity. Clin. Transl. Med. 2020, 10, e124. [Google Scholar] [CrossRef]
- Shabalala, S.; Muller, C.; Louw, J.; Johnson, R. Polyphenols, autophagy and doxorubicin-induced cardiotoxicity. Life Sci. 2017, 180, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sun, Z.; Lin, N.; Lu, W.; Huang, X.; Weng, J.; Sun, S.; Zhang, C.; Yang, Q.; Zhou, G.; et al. Fucoidan from Fucus vesiculosus attenuates doxorubicin-induced acute cardiotoxicity by regulating JAK2/STAT3-mediated apoptosis and autophagy. Biomed. Pharmacother. 2020, 130, 110534. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhang, F.; Sun, P. miR-140-3p impedes the proliferation of human cervical cancer cells by targeting RRM2 to induce cell-cycle arrest and early apoptosis. Bioorgan. Med. Chem. 2020, 28, 115283. [Google Scholar] [CrossRef] [PubMed]
- Hullin, R.; Métrich, M.; Sarre, A.; Basquin, D.; Maillard, M.; Regamey, J.; Martin, D. Diverging effects of enalapril or eplerenone in primary prevention against doxorubicin-induced cardiotoxicity. Cardiovasc. Res. 2017, 114, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Shang, Z.; Wang, P.; Li, S.; Zhang, Q.; Tian, H.; Ren, D.; Han, X. Protection of Luteolin-7-O-Glucoside Against Doxorubicin-Induced Injury Through PTEN/Akt and ERK Pathway in H9c2 Cells. Cardiovasc. Toxicol. 2015, 16, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Xu, Z.; Tian, J.; Liu, Q.; Dong, J.; Guo, L.; Hai, B.; Liu, X.; Yao, H.; Chen, Z.; et al. Pterostilbene inhibits hepatocellular carcinoma proliferation and HBV replication by targeting ribonucleo-tide reductase M2 protein. Am. J. Cancer Res. 2021, 11, 2975–2989. [Google Scholar] [PubMed]
- Shu, Z.; Li, Z.; Huang, H.; Chen, Y.; Fan, J.; Yu, L.; Wu, Z.; Tian, L.; Qi, Q.; Peng, S.; et al. Cell-cycle-dependent phosphorylation of RRM1 ensures efficient DNA replication and regulates cancer vulnerability to ATR inhibition. Oncogene 2020, 39, 1–13. [Google Scholar] [CrossRef]
- Jiang, K.; Zhi, T.; Xu, W.; Xu, X.; Wu, W.; Yu, T.; Nie, E.; Zhou, X.; Bao, Z.; Jin, X.; et al. Mi-croRNA-1468-5p inhibits glioma cell proliferation and induces cell cycle arrest by targeting RRM1. Am. J. Cancer Res. 2017, 7, 784–800. [Google Scholar]
- Zhang, X.; Taoka, R.; Liu, D.; Matsuoka, Y.; Tohi, Y.; Kakehi, Y.; Sugimoto, M. Knockdown of RRM1 with Adenoviral shRNA Vectors to Inhibit Tumor Cell Viability and Increase Chemotherapeutic Sensitivity to Gemcitabine in Bladder Cancer Cells. Int. J. Mol. Sci. 2021, 22, 4102. [Google Scholar] [CrossRef]
- Mazzu, Y.Z.; Armenia, J.; Chakraborty, G.; Yoshikawa, Y.; Coggins, S.A.; Nandakumar, S.; Gerke, T.A.; Pomerantz, M.M.; Qiu, X.; Zhao, H.; et al. A Novel Mechanism Driving Poor-Prognosis Prostate Cancer: Overexpression of the DNA Repair Gene, Ribonucleotide Reductase Small Subunit M2 (RRM2). Clin. Cancer Res. 2019, 25, 4480–4492. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.A.; Amin, A.R.; Wang, D.; Koenig, L.; Nannapaneni, S.; Chen, Z.; Wang, Z.; Sica, G.; Deng, X.; Chen, Z. (Georgia); et al. RRM2 Regulates Bcl-2 in Head and Neck and Lung Cancers: A Potential Target for Cancer Therapy. Clin. Cancer Res. 2013, 19, 3416–3428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammadi, M.; Arabi, L.; Alibolandi, M. Doxorubicin-loaded composite nanogels for cancer treatment. J. Control. Release 2020, 328, 171–191. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, D.; Colombo, A.; Bacchiani, G.; Tedeschi, I.; Meroni, C.A.; Veglia, F.; Civelli, M.; Lamantia, G.; Colombo, N.; Curigliano, G.; et al. Early Detection of Anthracycline Cardiotoxicity and Improvement with Heart Failure Therapy. Circulation 2015, 131, 1981–1988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, S.; Spilinga, M.; Albano, R.; Ferrauto, G.; Di Gregorio, E.; Casanova, E.; Balmativola, D.; Bonzano, A.; Boccaccio, C.; Sapino, A.; et al. Activation of the MET receptor attenuates doxorubicin-induced cardiotoxicity in vivo and in vitro. J. Cereb. Blood Flow Metab. 2020, 177, 3107–3122. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Gong, Y.; Hu, Y.; You, Y.; Wang, J.; Zhang, Z.; Wei, Z.; Tang, C. Peroxiredoxin-1 Overexpression Attenuates Doxorubicin-Induced Cardiotoxicity by Inhibiting Oxidative Stress and Cardiomyocyte Apoptosis. Oxidative Med. Cell. Longev. 2020, 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Korte, F.; Dai, J.; Buckley, K.; Feest, E.R.; Adamek, N.; Geeves, M.A.; Murry, C.E.; Regnier, M. Upregulation of cardiomyocyte ribonucleotide reductase increases intracellular 2 deoxy-ATP, contractility, and relaxation. J. Mol. Cell. Cardiol. 2011, 51, 894–901. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Huang, Y.; Que, B.; Chang, C.; Liu, W.; Hu, H.; Liu, L.; Shi, Y.; Wang, Y.; Wang, M.; et al. Interleukin-12p35 Knock Out Aggravates Doxorubicin-Induced Cardiac Injury and Dysfunction by Aggravating the Inflammatory Response, Oxidative Stress, Apoptosis and Autophagy in Mice. EBioMedicine 2018, 35, 29–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Yang, L.; Ma, J.; Lu, L.; Wang, X.; Ren, J.; Yang, J. Rutin attenuates doxorubicin-induced cardiotoxicity via regulating autophagy and apoptosis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 1904–1911. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.-M.; Li, C.-B.; Liu, Q.-L.; Li, P.; Yang, H. Ginsenoside Rg1 Prevents Doxorubicin-Induced Cardiotoxicity through the Inhibition of Autophagy and Endoplasmic Reticulum Stress in Mice. Int. J. Mol. Sci. 2018, 19, 3658. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.-N.; Shu, Y.; Chen, C.-G.; Li, X.-Q.; Li, M.-Y.; Zhao, X.-H.; Wang, S.; Li, J. Acquired tamoxifen resistance is surmounted by GW8510 through ribonucleotide reductase M2 downregulation-mediated autophagy induction. Biochem. Biophys. Res. Commun. 2020, 528, 554–560. [Google Scholar] [CrossRef]
- Zhang, S.; Yan, L.; Cui, C.; Wang, Z.; Wu, J.; Lv, A.; Zhao, M.; Dong, B.; Zhang, W.; Guan, X.; et al. Downregulation of RRM2 Attenuates Retroperitoneal Liposarcoma Progression via the Akt/mTOR/4EBP1 Pathway: Clinical, Biological, and Therapeutic Significance. OncoTargets Ther. 2020, 13, 6523–6537. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiao, Y.; Li, Y.; Zhang, J.; Zhang, S.; Zha, Y.; Wang, J. RRM2 Alleviates Doxorubicin-Induced Cardiotoxicity through the AKT/mTOR Signaling Pathway. Biomolecules 2022, 12, 299. https://doi.org/10.3390/biom12020299
Jiao Y, Li Y, Zhang J, Zhang S, Zha Y, Wang J. RRM2 Alleviates Doxorubicin-Induced Cardiotoxicity through the AKT/mTOR Signaling Pathway. Biomolecules. 2022; 12(2):299. https://doi.org/10.3390/biom12020299
Chicago/Turabian StyleJiao, Yuheng, Yanyan Li, Jiayan Zhang, Song Zhang, Yafang Zha, and Jian Wang. 2022. "RRM2 Alleviates Doxorubicin-Induced Cardiotoxicity through the AKT/mTOR Signaling Pathway" Biomolecules 12, no. 2: 299. https://doi.org/10.3390/biom12020299
APA StyleJiao, Y., Li, Y., Zhang, J., Zhang, S., Zha, Y., & Wang, J. (2022). RRM2 Alleviates Doxorubicin-Induced Cardiotoxicity through the AKT/mTOR Signaling Pathway. Biomolecules, 12(2), 299. https://doi.org/10.3390/biom12020299