
Interaction with the Assembly Chaperone Ump1 Promotes Incorporation of the β7 Subunit into Half-Proteasome Precursor Complexes Driving Their Dimerization


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Yeast Methods
2.2. Preparation and Analysis of Yeast Protein Extracts
2.3. Production and Purification of Proteins in Escherichia coli
2.4. In Vitro Binding Assay Using Ni-NTA Pulldown
2.5. Fluorescence Microscopy-Based On-Bead Binding Assay
3. Results
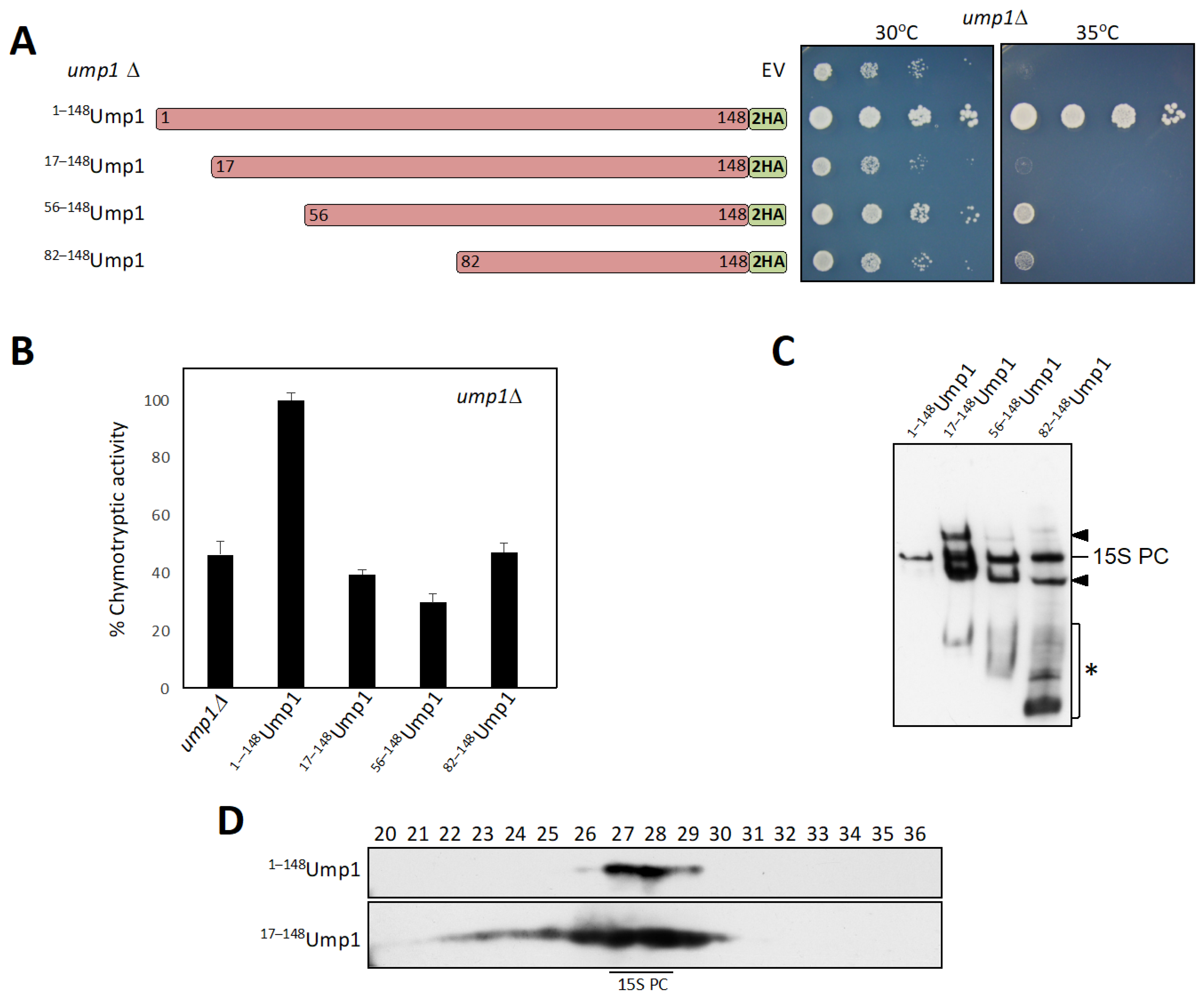
3.1. N-Terminally Truncated Ump1 Incorporates into Proteasome Assembly Intermediates
3.2. Autocatalytic Processing of β5 Is Incomplete in 17–148Ump1 Cells
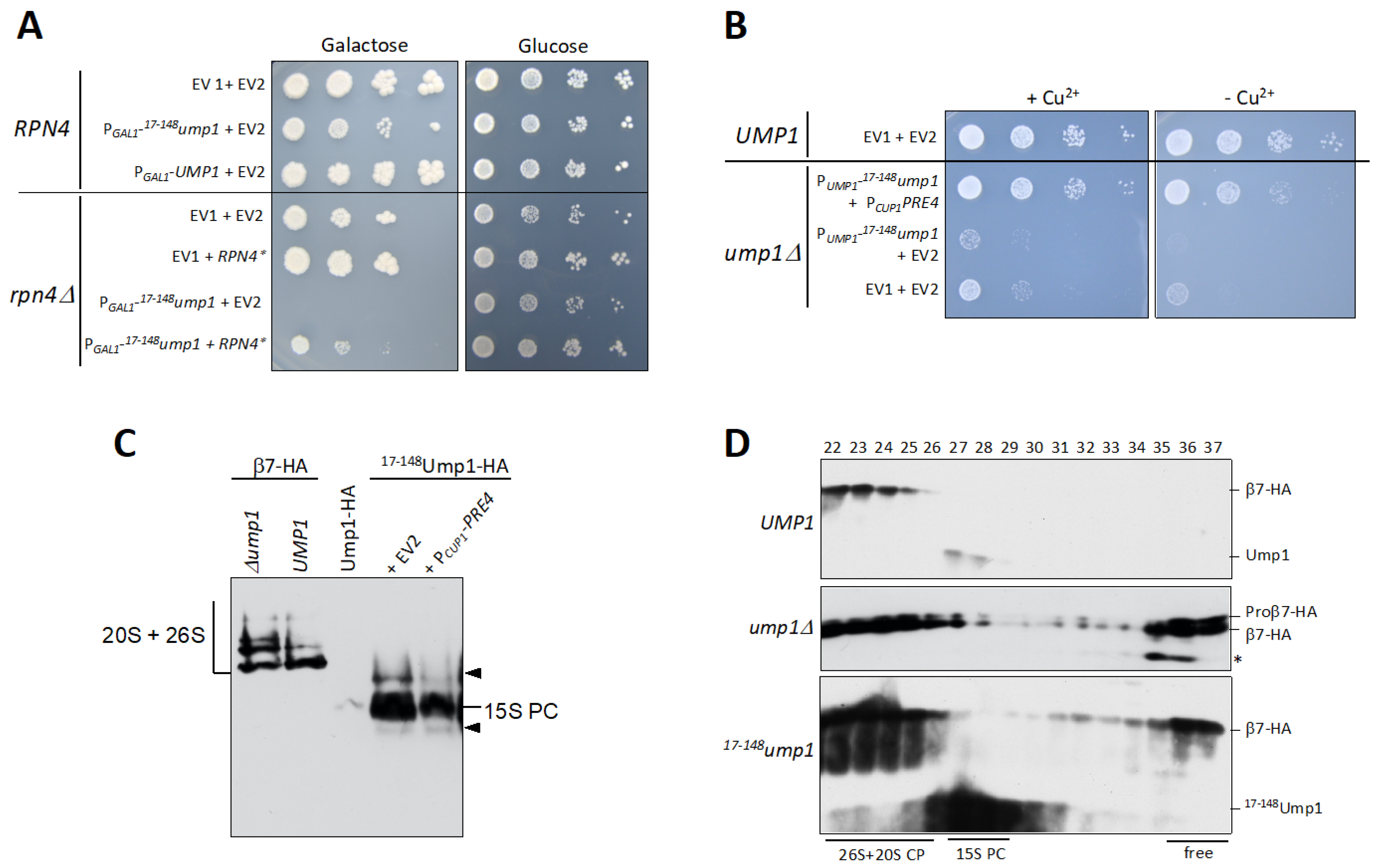
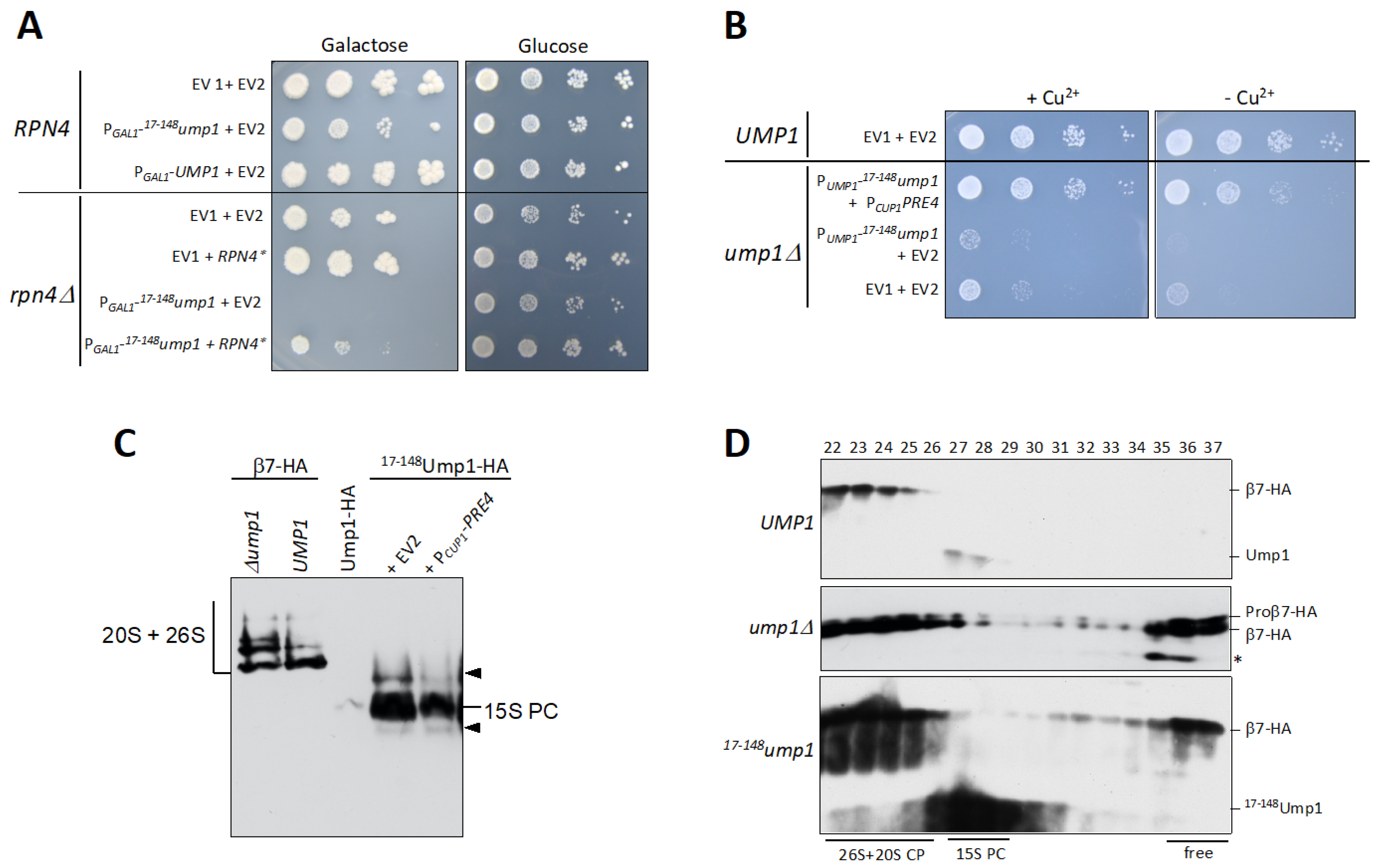
3.3. RPN4 Is Required to Tolerate N-Terminally Truncated Ump1
3.4. 17–148Ump1 Phenotypes Are Suppressed by Increased Levels of the β7 Proteasome Subunit
3.5. Free β7 Subunits Accumulate in ump1∆ and 17–148ump1 Cells
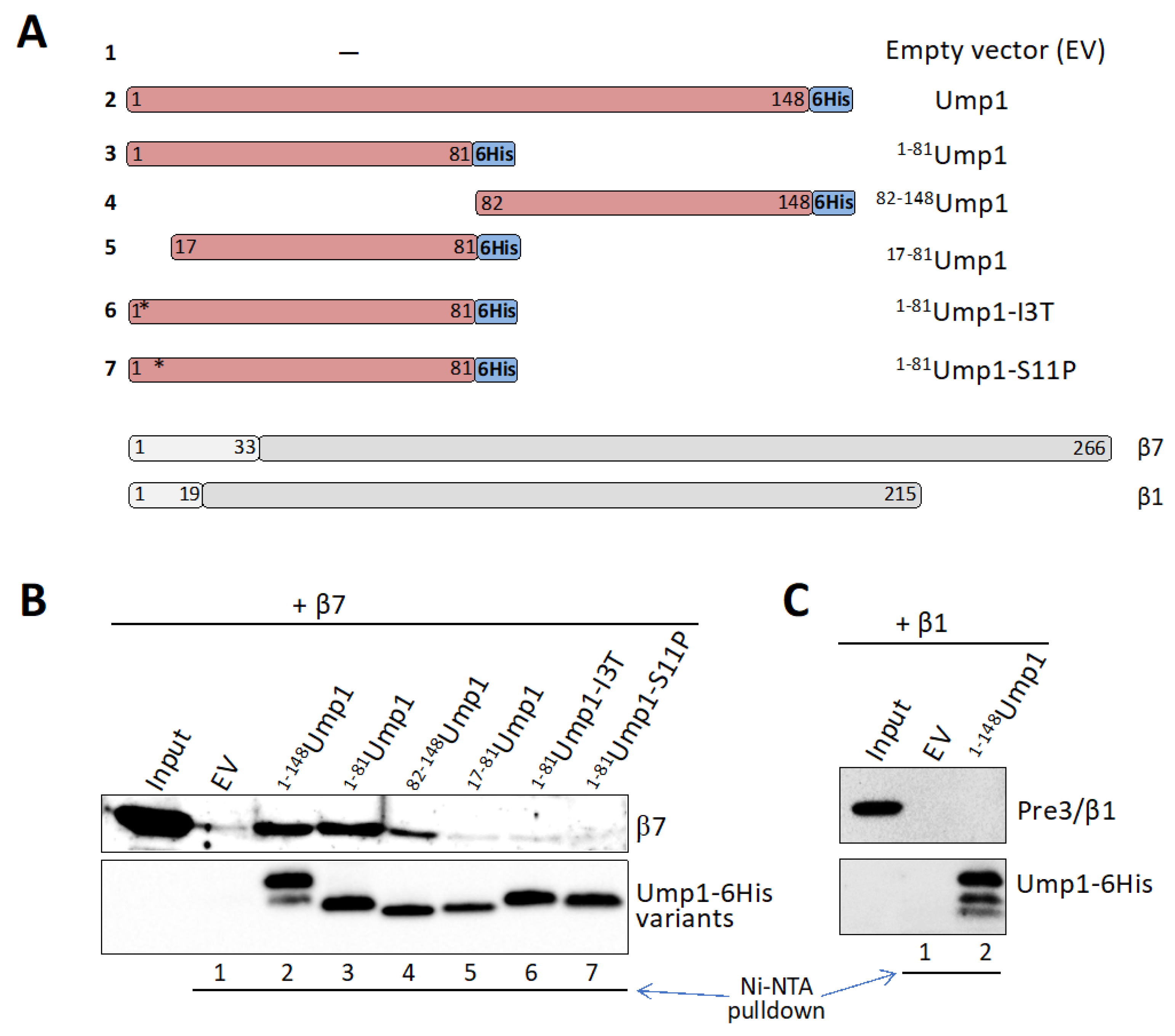
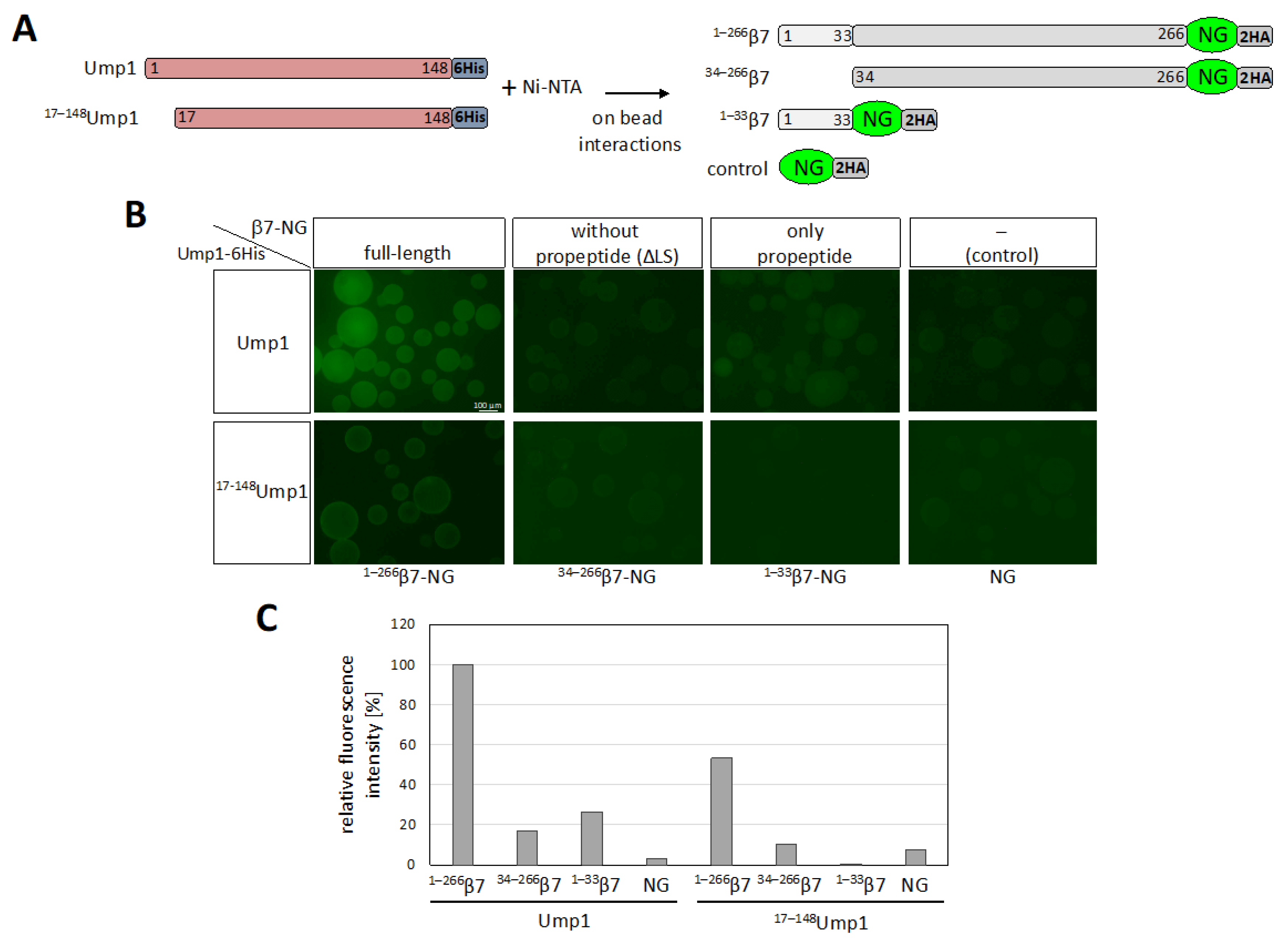
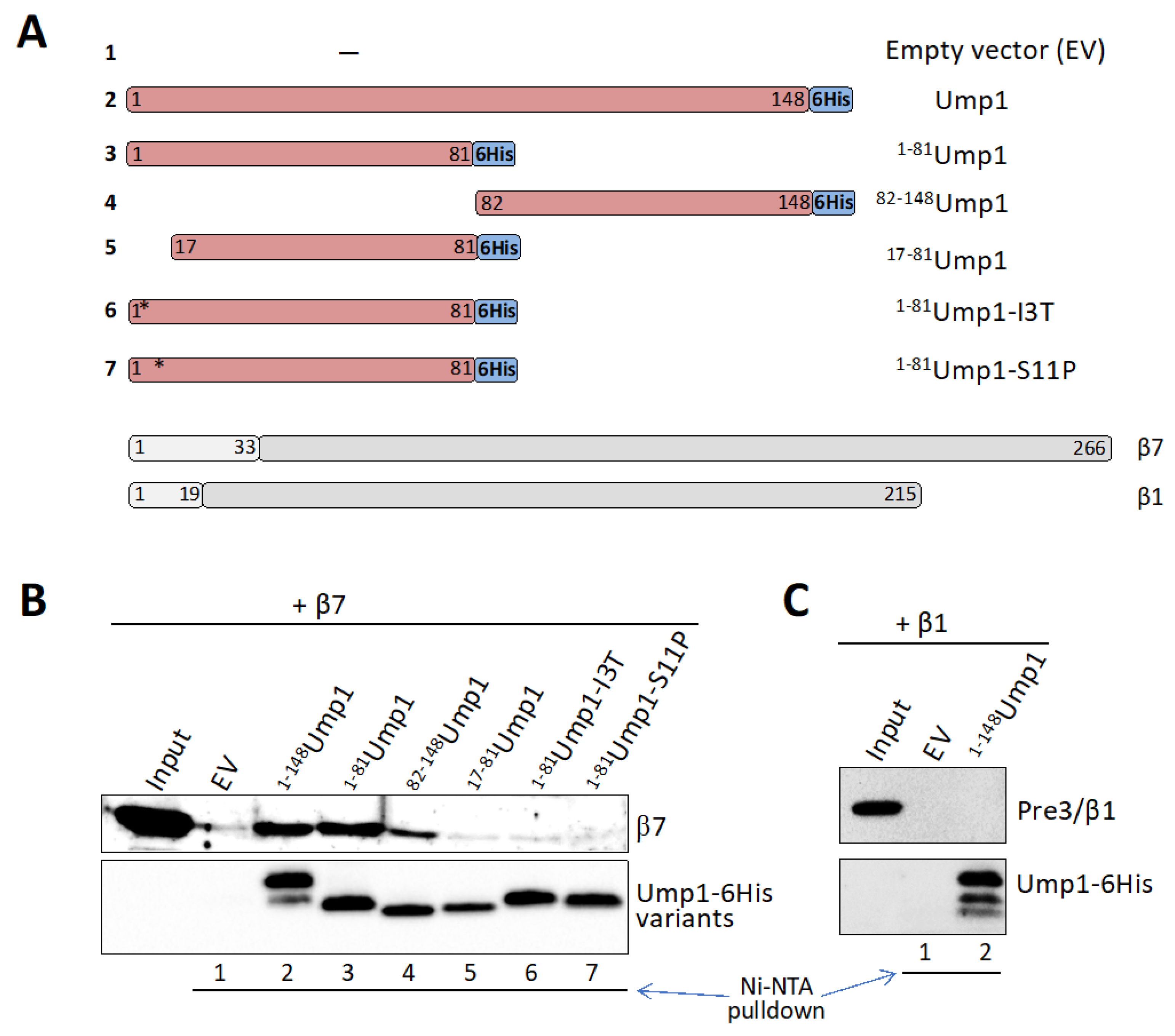
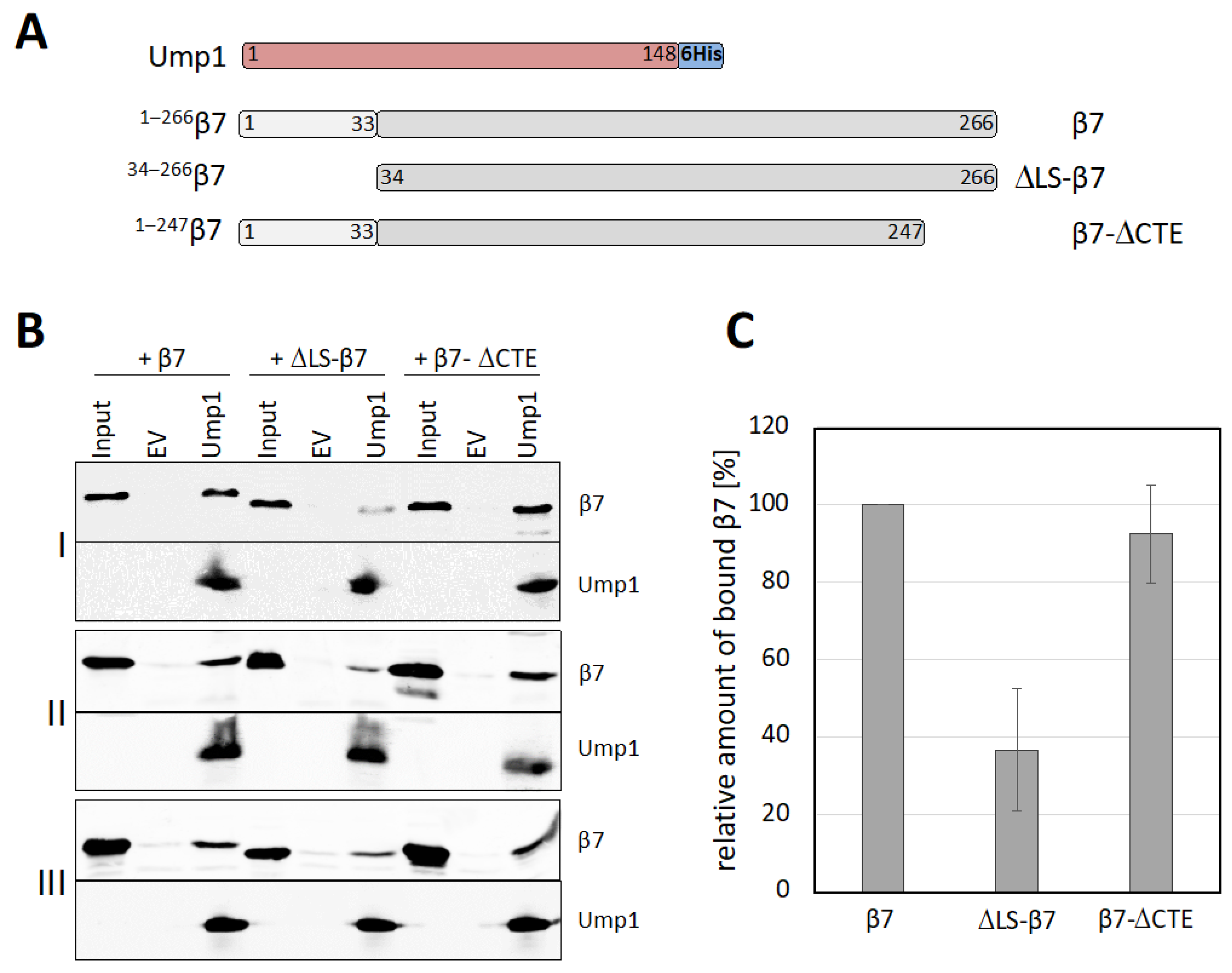
3.6. β7 Binds to Ump1 In Vitro
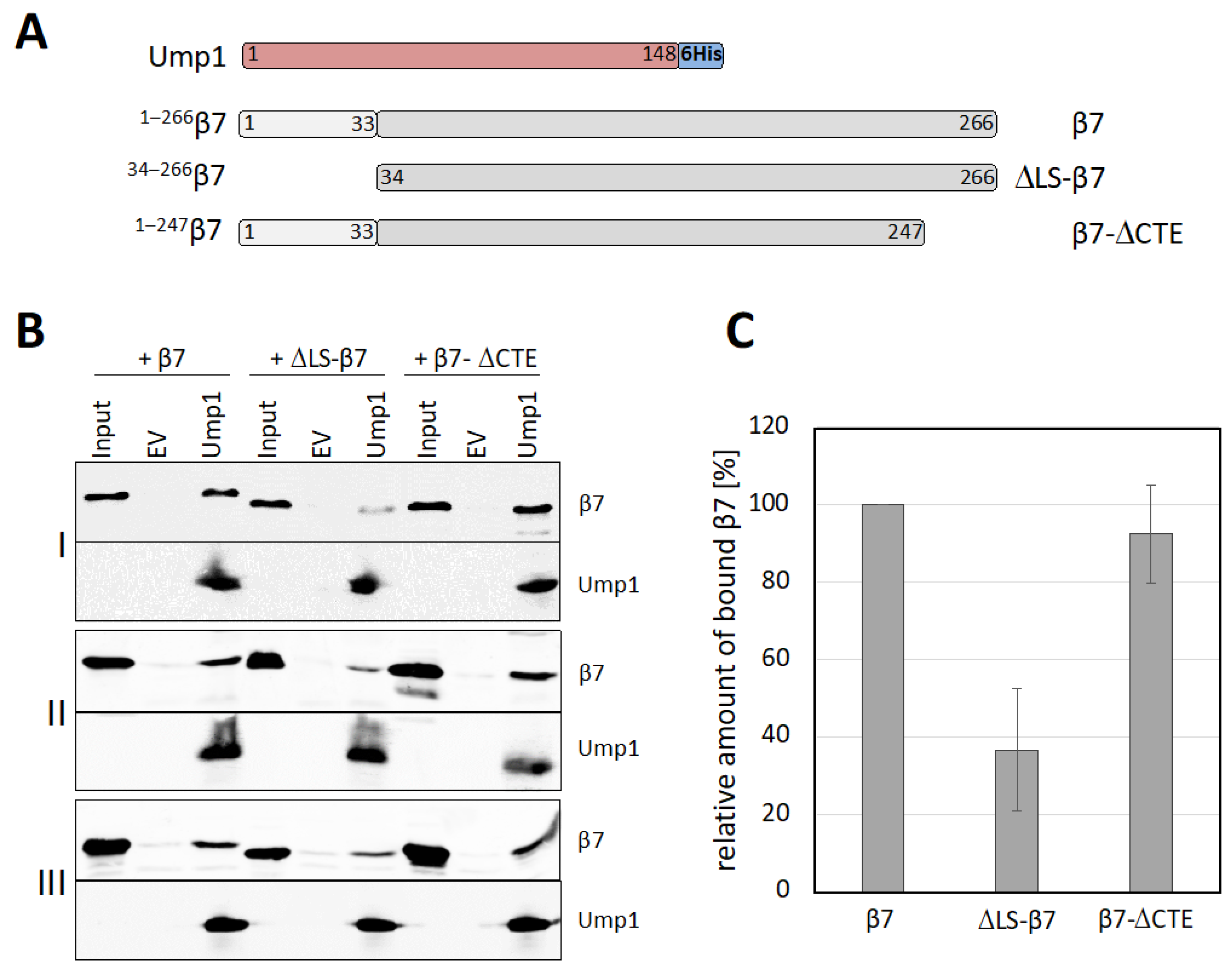
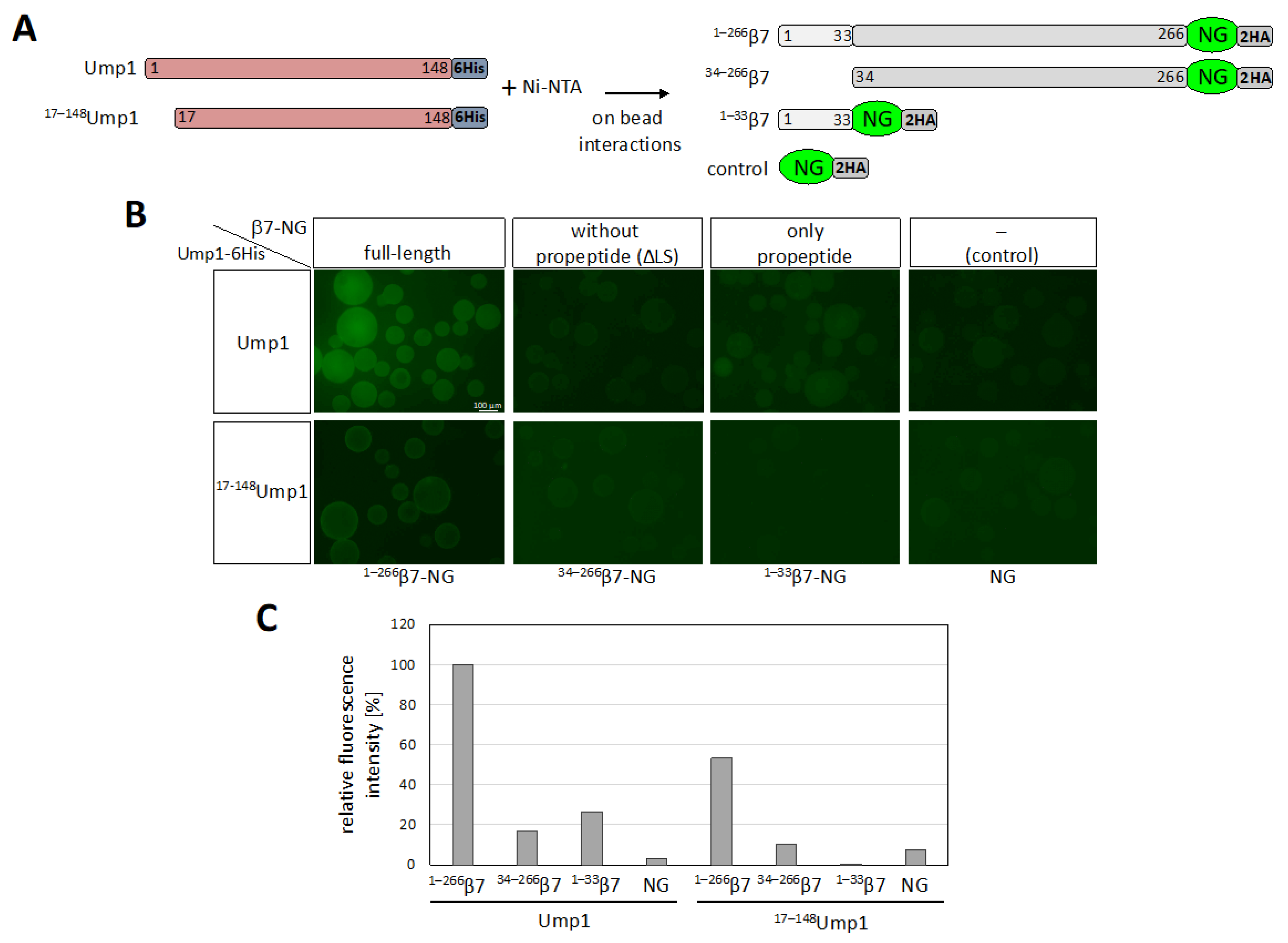
3.7. β7 Propeptide Promotes Binding to Ump1 In Vitro
4. Discussion
4.1. Ump1 N-Terminal Domain Is Required for Efficient Dimerization of 15S PCs
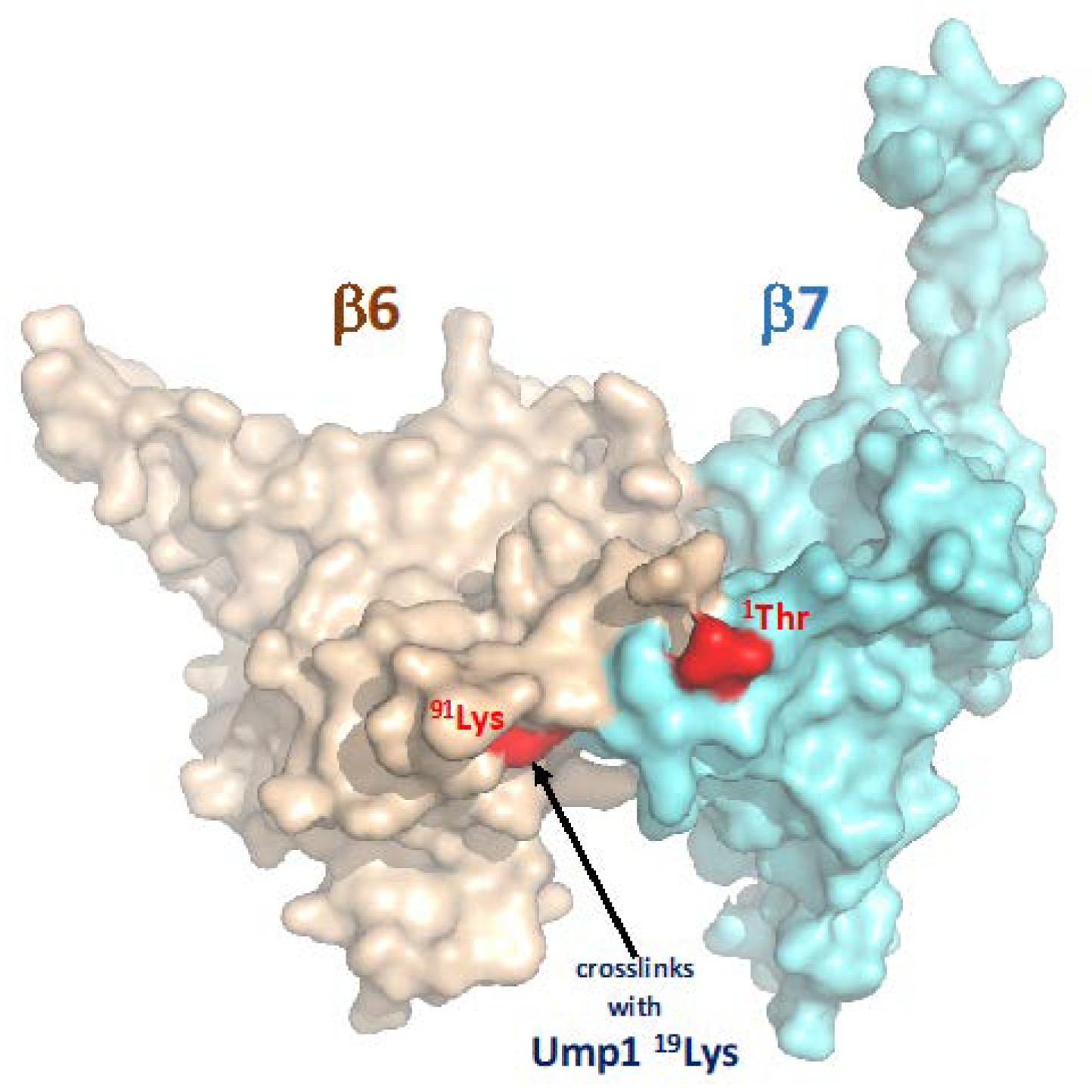
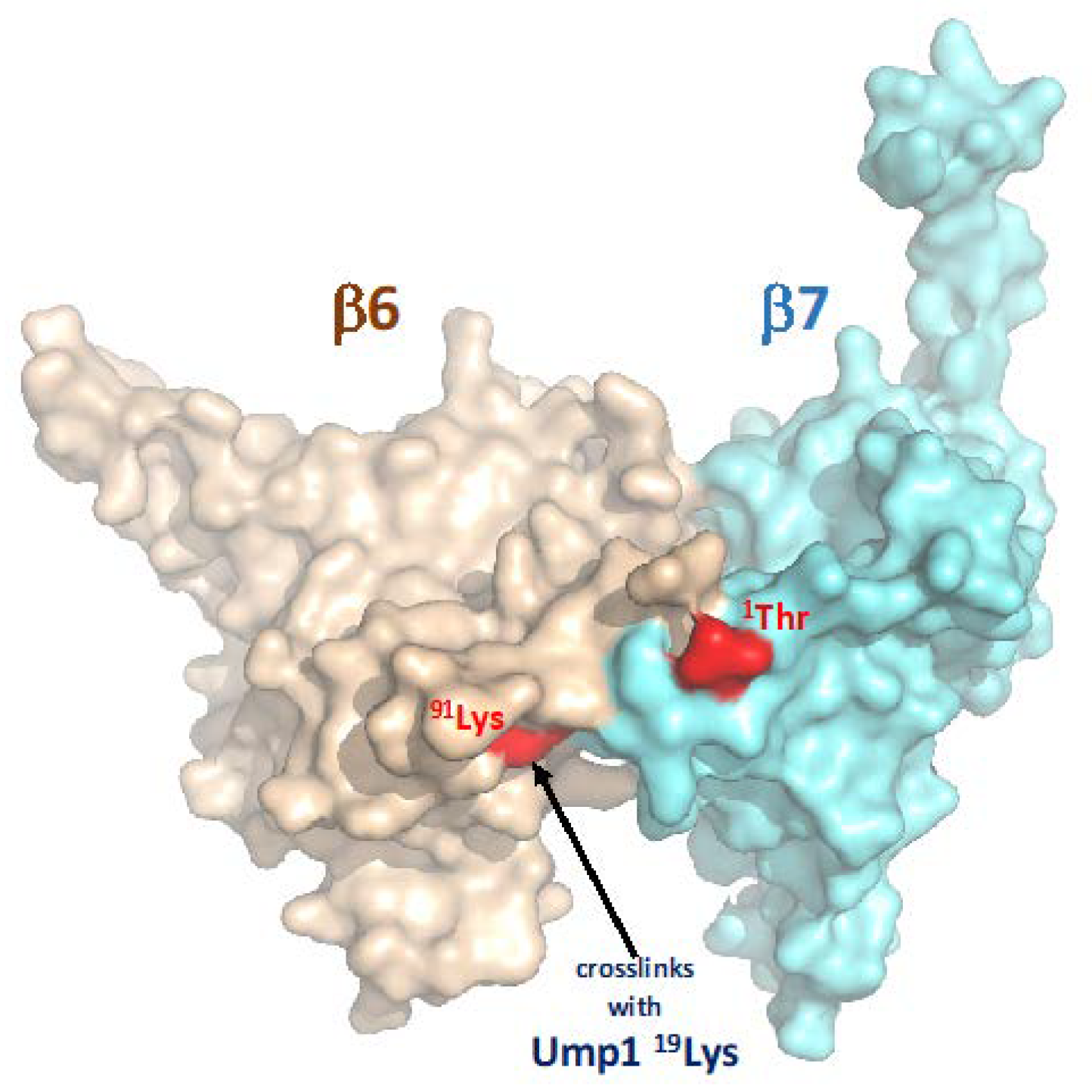
4.2. Ump1 N-Terminal Domain Interacts with Pro-β7 to Promote 15S PC Dimerization
4.3. Possible Additional Functions of the Ump1 N-Terminal Domain
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kato, K.; Satoh, T. Structural insights on the dynamics of proteasome formation. Biophys. Rev. 2018, 10, 597–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budenholzer, L.; Cheng, C.L.; Li, Y.; Hochstrasser, M. Proteasome structure and assembly. J. Mol. Biol. 2017, 429, 3500–3524. [Google Scholar] [CrossRef] [PubMed]
- Collins, G.A.; Goldberg, A.L. The logic of the 26S proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, A.J.; Palanimurugan, R.; Matias, A.C.; Ramos, P.C.; Dohmen, R.J. Catalytic mechanism and assembly of the proteasome. Chem. Rev. 2009, 109, 1509–1536. [Google Scholar] [CrossRef]
- Ramos, P.C.; Höckendorff, J.; Johnson, E.S.; Varshavsky, A.; Dohmen, R.J. Ump1p is required for proper maturation of the 20S proteasome and becomes its substrate upon completion of the assembly. Cell 1998, 92, 489–499. [Google Scholar] [CrossRef] [Green Version]
- Kock, M.; Nunes, M.M.; Hemann, M.; Kube, S.; Dohmen, R.J.; Herzog, F.; Ramos, P.C.; Wendler, P. Proteasome assembly from 15S precursors involves major conformational changes and recycling of the Pba1–Pba2 chaperone. Nat. Commun. 2015, 6, 6123. [Google Scholar] [CrossRef] [Green Version]
- Heink, S.; Ludwig, D.; Kloetzel, P.M.; Krüger, E. IFN-gamma-induced immune adaptation of the proteasome system is an accelerated and transient response. Proc. Natl. Acad. Sci. USA 2005, 102, 9241–9246. [Google Scholar] [CrossRef] [Green Version]
- Dahlqvist, J.; Klar, J.; Tiwari, N.; Schuster, J.; Törmä, H.; Badhai, J.; Pujol, R.; van Steensel, M.A.; Brinkhuizen, T.; Gijezen, L.; et al. A single-nucleotide deletion in the POMP 5′ UTR causes a transcriptional switch and altered epidermal proteasome distribution in KLICK genodermatosis. Am. J. Hum. Genet. 2010, 86, 596–603. [Google Scholar] [CrossRef] [Green Version]
- Brehm, A.; Liu, Y.; Sheikh, A.; Marrero, B.; Omoyinmi, E.; Zhou, Q.; Montealegre, G.; Biancotto, A.; Reinhardt, A.; De Jesus, A.A.; et al. Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type-I-IFN production. J. Clin. Investig. 2015, 125, 4196–4211. [Google Scholar] [CrossRef]
- Gatz, S.A.; Salles, D.; Jacobsen, E.M.; Dörk, T.; Rausch, T.; Aydin, S.; Surowy, H.; Volcic, M.; Vogel, W.; Debatin, K.M.; et al. MCM3AP and POMP mutations cause a DNA-repair and DNA-damage-signaling defect in an immunodeficient child. Hum. Mutat. 2016, 37, 257–268. [Google Scholar] [CrossRef]
- Zieba, B.A.; Henry, L.; Lacroix, M.; Jemaà, M.; Lavabre-Bertrand, T.; Meunier, L.; Coux, O.; Stoebner, P.E. The proteasome maturation protein POMP increases proteasome assembly and activity in psoriatic lesional skin. J. Dermatol. Sci. 2017, 88, 10–19. [Google Scholar] [CrossRef] [Green Version]
- Poli, M.C.; Ebstein, F.; Nicholas, S.K.; de Guzman, M.M.; Forbes, L.R.; Chinn, I.K.; Mace, E.M.; Vogel, T.P.; Carisey, A.F.; Benavides, F.; et al. Heterozygous truncating variants in POMP escape nonsense-mediated decay and cause a unique immune dysregulatory syndrome. Am. J. Hum. Genet. 2018, 102, 1126–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meinhardt, A.; Ramos, P.C.; Dohmen, R.J.; Lucas, N.; Lee-Kirsch, M.A.; Becker, B.; de Laffolie, J.; Cunha, T.; Niehues, T.; Salzer, U.; et al. Curative Treatment of POMP-related autoinflammation and immune dysregulation (PRAID) by hematopoietic stem sell transplantation. J. Clin. Immunol. 2021, 41, 1664–1667. [Google Scholar] [CrossRef]
- Zhang, X.; Schulz, R.; Edmunds, S.; Krüger, E.; Markert, E.; Gaedcke, J.; Cormet-Boyaka, E.; Ghadimi, M.; Beissbarth, T.; Levine, A.J.; et al. MicroRNA-101 suppresses tumor cell proliferation by acting as an endogenous proteasome inhibitor via targeting the proteasome assembly factor POMP. Mol. Cell 2015, 59, 243–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sá-Moura, B.; Simões, A.M.; Fraga, J.; Fernandes, H.; Abreu, I.A.; Botelho, H.M.; Gomes, C.M.; Marques, A.J.; Dohmen, R.J.; Ramos, P.C.; et al. Biochemical and biophysical characterization of recombinant yeast proteasome maturation factor Ump1. Comput. Struct. Biotechnol. J. 2013, 7, e201304006. [Google Scholar] [CrossRef] [Green Version]
- Uekusa, Y.; Okawa, K.; Yagi-Utsumi, M.; Serve, O.; Nakagawa, Y.; Mizushima, T.; Yagi, H.; Saeki, Y.; Tanaka, K.; Kato, K. Backbone 1H, 13C, and 15N assignments of yeast Ump1, an intrinsically disordered protein that functions as a proteasome assembly chaperone. Biomol. NMR Assign. 2014, 8, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Schnell, H.M.; Walsh, R.M., Jr.; Rawson, S.; Kaur, M.; Bhanu, M.K.; Tian, G.; Prado, M.A.; Guerra-Moreno, A.; Paulo, J.A.; Gygi, S.P.; et al. Structures of chaperone-associated assembly intermediates reveal coordinated mechanisms of proteasome biogenesis. Nat. Struct. Mol. Biol. 2021, 28, 418–425. [Google Scholar] [CrossRef]
- Burri, L.; Höckendorff, J.; Boehm, U.; Klamp, T.; Dohmen, R.J.; Lévy, F. Identification and characterization of a mammalian protein interacting with 20S proteasome precursors. Proc. Natl. Acad. Sci. USA 2000, 97, 10348–10353. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Varshavsky, A. RPN4 is a ligand, substrate, and transcriptional regulator of the 26S proteasome: A negative feedback circuit. Proc. Natl. Acad. Sci. USA 2001, 98, 3056–3061. [Google Scholar] [CrossRef] [Green Version]
- London, M.K.; Keck, B.I.; Ramos, P.C.; Dohmen, R.J. Regulatory mechanisms controlling biogenesis of ubiquitin and the proteasome. FEBS Lett. 2004, 567, 259–264. [Google Scholar] [CrossRef] [Green Version]
- Dohmen, R.J.; Stappen, R.; McGrath, J.P.; Forrová, H.; Kolarov, J.; Goffeau, A.; Varshavsky, A. An essential yeast gene encoding a homolog of ubiquitin-activating enzyme. J. Biol. Chem. 1995, 270, 18099–18109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohmen, R.J.; London, M.K.; Glanemann, C.; Ramos, P.C. Assays for proteasome assembly and maturation. In Ubiquitin-Proteasome Protocols; Humana Press: Totowa, NJ, USA, 2005; Volume 301, pp. 243–254. [Google Scholar] [CrossRef] [PubMed]
- Hermanns, T.; Pichlo, C.; Woiwode, I.; Klopffleisch, K.; Witting, K.F.; Ovaa, H.; Baumann, U.; Hofmann, K. A family of unconventional deubiquitinases with modular chain specificity determinants. Nat. Commun. 2018, 9, 799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaner, N.C.; Lambert, G.G.; Chammas, A.; Ni, Y.; Cranfill, P.J.; Baird, M.A.; Sell, B.R.; Allen, J.R.; Day, R.N.; Israelsson, M.; et al. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Methods 2013, 10, 407–409. [Google Scholar] [CrossRef]
- Wang, X.; Xu, H.; Ha, S.-W.; Ju, D.; Xie, Y. Proteasomal degradation of Rpn4 in Saccharomyces cerevisiae is critical for cell viability under stressed conditions. Genetics 2010, 184, 335–342. [Google Scholar] [CrossRef] [Green Version]
- Marques, A.J.; Glanemann, C.; Ramos, P.C.; Dohmen, R.J. The C-terminal extension of the β7 subunit and activator complexes stabilize nascent 20 S proteasomes and promote their maturation. J. Biol. Chem. 2007, 282, 34869–34876. [Google Scholar] [CrossRef] [Green Version]
- Ramos, P.C.; Marques, A.J.; London, M.K.; Dohmen, R.J. Role of C-terminal extensions of subunits β2 and β7 in assembly and activity of eukaryotic proteasomes. J. Biol. Chem. 2004, 279, 14323–14330. [Google Scholar] [CrossRef] [Green Version]
- Hirano, Y.; Kaneko, T.; Okamoto, K.; Bai, M.; Yashiroda, H.; Furuyama, K.; Kato, K.; Tanaka, K.; Murata, S. Dissecting β-ring assembly pathway of the mammalian 20S proteasome. EMBO J. 2008, 27, 2204–2213. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Kusmierczyk, A.R.; Wong, P.; Emili, A.; Hochstrasser, M. β-Subunit appendages promote 20S proteasome assembly by overcoming an Ump1-dependent checkpoint. EMBO J. 2007, 26, 2339–2349. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, A.; Janek, K.; Braun, B.; Kloetzel, P.M.; Enenkel, C. 20 S proteasomes are imported as precursor complexes into the nucleus of yeast. J. Mol. Biol. 2002, 317, 401–413. [Google Scholar] [CrossRef]
- Park, S.; Kim, W.; Tian, G.; Gygi, S.P.; Finley, D. Structural defects in the regulatory particle-core particle interface of the proteasome induce a novel proteasome stress response. J. Biol. Chem. 2011, 286, 36652–36666. [Google Scholar] [CrossRef] [Green Version]
- Wani, P.S.; Rowland, M.A.; Ondracek, A.; Deeds, E.J.; Roelofs, J. Maturation of the proteasome core particle induces an affinity switch that controls regulatory particle association. Nat. Commun. 2015, 6, 6384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Li, Y.; Arendt, C.S.; Hochstrasser, M. Distinct elements in the proteasomal β5 subunit propeptide required for autocatalytic processing and proteasome assembly. J. Biol. Chem. 2016, 291, 1991–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, P.C.; Dohmen, R.J. PACemakers of proteasome core particle assembly. Structure 2008, 16, 1296–1304. [Google Scholar] [CrossRef] [Green Version]
- Groll, M.; Ditzel, L.; Löwe, J.; Stock, D.; Bochtler, M.; Bartunik, H.D.; Huber, R. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 1997, 386, 463–471. [Google Scholar] [CrossRef]
- Schrödinger, L.; DeLano, W. PyMOL. Available online: http://www.pymol.org/pymol (accessed on 13 January 2018).
- Matias, A.C.; Ramos, P.C.; Dohmen, R.J. Chaperone-assisted assembly of the proteasome core particle. Biochem. Soc. Trans. 2010, 38, 29–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gietz, R.D.; Sugino, A. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 1988, 74, 527–534. [Google Scholar] [CrossRef]
- Sikorski, R.S.; Hieter, P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 1989, 122, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, P.; Thomas, L.R.; Thorburn, A.; Stillman, D.J. pRS yeast vectors with a LYS2 marker. BioTechniques 2004, 36, 212–213. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zimmermann, J.; Ramos, P.C.; Dohmen, R.J. Interaction with the Assembly Chaperone Ump1 Promotes Incorporation of the β7 Subunit into Half-Proteasome Precursor Complexes Driving Their Dimerization. Biomolecules 2022, 12, 253. https://doi.org/10.3390/biom12020253
Zimmermann J, Ramos PC, Dohmen RJ. Interaction with the Assembly Chaperone Ump1 Promotes Incorporation of the β7 Subunit into Half-Proteasome Precursor Complexes Driving Their Dimerization. Biomolecules. 2022; 12(2):253. https://doi.org/10.3390/biom12020253
Chicago/Turabian StyleZimmermann, Jessica, Paula C. Ramos, and R. Jürgen Dohmen. 2022. "Interaction with the Assembly Chaperone Ump1 Promotes Incorporation of the β7 Subunit into Half-Proteasome Precursor Complexes Driving Their Dimerization" Biomolecules 12, no. 2: 253. https://doi.org/10.3390/biom12020253
APA StyleZimmermann, J., Ramos, P. C., & Dohmen, R. J. (2022). Interaction with the Assembly Chaperone Ump1 Promotes Incorporation of the β7 Subunit into Half-Proteasome Precursor Complexes Driving Their Dimerization. Biomolecules, 12(2), 253. https://doi.org/10.3390/biom12020253