
Genotype-Phenotype Correlations of Pathogenic COCH Variants in DFNA9: A HuGE Systematic Review and Audiometric Meta-Analysis

 ,
,  ,
,  , ,
, ,  , and
, and 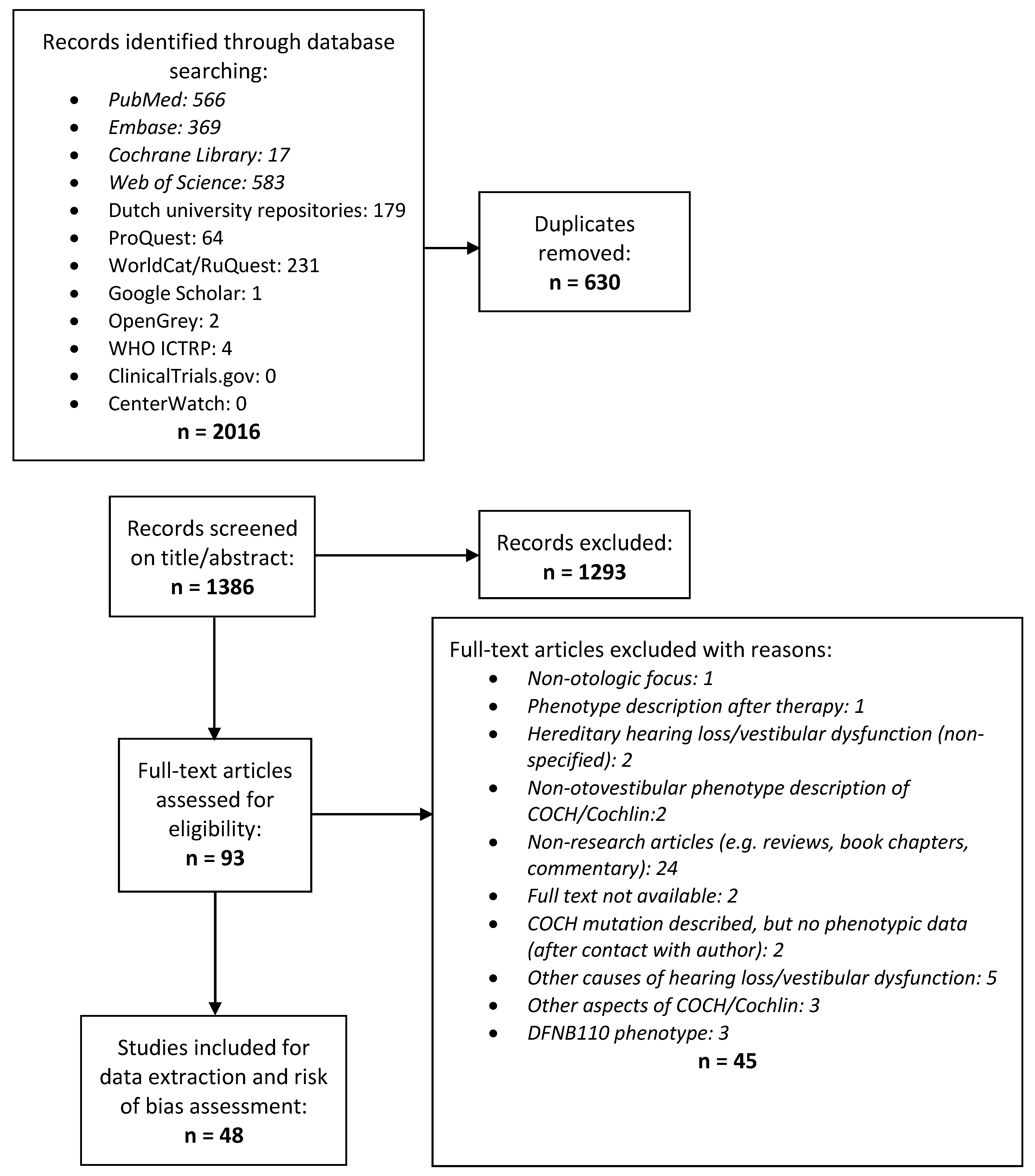{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Systematic Review
2.2. Meta-Analysis
3. Results
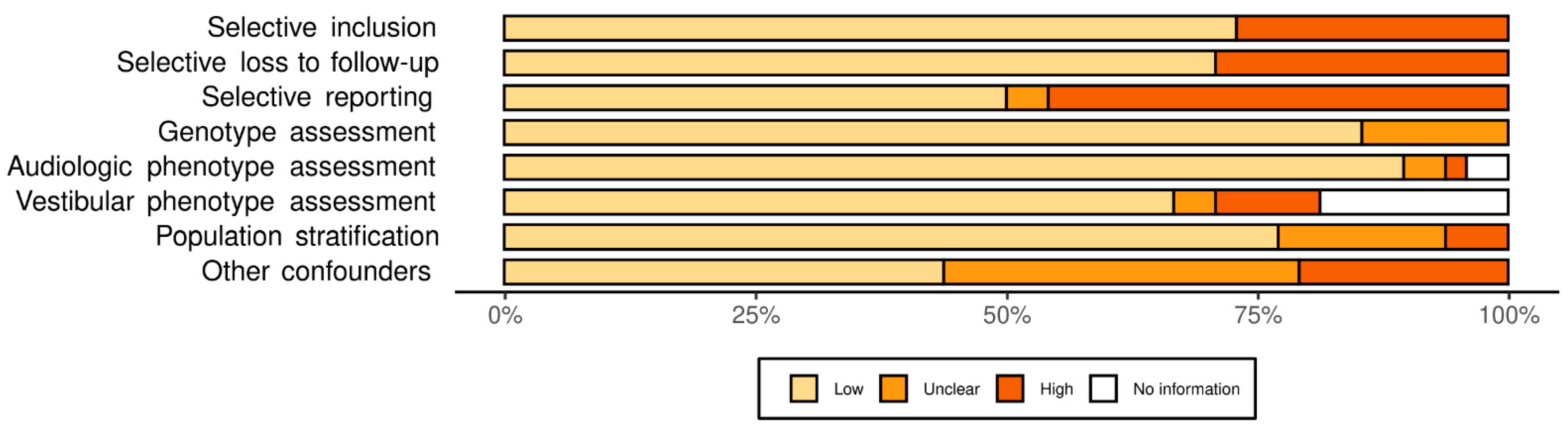
3.1. Systematic Review
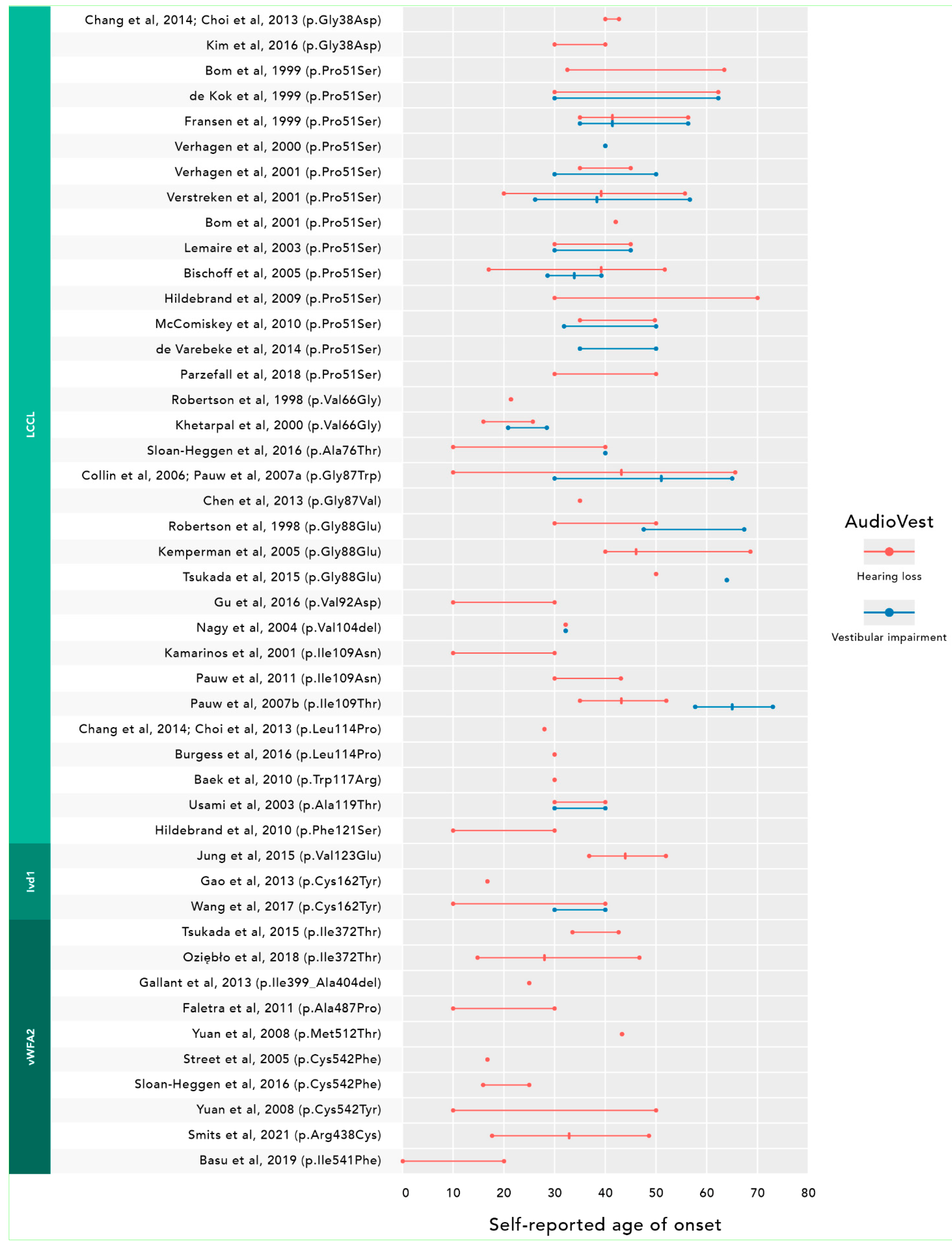
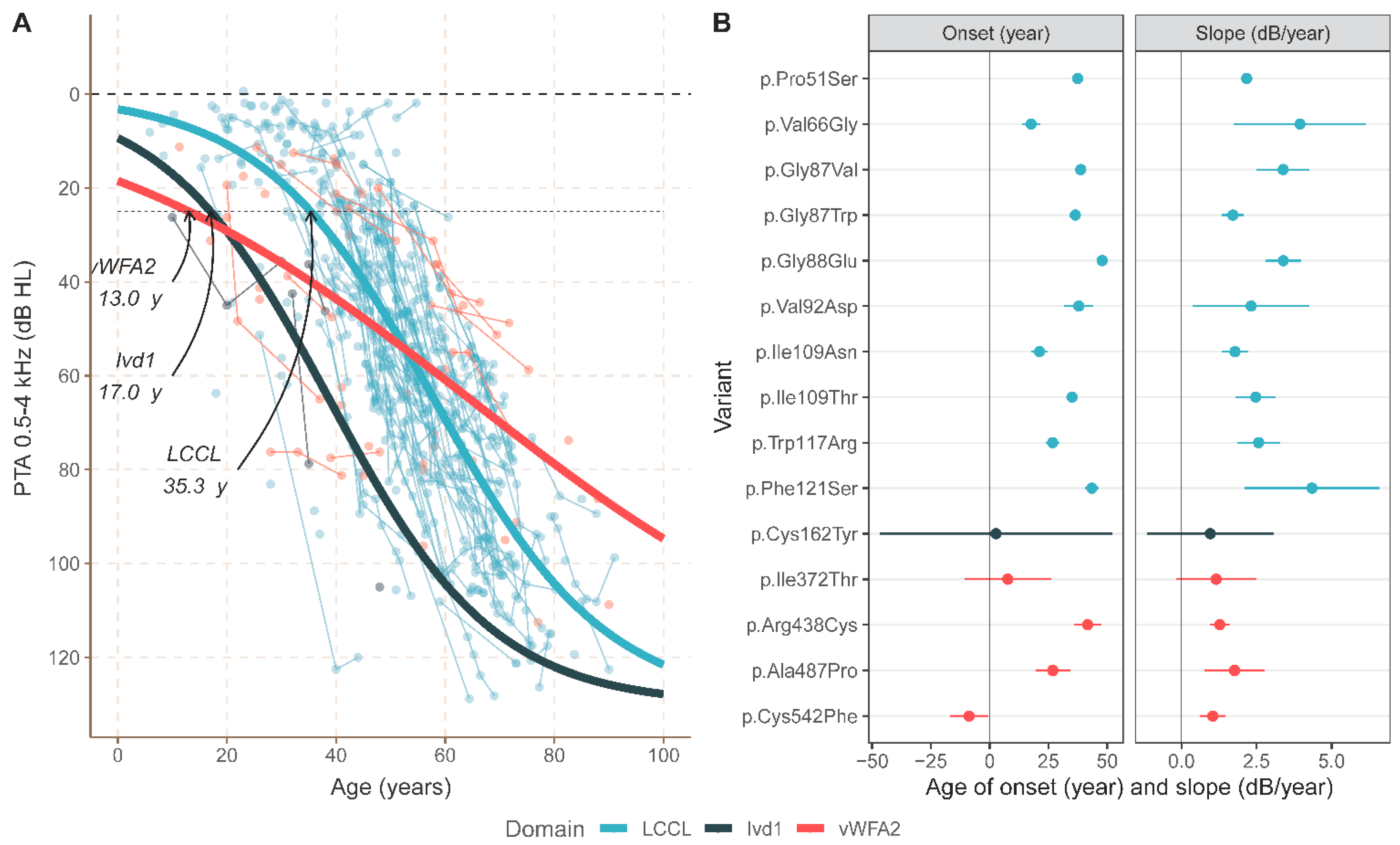
3.2. Meta-Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| HL | hearings loss |
| MIM | Mendelian Inheritance in Man number |
| DFNA | DeaFNess autosomal dominant |
| DFNB | DeaFNess autosomal recessive |
| vHIT | video Head Impulse Tests |
| ARTA | Age-Related Typical Audiograms |
| VEMP | Vestibular Evoked Myogenic Potential |
| ANOVA | analysis of variance |
References
- World Health Organization. Deafness and Hearing Loss. Available online: http://www.who.int/news-room/fact-sheets/detail/deafness-and-hearing-loss (accessed on 1 February 2021).
- Tu, N.C.; Friedman, R.A. Age-related hearing loss: Unraveling the pieces. Laryngoscope Investig. Otolaryngol. 2018, 3, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Oonk, A.M.; Leijendeckers, J.M.; Lammers, E.M.; Weegerink, N.J.; Oostrik, J.; Beynon, A.J.; Huygen, P.L.; Kunst, H.P.; Kremer, H.; Snik, A.F.; et al. Progressive hereditary hearing impairment caused by a MYO6 mutation resembles presbyacusis. Hear. Res. 2013, 299, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Pauw, R.J.; van Drunen, F.J.; Collin, R.W.; Huygen, P.L.; Kremer, H.; Cremers, C.W. Audiometric characteristics of a Dutch family linked to DFNA15 with a novel mutation (p.L289F) in POU4F3. Arch. Otolaryng Head Neck Surg. 2008, 134, 294–300. [Google Scholar] [CrossRef][Green Version]
- O’Neill, M.E.; Marietta, J.; Nishimura, D.; Wayne, S.; Van Camp, G.; Van Laer, L.; Negrini, C.; Wilcox, E.R.; Chen, A.; Fukushima, K.; et al. A gene for autosomal dominant late-onset progressive non-syndromic hearing loss, DFNA10, maps to chromosome 6. Hum. Mol. Genet. 1996, 5, 853–856. [Google Scholar] [CrossRef]
- de Bruijn, S.E.; Smits, J.J.; Liu, C.; Lanting, C.P.; Beynon, A.J.; Blankevoort, J.; Oostrik, J.; Koole, W.; de Vrieze, E.; Cremers, C.; et al. A RIPOR2 in-frame deletion is a frequent and highly penetrant cause of adult-onset hearing loss. J. Med. Genet. 2021, 58, 96–104. [Google Scholar] [CrossRef]
- JanssensdeVarebeke, S.P.F.; Van Camp, G.; Peeters, N.; Elinck, E.; Widdershoven, J.; Cox, T.; Deben, K.; Ketelslagers, K.; Crins, T.; Wuyts, W. Bi-allelic inactivating variants in the COCH gene cause autosomal recessive prelingual hearing impairment. Eur. J. Hum. Genet. 2018, 26, 587–591. [Google Scholar] [CrossRef]
- Bae, S.H.; Robertson, N.G.; Cho, H.J.; Morton, C.C.; Jung, D.J.; Baek, J.I.; Choi, S.Y.; Lee, J.; Lee, K.Y.; Kim, U.K. Identification of Pathogenic Mechanisms of COCH Mutations, Abolished Cochlin Secretion, and Intracellular Aggregate Formation: Genotype-Phenotype Correlations in DFNA9 Deafness and Vestibular Disorder. Hum. Mutat. 2014, 35, 1506–1513. [Google Scholar] [CrossRef]
- Robertson, N.G.; Lu, L.; Heller, S.; Merchant, S.N.; Eavey, R.D.; McKenna, M.; Nadol, J.B.; Miyamoto, R.T.; Linthicum, F.H.; Neto, J.F.L.; et al. Mutations in a novel cochlear gene cause DFNA9, a human nonsyndromic deafness with vestibular dysfunction. Nat. Genet. 1998, 20, 299–303. [Google Scholar] [CrossRef]
- Boulassel, M.R.; Tomasi, J.P.; Deggouj, N.; Gersdorff, M. COCH5B2 is a target antigen of anti-inner ear antibodies in autoimmune inner ear diseases. Otol. Neurotol. 2001, 22, 614–618. [Google Scholar] [CrossRef][Green Version]
- Jung, J.; Yoo, J.E.; Choe, Y.H.; Park, S.C.; Lee, H.J.; Lee, H.J.; Noh, B.; Kim, S.H.; Kang, G.Y.; Lee, K.M.; et al. Cleaved Cochlin Sequesters Pseudomonas aeruginosa and Activates Innate Immunity in the Inner Ear. Cell Host Microbe 2019, 25, 513–525.e6. [Google Scholar] [CrossRef]
- Rhyu, H.J.; Bae, S.H.; Jung, J.; Hyun, Y.M. Cochlin-cleaved LCCL is a dual-armed regulator of the innate immune response in the cochlea during inflammation. BMB Rep. 2020, 53, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Verdoodt, D.; Van Camp, G.; Ponsaerts, P.; Van Rompaey, V. On the pathophysiology of DFNA9: Effect of pathogenic variants in the COCH gene on inner ear functioning in human and transgenic mice. Hear. Res. 2021, 401, 108162. [Google Scholar] [CrossRef] [PubMed]
- Cochrane Reviews; Higgins, J.; Green, S. Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0; The Cochrane Collaboration: London, UK, 2011; Available online: https://handbook-5-1.cochrane.org/ (accessed on 2 June 2019).
- University of York: Centre for Reviews and Dissemination. Chapters 1-6. CRD’s Guidance for Undertaking Reviews in Health Care. Available online: https://www.york.ac.uk/media/crd/Systematic_Reviews.pdf (accessed on 2 June 2019).
- Little, J.; Higgins, J. The HuGENet™ HuGE Review Handbook, Version 1.0; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2006. [Google Scholar]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. Ann. Intern. Med. 2009, 151, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Bom, S.J.H.; Kemperman, M.H.; De Kok, Y.J.M.; Huygen, P.L.M.; Verhagen, W.I.M.; Cremers, F.P.M.; Cremers, C. Progressive cochleovestibular impairment caused by a point mutation in the COCH gene at DFNA9. Laryngoscope 1999, 109, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- de Kok, Y.J.M.; Bom, S.J.H.; Brunt, T.M.; Kemperman, M.H.; van Beusekom, E.; van der Velde-Visser, S.D.; Robertson, N.G.; Morton, C.C.; Huygen, P.L.M.; Verhagen, W.I.M.; et al. A Pro51Ser mutation in the COCH gene is associated with late onset autosomal dominant progressive sensorineural hearing loss with vestibular defects. Hum. Mol. Genet. 1999, 8, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Bom, S.J.H.; Kemperman, M.H.; Huygen, P.L.M.; Luijendijk, M.W.J.; Cremers, C. Cross-sectional analysis of hearing threshold in relation to age in a large family with cochleovestibular impairment thoroughly genotyped for DFNA9/COCH. Ann. Otol. Rhinol. Laryngol. 2003, 112, 280–286. [Google Scholar] [CrossRef]
- Bom, S.J.H.; De Leenheer, E.M.R.; Lemaire, F.X.; Kemperman, M.H.; Verhagen, W.I.M.; Marres, H.A.M.; Kunst, H.P.M.; Ensink, R.J.H.; Bosman, A.J.; Van Camp, G.; et al. Speech recognition scores related to age and degree of hearing impairment in DFNA2/KCNQ4 and DFNA9/COCH. Arch. Otolaryngol. Head Neck Surg. 2001, 127, 1045–1048. [Google Scholar] [CrossRef]
- Bischoff, A.; Huygen, P.L.M.; Kemperman, M.H.; Pennings, R.J.E.; Bom, S.J.H.; Verhagen, W.I.M.; Admiraal, R.J.C.; Kremer, H.; Cremers, C. Vestibular deterioration precedes hearing deterioration in the P51SCOCH mutation (DFNA9): An analysis in 74 mutation carriers. Otol. Neurotol. 2005, 26, 918–925. [Google Scholar] [CrossRef]
- Verhagen, W.I.M.; Bom, S.J.H.; Fransen, E.; Van Camp, G.; Huygen, P.L.M.; Theunissen, E.; Cremers, C. Hereditary cochleovestibular dysfunction due to a COCH gene mutation (DFNA9): A follow-up study of a family. Clin. Otolaryngol. 2001, 26, 477–483. [Google Scholar] [CrossRef]
- Alberts, B.; Selen, L.P.J.; Verhagen, W.I.M.; Pennings, R.J.E.; Medendorp, W.P. Bayesian quantification of sensory reweighting in a familial bilateral vestibular disorder (DFNA9). J. Neurophysiol. 2018, 119, 1209–1221. [Google Scholar] [CrossRef]
- Collin, R.W.J.; Pauw, R.J.; Schoots, J.; Huygen, P.L.M.; Hoefsloot, L.H.; Cremers, C.; Kremer, H. Identification of a novel COCH mutation, G87W, causing autosomal dominant hearing impairment (DFNA9). Am. J. Med. Genet. A. 2006, 140A, 1791–1794. [Google Scholar] [CrossRef]
- Pauw, R.J.; Huygen, P.L.M.; Collin, R.W.J.; Cruysberg, J.R.M.; Hoefsloot, L.H.; Kremer, H.; Cremers, C. Phenotype description of a novel DFNA9/COCH mutation, I109T. Ann. Otol. Rhinol. Laryngol. 2007, 116, 349–357. [Google Scholar] [CrossRef]
- Smits, J.J.; van Beelen, E.; Weegerink, N.J.D.; Oostrik, J.; Huygen, P.L.M.; Beynon, A.J.; Lanting, C.P.; Kunst, H.P.M.; Schraders, M.; Kremer, H.; et al. A Novel COCH Mutation Affects the vWFA2 Domain and Leads to a Relatively Mild DFNA9 Phenotype. Otol. Neurotol. 2021, 42, e399–e407. [Google Scholar] [CrossRef]
- Huygen, P.L.M.; Pennings, R.J.E.; Cremers, C. Characterizing and distinguishing progressive phenotypes in nonsyndromic autosomal dominant hearing impairment. Audiol. Med. 2003, 1, 37–46. [Google Scholar] [CrossRef]
- Pauw, R.J.; Huygen, P.L.; Colditz, G.M.; Cremers, C.W. Phenotype analysis of an Australian DFNA9 family with the 1109N COCH mutation. Ann. Otol. Rhinol. Laryngol. 2011, 120, 414–421. [Google Scholar] [CrossRef]
- Kim, B.J.; Kim, A.R.; Han, K.H.; Rah, Y.C.; Hyun, J.; Ra, B.S.; Koo, J.W.; Choi, B.Y. Distinct vestibular phenotypes in DFNA9 families with COCH variants. Eur. Arch. Oto-Rhino-Laryn. 2016, 273, 2993–3002. [Google Scholar] [CrossRef]
- Hildebrand, M.S.; Gandolfo, L.; Shearer, A.E.; Webster, J.A.; Jensen, M.; Kimberling, W.J.; Stephan, D.; Huygen, P.L.M.; Smith, R.J.H.; Bahlo, M. A Novel Mutation in COCH-Implications for Genotype-Phenotype Correlations in DFNA9 Hearing Loss. Laryngoscope 2010, 120, 2489–2493. [Google Scholar] [CrossRef]
- Chen, D.Y.; Chai, Y.C.; Yang, T.; Wu, H. Clinical characterization of a novel COCH mutation G87V in a Chinese DFNA9 family. Int. J. Pediatr. Otorhinolaryngol. 2013, 77, 1711–1715. [Google Scholar] [CrossRef]
- Fransen, E.; Verstreken, M.; Verhagen, W.I.M.; Wuyts, F.L.; Huygen, P.L.M.; D’Haese, P.; Robertson, N.G.; Morton, C.C.; McGuirt, W.T.; Smith, R.J.H.; et al. High prevalence of symptoms of Meniere’s disease in three families with a mutation in the COCH gene. Hum. Mol. Genet. 1999, 8, 1425–1429. [Google Scholar] [CrossRef]
- de Varebeke, S.P.J.; Termote, B.; Van Camp, G.; Govaerts, P.J.; Schepers, S.; Cox, T.; Deben, K.; Ketelslagers, K.; Souverijns, G. Focal Sclerosis of Semicircular Canals With Severe DFNA9 Hearing Impairment Caused by a P51S COCH-Mutation: Is There a Link? Otol. Neurotol. 2014, 35, 1077–1086. [Google Scholar] [CrossRef][Green Version]
- Verhagen, W.I.M.; Bom, S.J.H.; Huygen, P.L.M.; Fransen, E.; Van Camp, G.; Cremers, C. Familial progressive vestibulocochlear dysfunction caused by a COCH mutation (DFNA9). Arch. Neurol. 2000, 57, 1045–1047. [Google Scholar] [CrossRef]
- Kemperman, M.H.; De Leenheer, E.M.R.; Huygen, P.L.M.; van Duijnhoven, G.; Morton, C.C.; Robertson, N.G.; Cremers, F.P.M.; Kremer, H.; Cremers, C. Audiometric, vestibular, and genetic aspects of a DFNA9 family with a G88E COCH mutation. Otol. Neurotol. 2005, 26, 926–933. [Google Scholar] [CrossRef]
- Verstreken, M.; Declau, F.; Wuyts, F.L.; D’Haese, P.; Van Camp, G.; Fransen, E.; Van den Hauwe, L.; Buyle, S.; Smets, R.E.M.; Feenstra, L.; et al. Hereditary otovestibular dysfunction and Meniere’s disease in a large Belgian family is caused by a missense mutation in the COCH gene. Otol. Neurotol. 2001, 22, 874–881. [Google Scholar] [CrossRef]
- Usami, S.; Takahashi, K.; Yuge, I.; Ohtsuka, A.; Namba, A.; Abe, S.; Fransen, E.; Patthy, L.; Otting, G.; Van Camp, G. Mutations in the COCH gene are a frequent cause of autosomal dominant progressive cochleo-vestibular dysfunction, but not of Meniere’s disease. Eur. J. Hum. Genet. 2003, 11, 744–748. [Google Scholar] [CrossRef]
- Lemaire, F.X.; Feenstra, L.; Huygen, P.L.M.; Fransen, E.; Devriendt, K.; Van Camp, G.; Vantrappen, G.; Cremers, C. Progressive late-onset sensorineural hearing loss and vestibular impairment with vertigo (DFNA9/COCH): Longitudinal analyses in a Belgian family. Otol. Neurotol. 2003, 24, 743–748. [Google Scholar] [CrossRef]
- Baek, J.I.; Cho, H.J.; Choi, S.J.; Kim, L.S.; Zhao, C.; Sagong, B.R.; Kim, U.K.; Jeong, S.W. The Trp117Arg mutation of the COCH gene causes deafness in Koreans. Clin. Genet. 2010, 77, 399–403. [Google Scholar] [CrossRef]
- Yuan, H.J.; Han, D.Y.; Sun, Q.; Yan, D.; Sun, H.J.; Tao, R.; Cheng, J.; Qin, W.; Angeli, S.; Ouyang, X.M.; et al. Novel mutations in the vWFA2 domain of COCH in two Chinese DFNA9 families. Clin. Genet. 2008, 73, 391–394. [Google Scholar] [CrossRef]
- Kamarinos, M.; McGill, J.; Lynch, M.; Dahl, H. Identification of a novel COCH mutation, I109N, highlights the similar clinical features observed in DFNA9 families. Hum. Mutat. 2001, 17, 351. [Google Scholar] [CrossRef]
- Hildebrand, M.S.; Tack, D.; DeLuca, A.; Hur, I.A.; Van Rybroek, J.M.; McMordie, S.J.; Muilenburg, A.; Hoskinson, D.P.; Van Camp, G.; Pensak, M.L.; et al. Mutation in the COCH Gene Is Associated With Superior Semicircular Canal Dehiscence. Am. J. Med. Genet. A 2009, 149A, 280–285. [Google Scholar] [CrossRef]
- Nagy, I.; Horvath, M.; Trexler, M.; Repassy, G.; Patthy, L. A novel COCH mutation, V104del, impairs folding of the LCCL domain of cochlin and causes progressive hearing loss. J. Med. Genet. 2004, 41, e9. [Google Scholar] [CrossRef][Green Version]
- Gallant, E.; Francey, L.; Fetting, H.; Kaur, M.; Hakonarson, H.; Clark, D.; Devoto, M.; Krantz, I.D. Novel COCH mutation in a family with autosomal dominant late onset sensorineural hearing impairment and tinnitus. Am. J. Otolaryngol. 2013, 34, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, K.; Ichinose, A.; Miyagawa, M.; Mori, K.; Hattori, M.; Nishio, S.; Naito, Y.; Kitajiri, S.; Usami, S. Detailed Hearing and Vestibular Profiles in the Patients with COCH Mutations. Ann. Otol. Rhinol. Laryngol. 2015, 124, 100S–110S. [Google Scholar] [CrossRef] [PubMed]
- Street, V.A.; Kallman, J.C.; Robertson, N.G.; Kuo, S.F.; Morton, C.C.; Phillips, J.O. A novel DFNA9 mutation in the vWFA2 domain of COCH alters a conserved cysteine residue and intrachain disulfide bond formation resulting in progressive hearing loss and site-specific vestibular and central oculomotor dysfunction. Am. J. Med. Genet. A 2005, 139A, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Pauw, R.J.; Collin, R.W.J.; Huygen, P.L.M.; Hoefsloot, L.H.; Kremer, H.; Cremers, C. Clinical characteristics of a Dutch DFNA9 family with a novel COCH mutation, G87W. Audiol. Neuro-Otol. 2007, 12, 77–84. [Google Scholar] [CrossRef]
- Faletra, F.; Pirastu, N.; Athanasakis, E.; Somaschini, A.; Pianigiani, G.; Gasparini, P. A novel mutation in the vWFA2 domain of the COCH gene in an Italian DFNA9 family. Audiol. Med. 2011, 9, 4–7. [Google Scholar] [CrossRef]
- McComiskey, D.A. Investigation of the Genetic Cause of Hearing Loss in 28 Autosomal Dominant Families within the Newfoundland Founder Population. Master’s Thesis, Memorial University of Newfoundland, St. John’s, NL, USA, 2010. [Google Scholar]
- Oziębło, D.; Tacikowska, G.; Skarżyński, H.; Ołdak, M. Postlingual sensorineural hearing loss due to a very rare coch pathogenic variant. J. Hear. Sci. 2018, 8, 31–37. [Google Scholar]
- Wang, Q.; Fei, P.; Gu, H.; Zhang, Y.; Ke, X.; Liu, Y. Different phenotypes of the two Chinese probands with the same c.889G>A (p.C162Y) mutation in COCH gene verify different mechanisms underlying autosomal dominant nonsyndromic deafness 9. PLoS ONE 2017, 12, e0170011. [Google Scholar] [CrossRef]
- Basu, A.; Boczek, N.J.; Robertson, N.G.; Nasr, S.H.; Jethanamest, D.; McPhail, E.D.; Kurtin, P.J.; Dasari, S.; Butz, M.; Morton, C.C.; et al. First Report of Bilateral External Auditory Canal Cochlin Aggregates (“Cochlinomas”) with Multifocal Amyloid-Like Deposits, Associated with Sensorineural Hearing Loss and a Novel Genetic Variant in COCH Encoding Cochlin. Head Neck Pathol. 2020, 14, 808–816. [Google Scholar] [CrossRef]
- Parzefall, T.; Frohne, A.; Koenighofer, M.; Kirchnawy, A.; Streubel, B.; Schoefer, C.; Gstoettner, W.; Frei, K.; Lucas, T. Identification of a rare COCH mutation by whole-exome sequencing Implications for personalized therapeutic rehabilitation in an Austrian family with non-syndromic autosomal dominant late-onset hearing loss. Wien. Klin. Wochen. 2018, 130, 299–306. [Google Scholar] [CrossRef]
- Gu, X.D.; Su, W.L.; Tang, M.L.; Guo, L.; Zhao, L.P.; Li, H.W. Massively Parallel Sequencing of a Chinese Family with DFNA9 Identified a Novel Missense Mutation in the LCCL Domain of COCH. Neural. Plast. 2016, 2016, 5310192. [Google Scholar] [CrossRef]
- Khetarpal, U. DFNA9 is a progressive audiovestibular dysfunction with a microfibrillar deposit in the inner ear. Laryngoscope 2000, 110, 1379–1384. [Google Scholar] [CrossRef]
- Kim, B.J.; Kim, A.R.; Park, G.; Park, W.Y.; Chang, S.O.; Oh, S.H.; Choi, B.Y. Targeted Exome Sequencing of Deafness Genes After Failure of Auditory Phenotype-Driven Candidate Gene Screening. Otol. Neurotol. 2015, 36, 1096–1102. [Google Scholar] [CrossRef]
- Burgess, B.J.; O’Malley, J.T.; Kamakura, T.; Kristiansen, K.; Robertson, N.G.; Morton, C.C.; Nadol, J.B. Histopathology of the Human Inner Ear in the p.L114P COCH Mutation (DFNA9). Audiol. Neuro-Otol. 2016, 21, 88–97. [Google Scholar] [CrossRef]
- Chang, M.Y.; Choi, B.Y. Strategy for the customized mass screening of genetic sensorineural hearing loss in koreans. Korean J. Audiol. 2014, 18, 45–49. [Google Scholar] [CrossRef]
- Wei, Q.J.; Zhu, H.M.; Qian, X.L.; Chen, Z.B.; Yao, J.; Lu, Y.J.; Cao, X.; Xing, G.Q. Targeted genomic capture and massively parallel sequencing to identify novel variants causing Chinese hereditary hearing loss. J. Transl. Med. 2014, 12, 8. [Google Scholar] [CrossRef]
- Choi, B.Y.; Park, G.; Gim, J.; Kim, A.R.; Kim, B.J.; Kim, H.S.; Park, J.H.; Park, T.; Oh, S.H.; Han, K.H.; et al. Diagnostic Application of Targeted Resequencing for Familial Nonsyndromic Hearing Loss. PLoS ONE 2013, 8, 8. [Google Scholar] [CrossRef]
- Cho, H.J.; Park, H.J.; Trexler, M.; Venselaar, H.; Lee, K.Y.; Robertson, N.G.; Baek, J.I.; Kang, B.S.; Morton, C.C.; Vriend, G.; et al. A novel COCH mutation associated with autosomal dominant nonsyndromic hearing loss disrupts the structural stability of the vWFA2 domain. J. Mol. Med. 2012, 90, 1321–1331. [Google Scholar] [CrossRef][Green Version]
- Gao, J.; Xue, J.; Chen, L.; Ke, X.; Qi, Y.; Liu, Y. Whole exome sequencing identifies a novel DFNA9 mutation, C162Y. Clin. Genet. 2013, 83, 477–481. [Google Scholar] [CrossRef]
- Jung, J.; Kim, H.S.; Lee, M.G.; Yang, E.J.; Choi, J.Y. Novel COCH p.V123E Mutation, Causative of DFNA9 Sensorineural Hearing Loss and Vestibular Disorder, Shows Impaired Cochlin Post-Translational Cleavage and Secretion. Hum. Mutat. 2015, 36, 1168–1175. [Google Scholar] [CrossRef]
- Sloan-Heggen, C.M.; Bierer, A.O.; Shearer, A.E.; Kolbe, D.L.; Nishimura, C.J.; Frees, K.L.; Ephraim, S.S.; Shibata, S.B.; Booth, K.T.; Campbell, C.A.; et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 2016, 135, 441–450. [Google Scholar] [CrossRef]
- Mehregan, H.; Mohseni, M.; Akbari, M.; Jalalvand, K.; Arzhangi, S.; Nikzat, N.; Kahrizi, K.; Najmabadi, H. Novel Mutations in KCNQ4, LHFPL5 and COCH Genes in Iranian Families with Hearing Impairment. Arch. Iran. Med. 2019, 22, 189–197. [Google Scholar] [PubMed]
- Danial-Farran, N.; Chervinsky, E.; Nadar-Ponniah, P.T.; Cohen Barak, E.; Taiber, S.; Khayat, M.; Avraham, K.B.; Shalev, S.A. Homozygote loss-of-function variants in the human COCH gene underlie hearing loss. Eur. J. Hum. Genet. 2021, 29, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Booth, K.T.; Ghaffar, A.; Rashid, M.; Hovey, L.T.; Hussain, M.; Frees, K.; Renkes, E.M.; Nishimura, C.J.; Shahzad, M.; Smith, R.J.; et al. Novel loss-of-function mutations in COCH cause autosomal recessive nonsyndromic hearing loss. Hum. Genet. 2020, 139, 1565–1574. [Google Scholar] [CrossRef]
- Dodson, K.M.; Georgolios, A.; Barr, N.; Nguyen, B.; Sismanis, A.; Arnos, K.S.; Norris, V.W.; Chapman, D.; Nance, W.E.; Pandya, A. Etiology of unilateral hearing loss in a national hereditary deafness repository. Am. J. Otolaryngol. 2012, 33, 590–594. [Google Scholar] [CrossRef]
- Janssens de Varebeke, S.; Topsakal, V.; Van Camp, G.; Van Rompaey, V. A systematic review of hearing and vestibular function in carriers of the Pro51Ser mutation in the COCH gene. Eur. Arch. Otorhinolaryngol. 2019, 276, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Bom, S.J.H.; Kunst, H.P.M.; Huygen, P.L.M.; Cremers, F.P.M.; Cremers, C. Non-syndromal autosomal dominant hearing impairment: Ongoing phenotypical characterization of genotypes. Br. J. Audiol. 1999, 33, 335–348. [Google Scholar] [CrossRef]
- Bischoff, A.; Pauw, R.J.; Huygen, P.L.M.; Aandekerk, A.L.; Kremer, H.; Cremers, C.; Cruysberg, J.R.M. Vertical corneal striae in families with authosomal dominant hearing loss: DFNA9/COCH. Am. J. Ophthalmol. 2007, 143, 847–852. [Google Scholar] [CrossRef]
- Kemperman, M.H.; Bom, S.J.; Lemaire, F.X.; Verhagen, W.I.; Huygen, P.L.; Cremers, C.W. DFNA9/COCH and its phenotype. Adv. Otorhinolaryngol. 2002, 61, 66–72. [Google Scholar]
- de Vrieze, E.; Canas Martin, J.; Peijnenborg, J.; Martens, A.; Oostrik, J.; van den Heuvel, S.; Neveling, K.; Pennings, R.; Kremer, H.; van Wijk, E. AON-based degradation of c.151C>T mutant COCH transcripts associated with dominantly inherited hearing impairment DFNA9. Mol. Ther. Nucleic Acids 2021, 24, 274–283. [Google Scholar] [CrossRef]
- Althubaiti, A. Information bias in health research: Definition, pitfalls, and adjustment methods. J. Multidiscip. Healthc. 2016, 9, 211–217. [Google Scholar] [CrossRef]
- Vermeire, K.; Brokx, J.P.L.; Wuyts, F.L.; Cochet, E.; Hofkens, A.; De Bodt, M.; Van de Heyning, P.H. Good speech recognition and quality-of-life scores after cochlear implantation in patients with DFNA9. Otol. Neurotol. 2006, 27, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Grabski, R.; Szul, T.; Sasaki, T.; Timpl, R.; Mayne, R.; Hicks, B.; Sztul, E. Mutations in COCH that result in non-syndromic autosomal dominant deafness (DFNA9) affect matrix deposition of cochlin. Hum. Genet. 2003, 113, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Robertson, N.G.; Hamaker, S.A.; Patriub, V.; Aster, J.C.; Morton, C.C. Subcellular localisation, secretion, and post-translational processing of normal cochlin, and of mutants causing the sensorineural deafness and vestibular disorder, DFNA9. J. Med. Genet. 2003, 40, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Py, B.F.; Zhu, H.; Bao, J.; Yuan, J. Role of protein misfolding in DFNA9 hearing loss. J. Biol. Chem. 2010, 285, 14909–14919. [Google Scholar] [CrossRef]
- Gibson, T.J.; Seiler, M.; Veitia, R.A. The transience of transient overexpression. Nat. Methods 2013, 10, 715–721. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robijn, S.M.M.; Smits, J.J.; Sezer, K.; Huygen, P.L.M.; Beynon, A.J.; van Wijk, E.; Kremer, H.; de Vrieze, E.; Lanting, C.P.; Pennings, R.J.E. Genotype-Phenotype Correlations of Pathogenic COCH Variants in DFNA9: A HuGE Systematic Review and Audiometric Meta-Analysis. Biomolecules 2022, 12, 220. https://doi.org/10.3390/biom12020220
Robijn SMM, Smits JJ, Sezer K, Huygen PLM, Beynon AJ, van Wijk E, Kremer H, de Vrieze E, Lanting CP, Pennings RJE. Genotype-Phenotype Correlations of Pathogenic COCH Variants in DFNA9: A HuGE Systematic Review and Audiometric Meta-Analysis. Biomolecules. 2022; 12(2):220. https://doi.org/10.3390/biom12020220
Chicago/Turabian StyleRobijn, Sybren M. M., Jeroen J. Smits, Kadriye Sezer, Patrick L. M. Huygen, Andy J. Beynon, Erwin van Wijk, Hannie Kremer, Erik de Vrieze, Cornelis P. Lanting, and Ronald J. E. Pennings. 2022. "Genotype-Phenotype Correlations of Pathogenic COCH Variants in DFNA9: A HuGE Systematic Review and Audiometric Meta-Analysis" Biomolecules 12, no. 2: 220. https://doi.org/10.3390/biom12020220
APA StyleRobijn, S. M. M., Smits, J. J., Sezer, K., Huygen, P. L. M., Beynon, A. J., van Wijk, E., Kremer, H., de Vrieze, E., Lanting, C. P., & Pennings, R. J. E. (2022). Genotype-Phenotype Correlations of Pathogenic COCH Variants in DFNA9: A HuGE Systematic Review and Audiometric Meta-Analysis. Biomolecules, 12(2), 220. https://doi.org/10.3390/biom12020220