
Integrative Analysis of Bulk RNA-Seq and Single-Cell RNA-Seq Unveils Novel Prognostic Biomarkers in Multiple Myeloma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Source and Processing
2.2. Correlation Analysis
2.3. Screening Hub Genes Associated with MM Prognosis
2.4. Relationship between Prognostic Biomarkers and Tumor Microenvironment
2.5. Patients and Samples
2.6. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.7. Statistical Analysis
3. Results
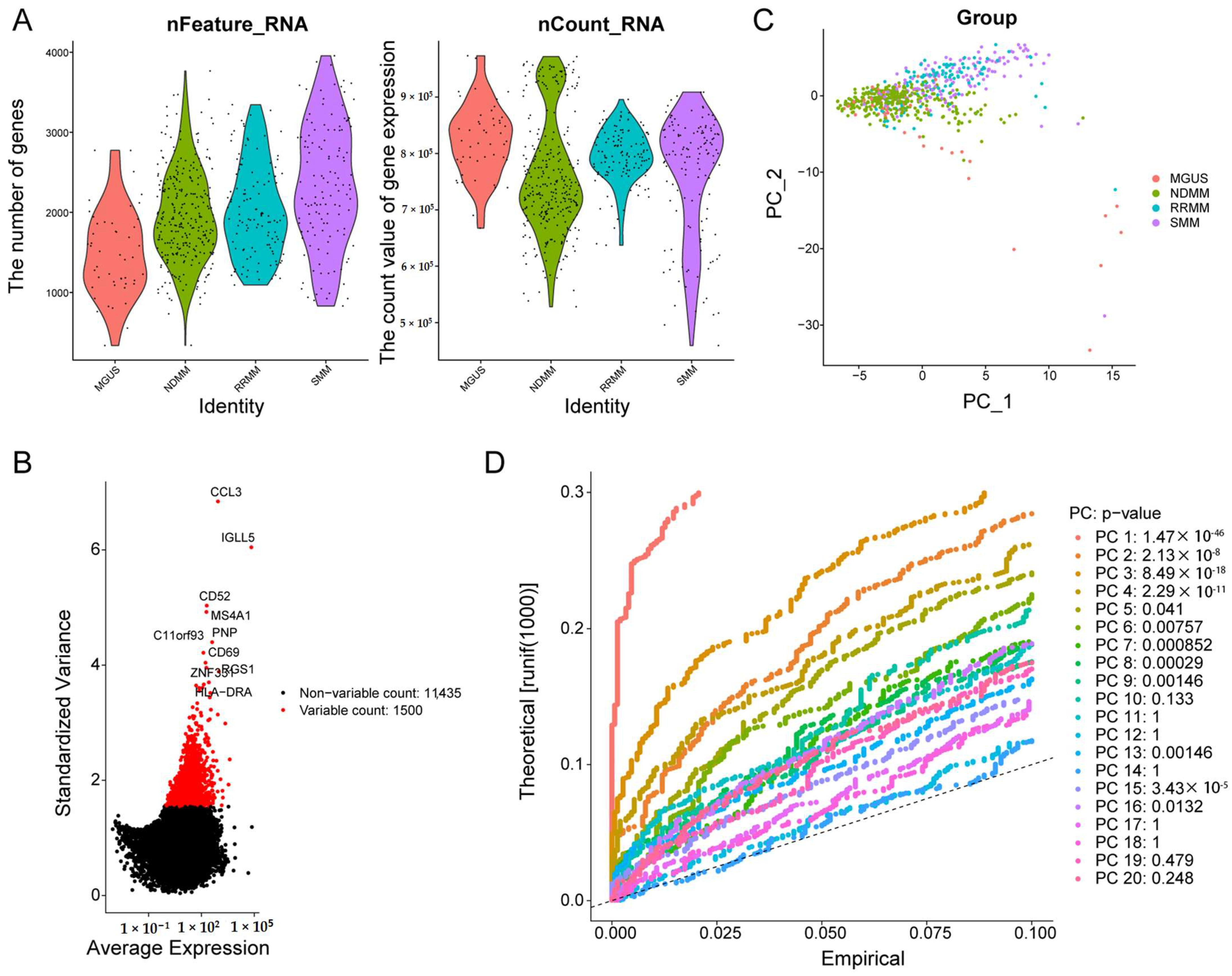
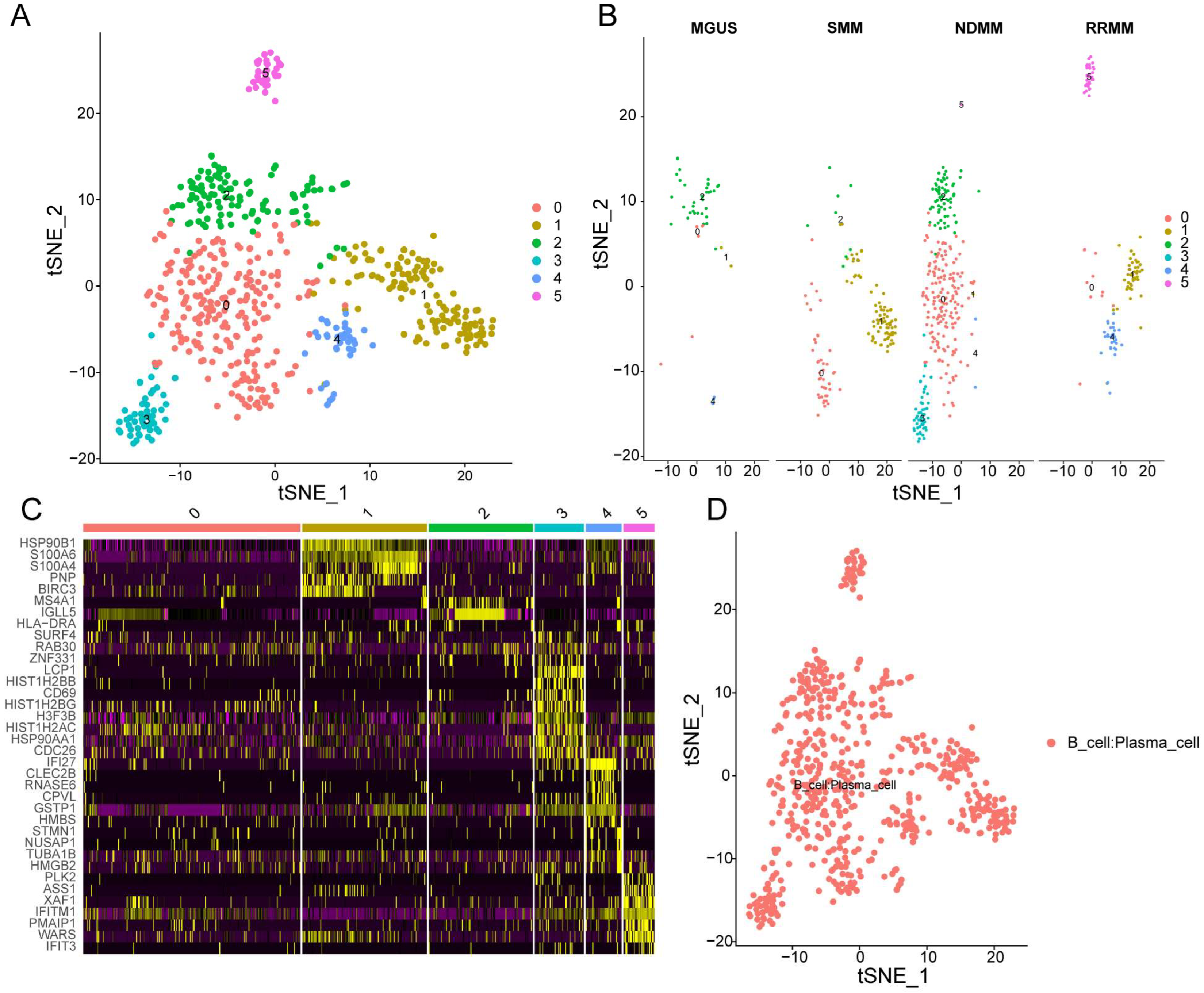
3.1. Single-Cell Sequencing Revealed the Cell Distribution of MM and Six Cell Clusters Were Identified in MM
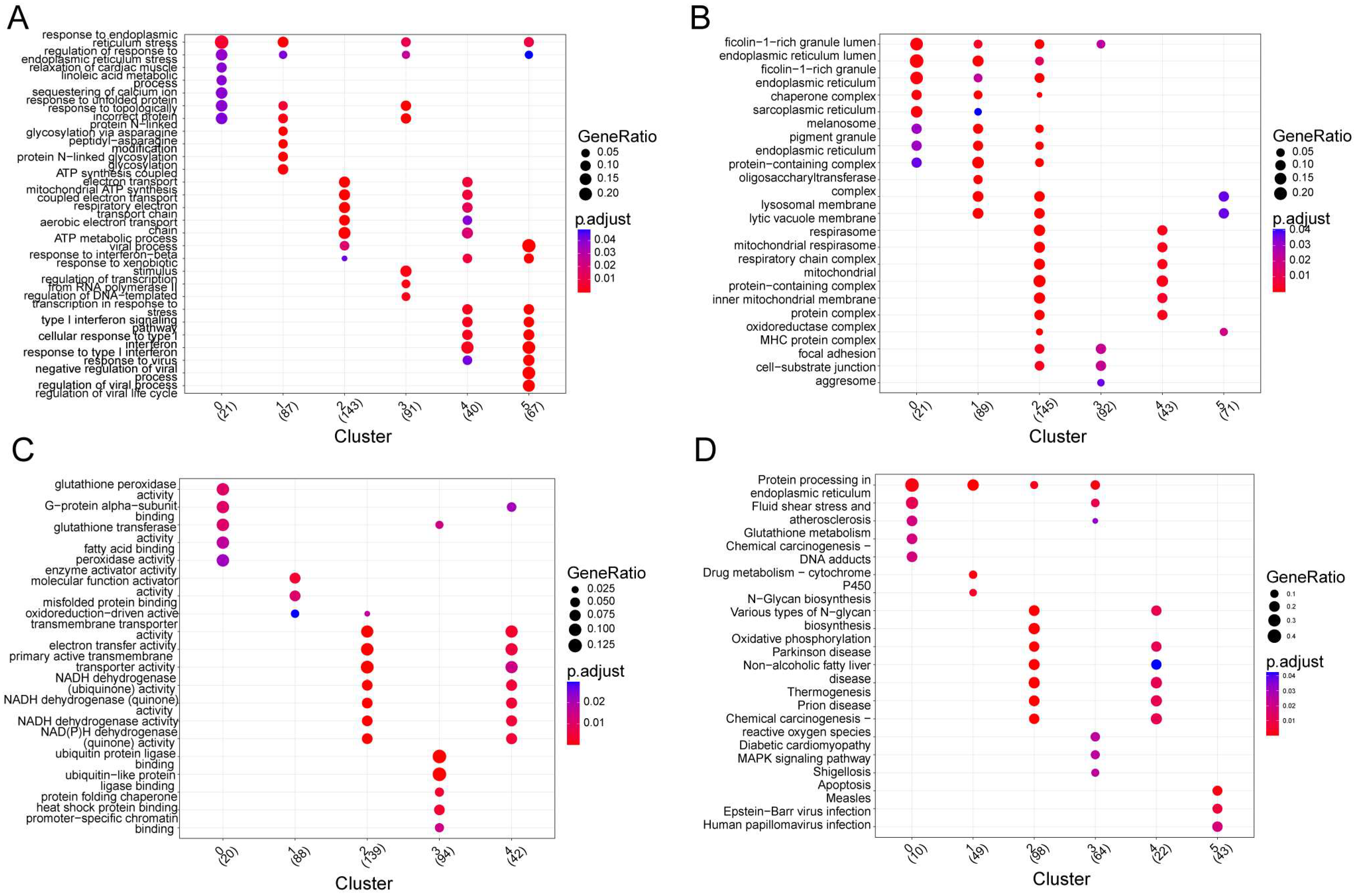
3.2. Investigation of the Functions of Marker Genes of the Six Cell Clusters
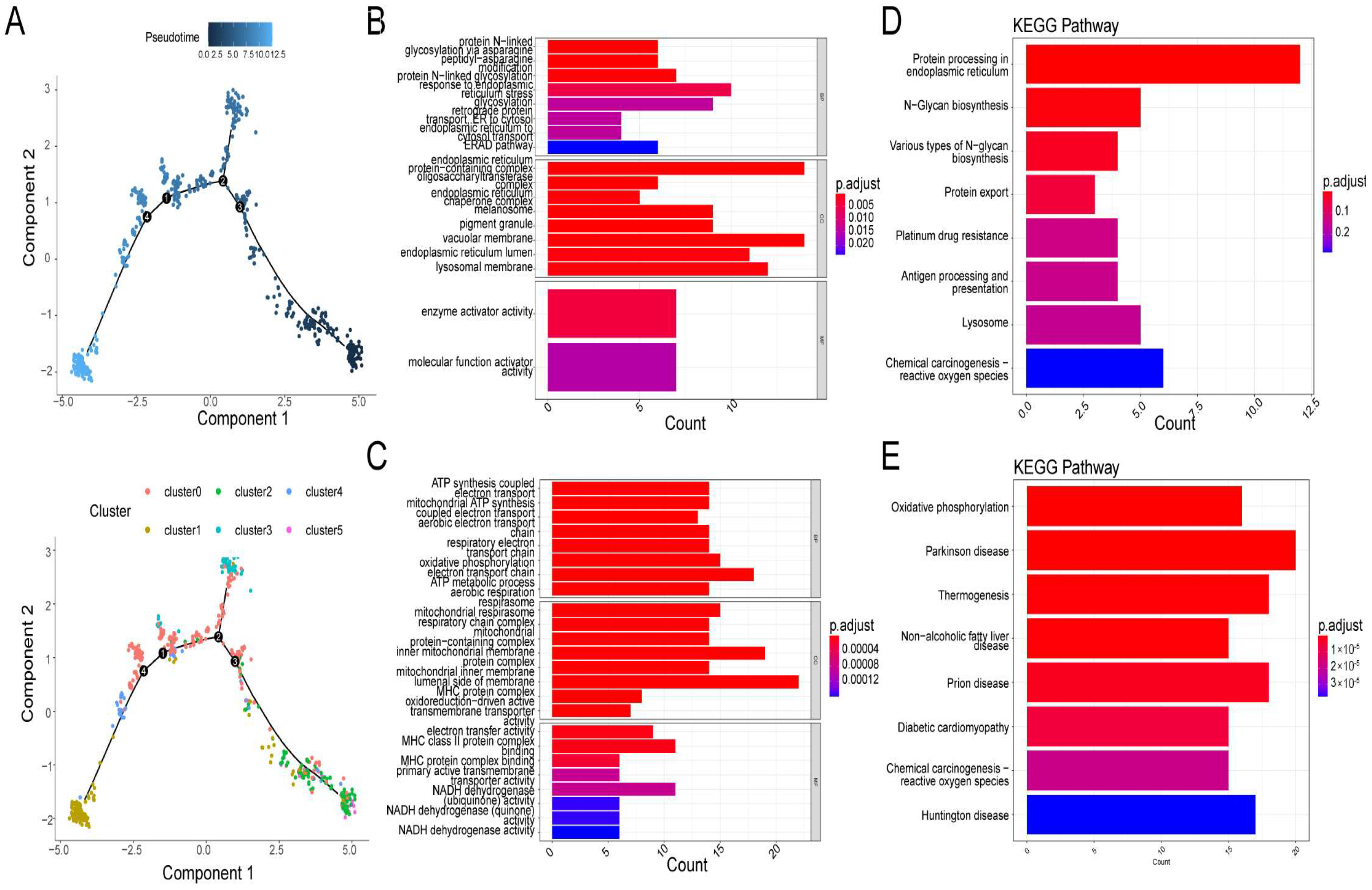
3.3. Two Different Differentiation Traces Were Observed in MM Cells
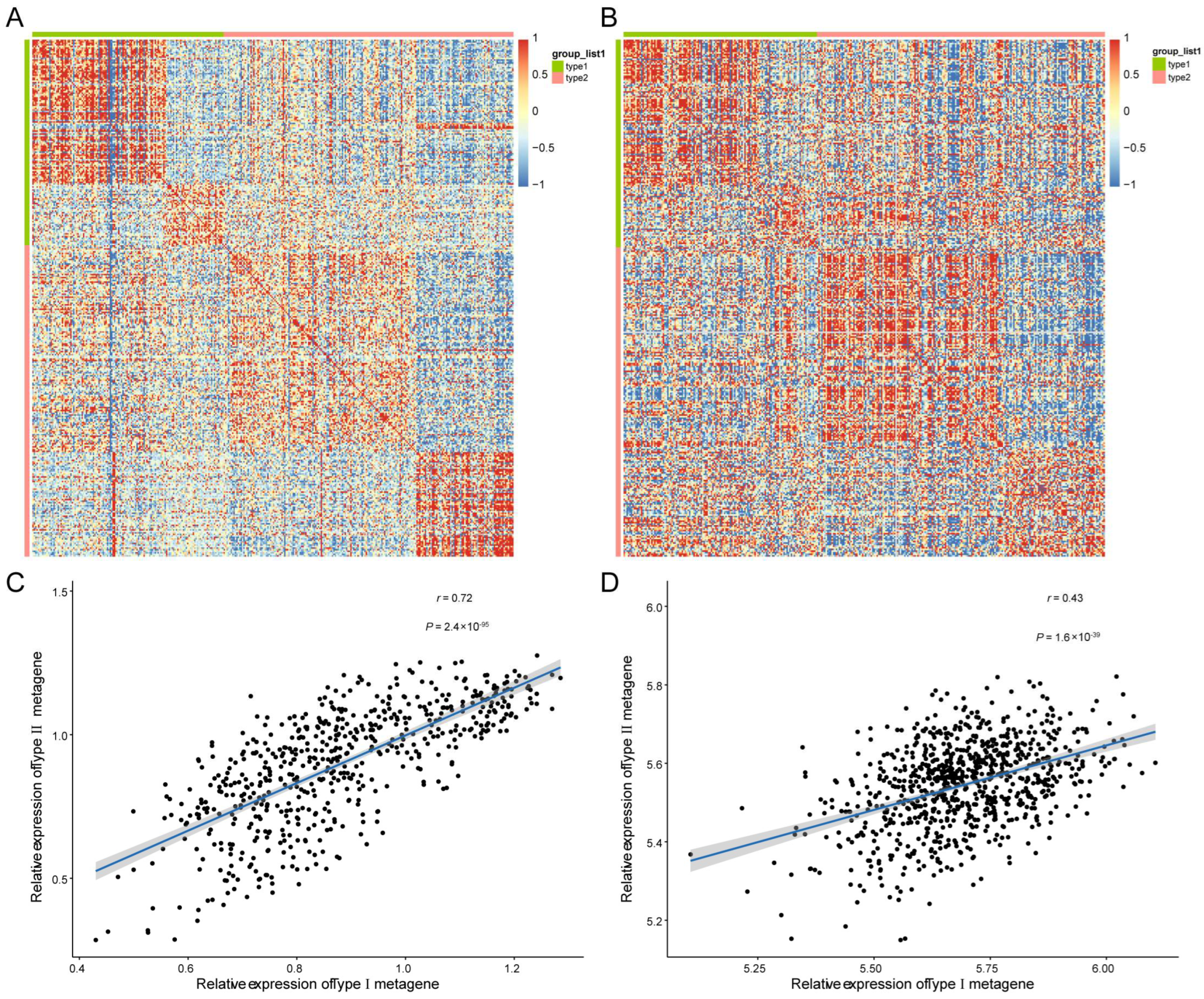
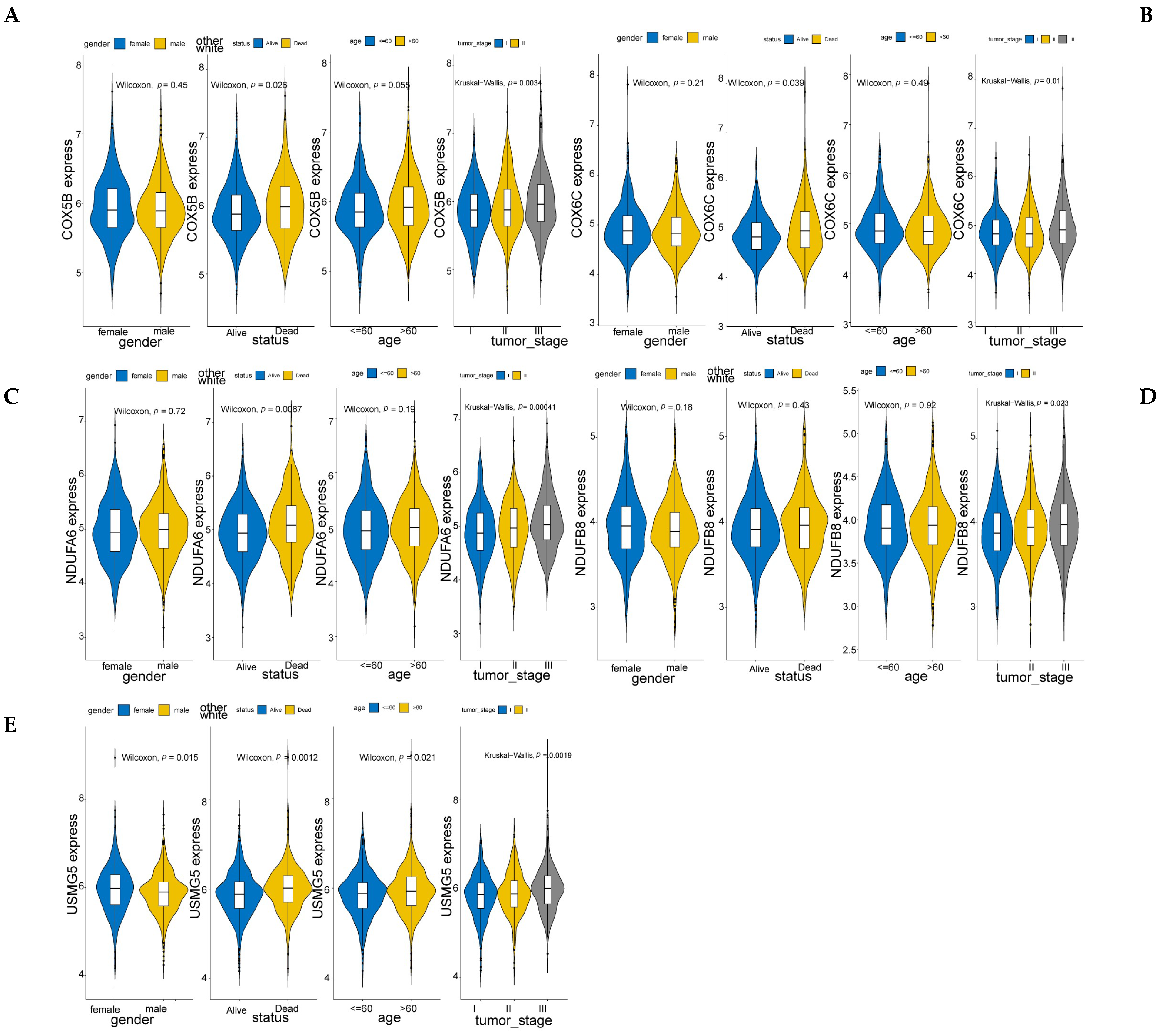
3.4. Five Candidate Prognostic Biomarkers Were Identified in MM
3.5. Prognostic Biomarkers Were Related to the MM Tumor Microenvironment
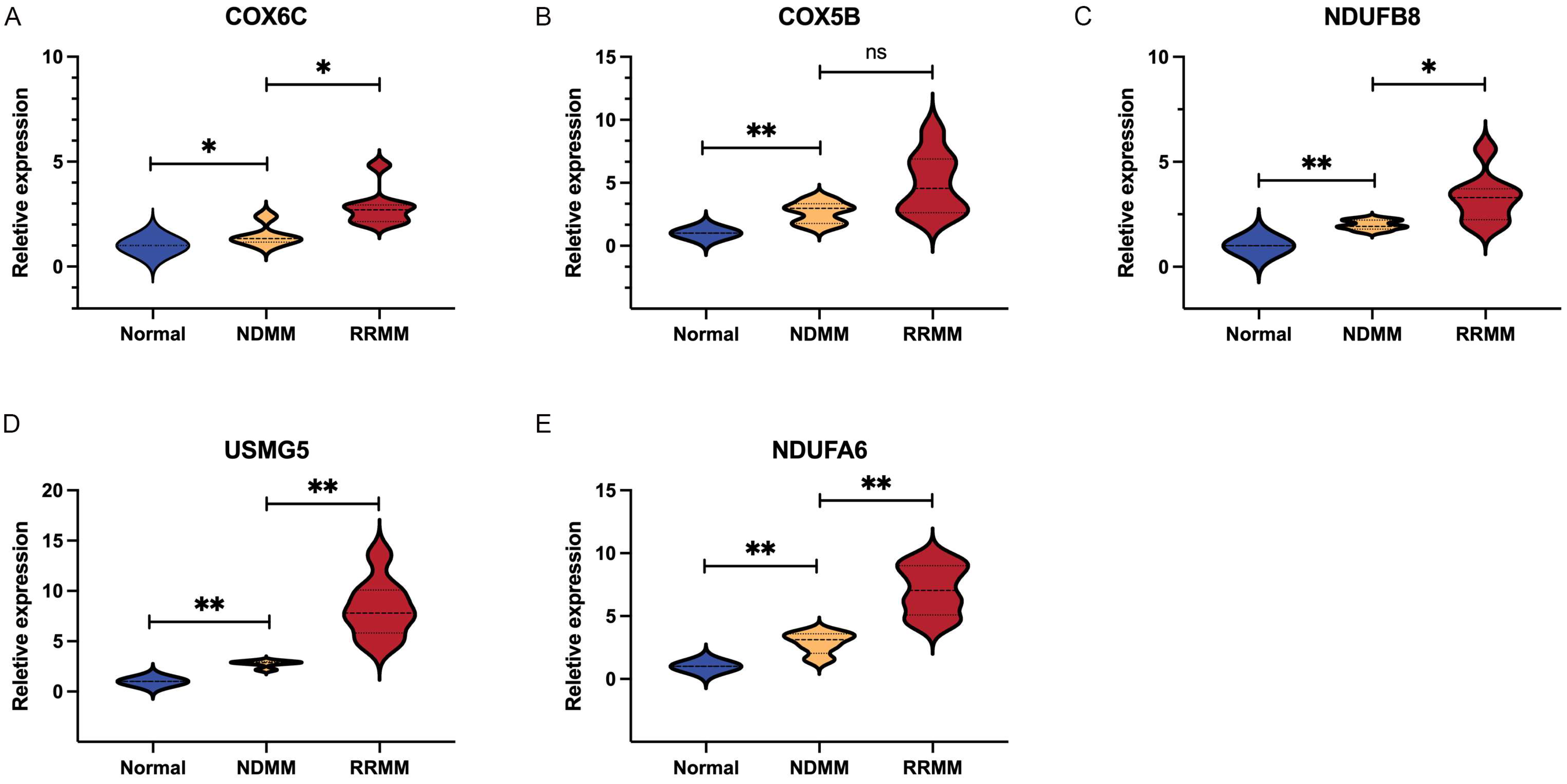
3.6. Verification of Diagnostic Markers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kyle, R.A.; Durie, B.G.; Rajkumar, S.V.; Landgren, O.; Blade, J.; Merlini, G.; Kröger, N.; Einsele, H.; Vesole, D.H.; Dimopoulos, M.; et al. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia 2010, 24, 1121–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, A.; Ghobrial, I.M. Monoclonal gammopathy of undetermined significance and smoldering multiple myeloma: A review of the current understanding of epidemiology, biology, risk stratification, and management of myeloma precursor disease. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 985–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van de Donk, N.; Pawlyn, C.; Yong, K.L. Multiple myeloma. Lancet 2021, 397, 410–427. [Google Scholar] [CrossRef] [PubMed]
- Tacchetti, P.; Pantani, L.; Patriarca, F.; Petrucci, M.T.; Zamagni, E.; Dozza, L.; Galli, M.; Di Raimondo, F.; Crippa, C.; Boccadoro, M.; et al. Bortezomib, thalidomide, and dexamethasone followed by double autologous haematopoietic stem-cell transplantation for newly diagnosed multiple myeloma (GIMEMA-MMY-3006): Long-term follow-up analysis of a randomised phase 3, open-label study. Lancet Haematol. 2020, 7, e861–e873. [Google Scholar] [CrossRef]
- Mikkilineni, L.; Kochenderfer, J.N. CAR T cell therapies for patients with multiple myeloma. Nat. Rev. Clin. Oncol. 2021, 18, 71–84. [Google Scholar] [CrossRef]
- Pawlyn, C.; Morgan, G.J. Evolutionary biology of high-risk multiple myeloma. Nat. Rev. Cancer 2017, 17, 543–556. [Google Scholar] [CrossRef]
- Maura, F.; Bolli, N.; Angelopoulos, N.; Dawson, K.J.; Leongamornlert, D.; Martincorena, I.; Mitchell, T.J.; Fullam, A.; Gonzalez, S.; Szalat, R.; et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat. Commun. 2019, 10, 3835. [Google Scholar] [CrossRef] [Green Version]
- Bolli, N.; Maura, F.; Minvielle, S.; Gloznik, D.; Szalat, R.; Fullam, A.; Martincorena, I.; Dawson, K.J.; Samur, M.K.; Zamora, J.; et al. Genomic patterns of progression in smoldering multiple myeloma. Nat. Commun. 2018, 9, 3363. [Google Scholar] [CrossRef] [Green Version]
- Boyle, E.M.; Deshpande, S.; Tytarenko, R.; Ashby, C.; Wang, Y.; Bauer, M.A.; Johnson, S.K.; Wardell, C.P.; Thanendrarajan, S.; Zangari, M.; et al. The molecular make up of smoldering myeloma highlights the evolutionary pathways leading to multiple myeloma. Nat. Commun. 2021, 12, 293. [Google Scholar] [CrossRef]
- Foltz, S.M.; Gao, Q.; Yoon, C.J.; Sun, H.; Yao, L.; Li, Y.; Jayasinghe, R.G.; Cao, S.; King, J.; Kohnen, D.R.; et al. Evolution and structure of clinically relevant gene fusions in multiple myeloma. Nat. Commun. 2020, 11, 2666. [Google Scholar] [CrossRef]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised International Staging System for Multiple Myeloma: A Report from International Myeloma Working Group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.; Lin, Y.; Navin, N. Advancing Cancer Research and Medicine with Single-Cell Genomics. Cancer Cell 2020, 37, 456–470. [Google Scholar] [CrossRef] [PubMed]
- Melchor, L.; Brioli, A.; Wardell, C.P.; Murison, A.; Potter, N.E.; Kaiser, M.F.; Fryer, R.A.; Johnson, D.C.; Begum, D.B.; Hulkki Wilson, S.; et al. Single-cell genetic analysis reveals the composition of initiating clones and phylogenetic patterns of branching and parallel evolution in myeloma. Leukemia 2014, 28, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- Merz, M.; Merz, A.M.A.; Wang, J.; Wei, L.; Hu, Q.; Hutson, N.; Rondeau, C.; Celotto, K.; Belal, A.; Alberico, R.; et al. Deciphering spatial genomic heterogeneity at a single cell resolution in multiple myeloma. Nat. Commun. 2022, 13, 807. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Li, Z.; Lu, J.; Qiang, W.; Jiang, S.; Xu, Y.; Fu, W.; Zhai, X.; Zhou, L.; Qian, M.; et al. Single-cell RNA-seq reveals clonal diversity and prognostic genes of relapsed multiple myeloma. Clin. Transl. Med. 2022, 12, e757. [Google Scholar] [CrossRef]
- Cohen, Y.C.; Zada, M.; Wang, S.Y.; Bornstein, C.; David, E.; Moshe, A.; Li, B.; Shlomi-Loubaton, S.; Gatt, M.E.; Gur, C.; et al. Identification of resistance pathways and therapeutic targets in relapsed multiple myeloma patients through single-cell sequencing. Nat. Med. 2021, 27, 491–503. [Google Scholar] [CrossRef]
- Schinke, C.; Poos, A.M.; Bauer, M.; John, L.; Johnson, S.; Deshpande, S.; Carrillo, L.; Alapat, D.; Rasche, L.; Thanendrarajan, S.; et al. Characterizing the role of the immune microenvironment in multiple myeloma progression at a single-cell level. Blood Adv. 2022, 6, 5873–5883. [Google Scholar] [CrossRef]
- Sklavenitis-Pistofidis, R.; Aranha, M.P.; Redd, R.A.; Baginska, J.; Haradhvala, N.J.; Hallisey, M.; Dutta, A.K.; Savell, A.; Varmeh, S.; Heilpern-Mallory, D.; et al. Immune biomarkers of response to immunotherapy in patients with high-risk smoldering myeloma. Cancer Cell 2022, 40, 1358–1373.e8. [Google Scholar] [CrossRef]
- Liu, R.; Gao, Q.; Foltz, S.M.; Fowles, J.S.; Yao, L.; Wang, J.T.; Cao, S.; Sun, H.; Wendl, M.C.; Sethuraman, S.; et al. Co-evolution of tumor and immune cells during progression of multiple myeloma. Nat. Commun. 2021, 12, 2559. [Google Scholar] [CrossRef]
- Qiu, X.; Mao, Q.; Tang, Y.; Wang, L.; Chawla, R.; Pliner, H.A.; Trapnell, C. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 2017, 14, 979–982. [Google Scholar] [CrossRef]
- Wagner, A.; Regev, A.; Yosef, N. Revealing the vectors of cellular identity with single-cell genomics. Nat. Biotechnol. 2016, 34, 1145–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, J.S.; Li, Y.; Mitra, A.K.; Bi, L.; Abyzov, A.; van Wijnen, A.J.; Baughn, L.B.; Van Ness, B.; Rajkumar, V.; Kumar, S.; et al. Molecular signatures of multiple myeloma progression through single cell RNA-Seq. Blood Cancer J. 2019, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Lan, Y.; Xu, J.; Quan, F.; Zhao, E.; Deng, C.; Luo, T.; Xu, L.; Liao, G.; Yan, M.; et al. CellMarker: A manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 2019, 47, D721–D728. [Google Scholar] [CrossRef] [Green Version]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef]
- Wang, Z.; Guo, X.; Gao, L.; Wang, Y.; Ma, W.; Xing, B. Glioblastoma cell differentiation trajectory predicts the immunotherapy response and overall survival of patients. Aging 2020, 12, 18297–18321. [Google Scholar] [CrossRef]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. 4), S11. [Google Scholar] [CrossRef] [Green Version]
- The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still Going strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef] [Green Version]
- Alameda, D.; Saez, B.; Lara-Astiaso, D.; Sarvide, S.; Lasa, M.; Alignani, D.; Rodriguez, I.; Garate, S.; Vilas, A.; Paiva, B.; et al. Characterization of freshly isolated bone marrow mesenchymal stromal cells from healthy donors and patients with multiple myeloma: Transcriptional modulation of the microenvironment. Haematologica 2020, 105, e470–e473. [Google Scholar] [CrossRef] [Green Version]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef]
- Serrano-Del Valle, A.; Anel, A.; Naval, J.; Marzo, I. Immunogenic Cell Death and Immunotherapy of Multiple Myeloma. Front. Cell Dev. Biol. 2019, 7, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marlein, C.R.; Piddock, R.E.; Mistry, J.J.; Zaitseva, L.; Hellmich, C.; Horton, R.H.; Zhou, Z.; Auger, M.J.; Bowles, K.M.; Rushworth, S.A. CD38-Driven Mitochondrial Trafficking Promotes Bioenergetic Plasticity in Multiple Myeloma. Cancer Res. 2019, 79, 2285–2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, P.; Wang, M.L. Mantle cell lymphoma in 2022-A comprehensive update on molecular pathogenesis, risk stratification, clinical approach, and current and novel treatments. Am. J. Hematol. 2022, 97, 638–656. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.A.; Anderson, E.A.; Jessen, E.; Nandakumar, B.; Atilgan, E.; Jevremovic, D.; Hitosugi, T.; Fonseca, R.; Kumar, S.K.; Gonsalves, W.I. Overexpression of the energy metabolism transcriptome within clonal plasma cells is associated with the pathogenesis and outcomes of patients with multiple myeloma. Am. J. Hematol. 2022, 97, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Fang, B.; Liu, Y.; Yan, S.; Cao, D.; Mei, H.; Wang, Q.; Hu, Y.; Guo, T. SR18292 exerts potent antitumor effects in multiple myeloma via inhibition of oxidative phosphorylation. Life Sci. 2020, 256, 117971. [Google Scholar] [CrossRef] [PubMed]
- Waldschmidt, J.M.; Kloeber, J.A.; Anand, P.; Frede, J.; Kokkalis, A.; Dimitrova, V.; Potdar, S.; Nair, M.S.; Vijaykumar, T.; Im, N.G.; et al. Single-Cell Profiling Reveals Metabolic Reprogramming as a Resistance Mechanism in BRAF-Mutated Multiple Myeloma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 6432–6444. [Google Scholar] [CrossRef] [PubMed]
- Sazanov, L.A. A giant molecular proton pump: Structure and mechanism of respiratory complex I. Nat. Rev. Mol. Cell Biol. 2015, 16, 375–388. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Cardona, L.R.; Kong, H.; Vasan, K.; McElroy, G.S.; Werner, M.; Kihshen, H.; Reczek, C.R.; Weinberg, S.E.; Gao, P.; et al. Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature 2020, 585, 288–292. [Google Scholar] [CrossRef]
- Urra, F.A.; Muñoz, F.; Lovy, A.; Cárdenas, C. The Mitochondrial Complex(I)ty of Cancer. Front. Oncol. 2017, 7, 118. [Google Scholar] [CrossRef]
- Bajpai, R.; Sharma, A.; Achreja, A.; Edgar, C.L.; Wei, C.; Siddiqa, A.A.; Gupta, V.A.; Matulis, S.M.; McBrayer, S.K.; Mittal, A.; et al. Electron transport chain activity is a predictor and target for venetoclax sensitivity in multiple myeloma. Nat. Commun. 2020, 11, 1228. [Google Scholar] [CrossRef]
- Belevich, I.; Verkhovsky, M.I. Molecular mechanism of proton translocation by cytochrome c oxidase. Antioxid. Redox Signal. 2008, 10, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.W.; Fan, H.C.; Liu, J.M.; Fan, T.P.; Jing, J.; Yang, C.L.; Hsu, R.J. Estrogen Enhances the Expression of the Multidrug Transporter Gene ABCG2-Increasing Drug Resistance of Breast Cancer Cells through Estrogen Receptors. Int. J. Mol. Sci. 2017, 18, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Li, Y.; Huang, Y.; Ouyang, Y.; Zhu, Y.; Wang, Y.; Guo, X.; Yuan, Y.; Gong, K. Long non-coding RNA MIF-AS1 promotes gastric cancer cell proliferation and reduces apoptosis to upregulate NDUFA4. Cancer Sci. 2018, 109, 3714–3725. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.C.; Crescitelli, R.; Cvjetkovic, A.; Belgrano, V.; Olofsson Bagge, R.; Sundfeldt, K.; Ochiya, T.; Kalluri, R.; Lötvall, J. Mitochondrial protein enriched extracellular vesicles discovered in human melanoma tissues can be detected in patient plasma. J. Extracell. Vesicles 2019, 8, 1635420. [Google Scholar] [CrossRef] [Green Version]
- Tian, B.X.; Sun, W.; Wang, S.H.; Liu, P.J.; Wang, Y.C. Differential expression and clinical significance of COX6C in human diseases. Am. J. Transl. Res. 2021, 13, 1–10. [Google Scholar]
- Wang, F.; Liu, R.; Yang, J.; Chen, B. New insights into genetic characteristics between multiple myeloma and COVID-19: An integrative bioinformatics analysis of gene expression omnibus microarray and the cancer genome atlas data. Int. J. Lab. Hematol. 2021, 43, 1325–1333. [Google Scholar] [CrossRef]
- Chu, Y.D.; Lim, S.N.; Yeh, C.T.; Lin, W.R. COX5B-Mediated Bioenergetic Alterations Modulate Cell Growth and Anticancer Drug Susceptibility by Orchestrating Claudin-2 Expression in Colorectal Cancers. Biomedicines 2021, 10, 60. [Google Scholar] [CrossRef]
- Chu, Y.D.; Lin, W.R.; Lin, Y.H.; Kuo, W.H.; Tseng, C.J.; Lim, S.N.; Huang, Y.L.; Huang, S.C.; Wu, T.J.; Lin, K.H.; et al. COX5B-Mediated Bioenergetic Alteration Regulates Tumor Growth and Migration by Modulating AMPK-UHMK1-ERK Cascade in Hepatoma. Cancers 2020, 12, 1646. [Google Scholar] [CrossRef]
- Stein, J.; Tenbrock, J.; Kristiansen, G.; Müller, S.C.; Ellinger, J. Systematic expression analysis of the mitochondrial respiratory chain protein subunits identifies COX5B as a prognostic marker in clear cell renal cell carcinoma. Int. J. Urol. 2019, 26, 910–916. [Google Scholar] [CrossRef]
- Kontro, H.; Cannino, G.; Rustin, P.; Dufour, E.; Kainulainen, H. DAPIT Over-Expression Modulates Glucose Metabolism and Cell Behaviour in HEK293T Cells. PLoS ONE 2015, 10, e0131990. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, J.; Wang, X.; Zhu, H.; Wei, S.; Zhang, H.; Ma, L.; He, P. Integrative Analysis of Bulk RNA-Seq and Single-Cell RNA-Seq Unveils Novel Prognostic Biomarkers in Multiple Myeloma. Biomolecules 2022, 12, 1855. https://doi.org/10.3390/biom12121855
Zhao J, Wang X, Zhu H, Wei S, Zhang H, Ma L, He P. Integrative Analysis of Bulk RNA-Seq and Single-Cell RNA-Seq Unveils Novel Prognostic Biomarkers in Multiple Myeloma. Biomolecules. 2022; 12(12):1855. https://doi.org/10.3390/biom12121855
Chicago/Turabian StyleZhao, Jing, Xiaoning Wang, Huachao Zhu, Suhua Wei, Hailing Zhang, Le Ma, and Pengcheng He. 2022. "Integrative Analysis of Bulk RNA-Seq and Single-Cell RNA-Seq Unveils Novel Prognostic Biomarkers in Multiple Myeloma" Biomolecules 12, no. 12: 1855. https://doi.org/10.3390/biom12121855
APA StyleZhao, J., Wang, X., Zhu, H., Wei, S., Zhang, H., Ma, L., & He, P. (2022). Integrative Analysis of Bulk RNA-Seq and Single-Cell RNA-Seq Unveils Novel Prognostic Biomarkers in Multiple Myeloma. Biomolecules, 12(12), 1855. https://doi.org/10.3390/biom12121855