

Entropy and Variability: A Second Opinion by Deep Learning


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
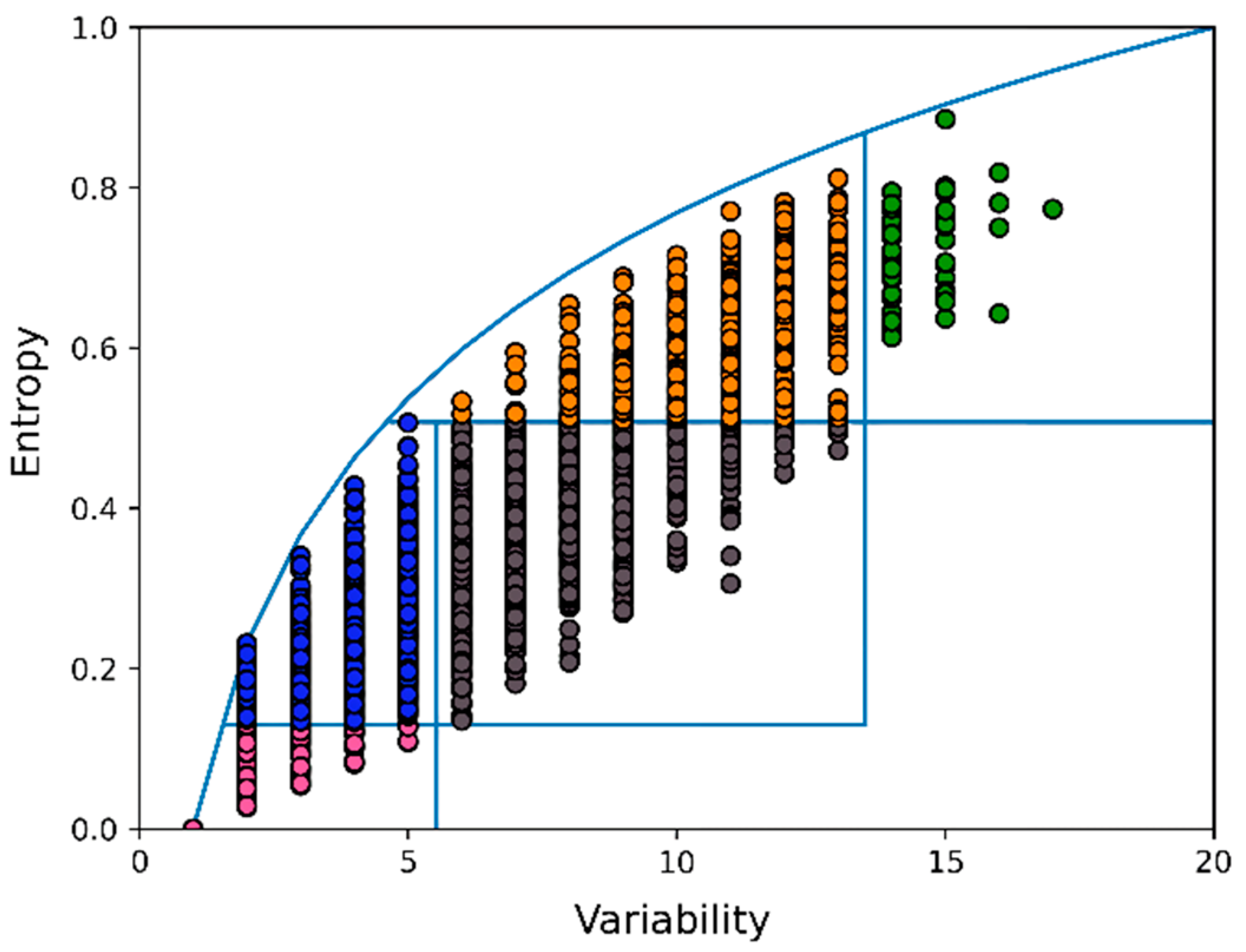
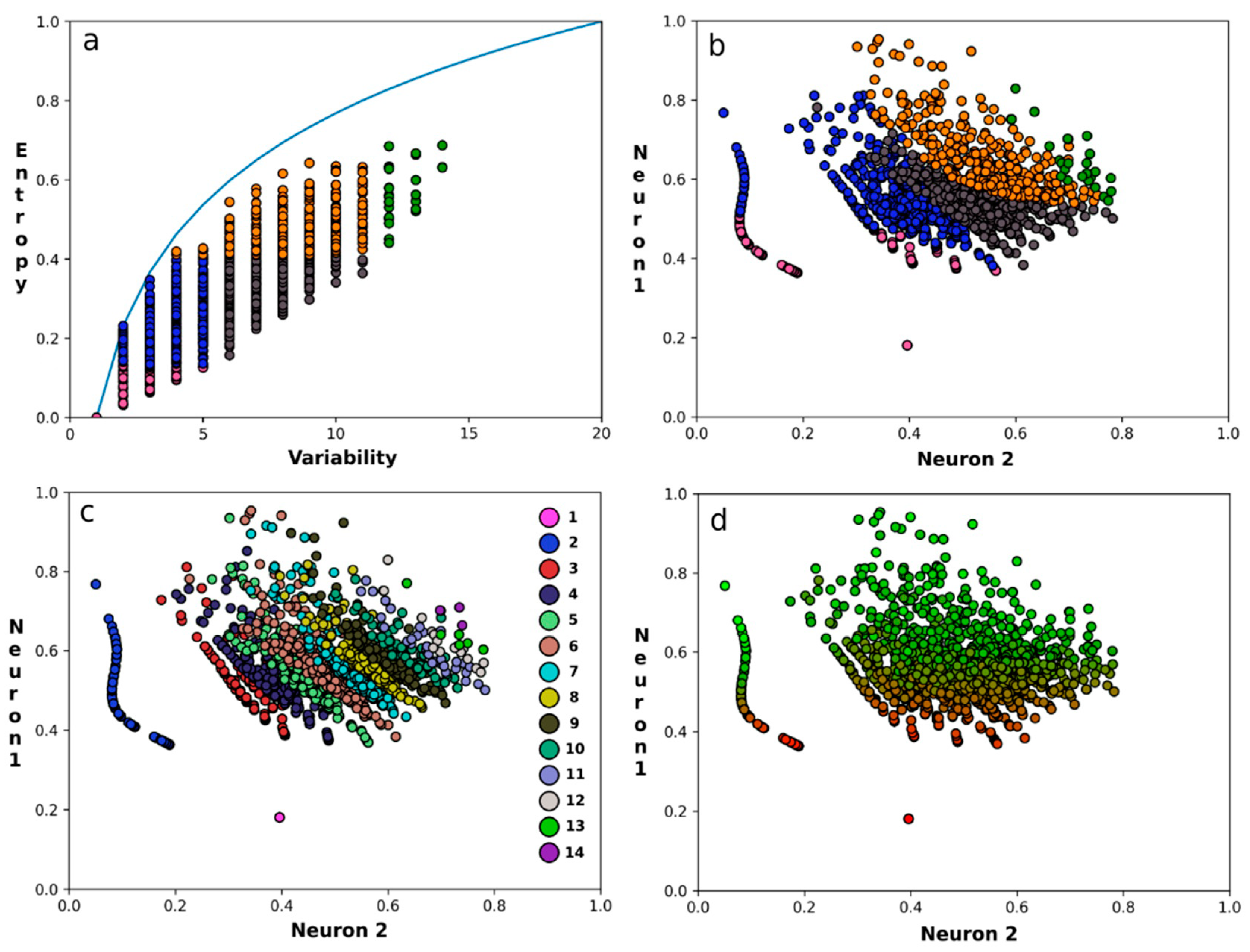
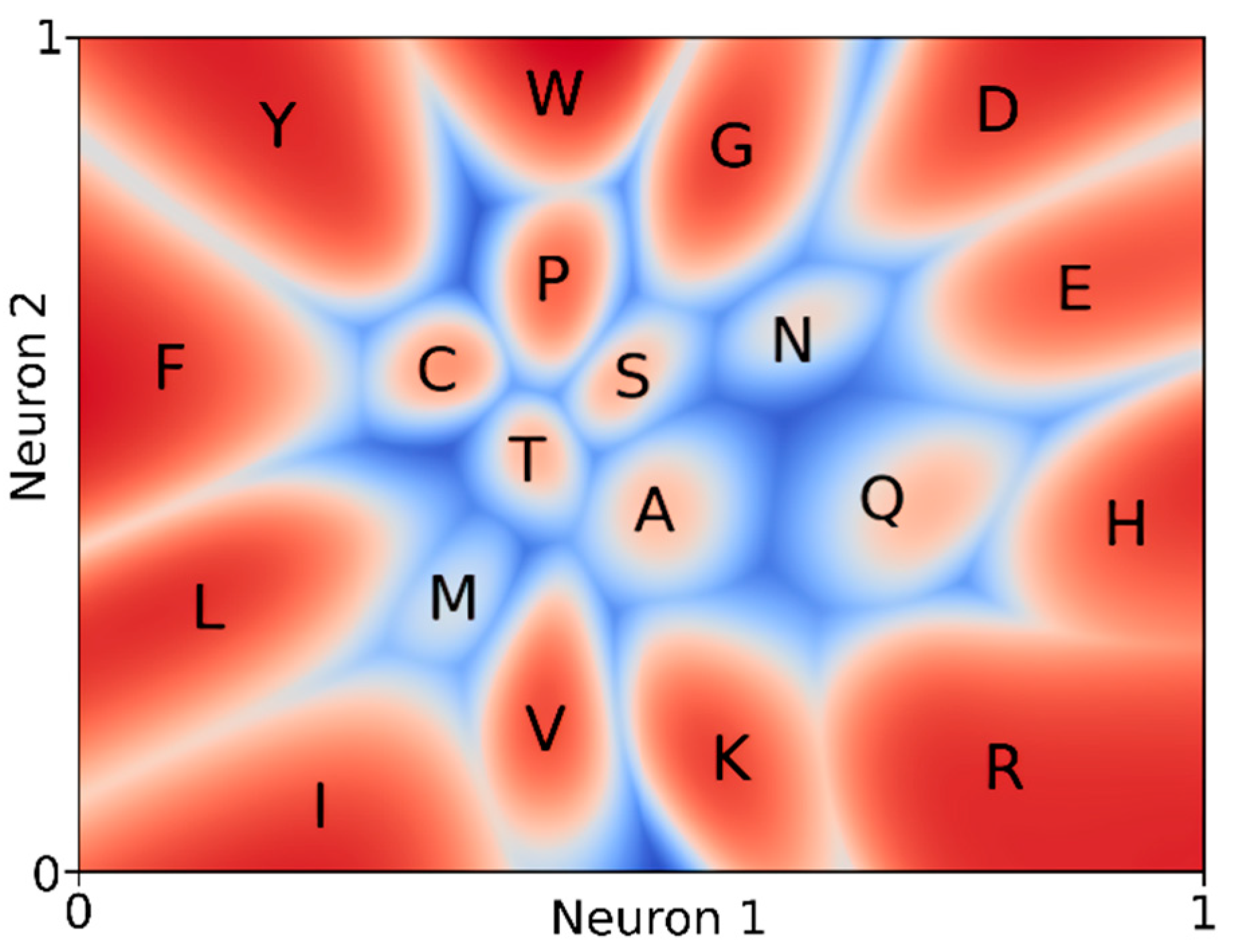
3. Results
4. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bourne, P.E. Is “bioinformatics” dead? PLoS Biol. 2021, 19, e3001165. [Google Scholar] [CrossRef] [PubMed]
- Markowetz, F. All biology is computational biology. PLoS Biol. 2017, 15, e2002050. [Google Scholar] [CrossRef] [PubMed]
- Wikipedia. The Fourth Paradigm. 19 March 2021. Available online: https://en.wikipedia.org/w/index.php?title=The_Fourth_Paradigm&oldid=1012968154 (accessed on 28 July 2022).
- Zubarev, D.Y.; Pitera, J.W. Cognitive materials discovery and onset of the 5th discovery paradigm. In ACS Symposium Series; Pyzer-Knapp, E.O., Laino, T., Eds.; American Chemical Society: Washington, DC, USA, 2019; Volume 1326, pp. 103–120. [Google Scholar] [CrossRef]
- Babbitt, P.C.; Bagos, P.G.; Bairoch, A.; Bateman, A.; Chatonnet, A.; Chen, M.J.; Craik, D.J.; Finn, R.D.; Gloriam, D.; Haft, D.H.; et al. Creating a specialist protein resource network: A meeting report for the protein bioinformatics and community resources retreat. Database 2015, 2015, bav063. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.S.; Burgess, A.E.; Bourne, P.E. Ten simple rules for starting (and sustaining) an academic data science initiative. PLoS Comput. Biol. 2021, 17, e1008628. [Google Scholar] [CrossRef]
- Wikipedia. FAIR Data. 30 June 2022. Available online: https://en.wikipedia.org/w/index.php?title=FAIR_data&oldid=1095813033 (accessed on 28 July 2022).
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Marks, D.S.; Hopf, T.A.; Sander, C. Protein structure prediction from sequence variation. Nat. Biotechnol. 2012, 30, 1072–1080. [Google Scholar] [CrossRef]
- Jones, D.T.; Buchan, D.W.A.; Cozzetto, D.; Pontil, M. PSICOV: Precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments. Bioinformatics 2012, 28, 184–190. [Google Scholar] [CrossRef]
- Wang, J.; Lisanza, S.; Juergens, D.; Tischer, D.; Watson, J.L.; Castro, K.M.; Ragotte, R.; Saragovi, A.; Milles, L.F.; Baek, M.; et al. Scaffolding protein functional sites using deep learning. Science 2022, 377, 387–394. [Google Scholar] [CrossRef]
- Mirhoseini, A.; Goldie, A.; Yazgan, M.; Jiang, J.W.; Songhori, E.; Wang, S.; Lee, Y.-J.; Johnson, E.; Pathak, O.; Nazi, A.; et al. A graph placement methodology for fast chip design. Nature 2021, 594, 207–212. [Google Scholar] [CrossRef]
- Renaud, N.; Geng, C.; Georgievska, S.; Ambrosetti, F.; Ridder, L.; Marzella, D.F.; Réau, M.F.; Bonvin, A.M.J.J.; Xue, L.C. DeepRank: A deep learning framework for data mining 3D protein-protein interfaces. Nat. Commun. 2021, 12, 7068. [Google Scholar] [CrossRef]
- Oliveira, L.; Paiva, A.C.M.; Vriend, G. Correlated Mutation Analyses on Very Large Sequence Families. ChemBioChem 2002, 3, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.; Paiva, P.B.; Paiva, A.C.M.; Vriend, G. Identification of functionally conserved residues with the use of entropy-variability plots. Proteins 2003, 52, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Pándy-Szekeres, G.; Munk, C.; Tsonkov, T.M.; Mordalski, S.; Harpsøe, K.; Hauser, A.S.; Bojarski, A.J.; Gloriam, D.E. GPCRdb in 2018: Adding GPCR structure models and ligands. Nucleic Acids Res. 2018, 46, D440–D446. [Google Scholar] [CrossRef] [PubMed]
- Munk, C.; Isberg, V.; Mordalski, S.; Harpsøe, K.; Rataj, K.; Hauser, A.S.; Kolb, P.; Bojarski, A.J.; Vriend, G.; E Gloriam, D. GPCRdb: The G protein-coupled receptor database—An introduction. Br. J. Pharmacol. 2016, 173, 2195–2207. [Google Scholar] [CrossRef]
- Kuipers, R.K.; Joosten, H.-J.; van Berkel, W.; Leferink, N.; Rooijen, E.; Ittmann, E.; van Zimmeren, F.; Jochens, H.; Bornscheuer, U.; Vriend, G.; et al. 3DM: Systematic analysis of heterogeneous superfamily data to discover protein functionalities. Proteins Struct. Funct. Bioinform. 2010, 78, 2101–2113. [Google Scholar] [CrossRef] [PubMed]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Protein structure prediction using multiple deep neural networks in the 13th Critical Assessment of Protein Structure Prediction (CASP13). Proteins Struct. Funct. Bioinform. 2019, 87, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.M.; Liu, J.; Verkuil, R.; Meier, J.; Canny, J.; Abbeel, P.; Sercu, T.; Rives, A. MSA transformer. In Proceedings of the 38th International Conference on Machine Learning, Virtual, 18–24 July 2021; pp. 8844–8856. Available online: https://proceedings.mlr.press/v139/rao21a.html (accessed on 30 August 2022).
- Mooney, S.D.; Klein, T.E. The functional importance of disease-associated mutation. BMC Bioinform. 2002, 3, 24. [Google Scholar] [CrossRef]
- Vollan, H.S.; Tannæs, T.; Vriend, G.; Bukholm, G. In Silico Structure and Sequence Analysis of Bacterial Porins and Specif-ic Diffusion Channels for Hydrophilic Molecules: Conservation, Multimericity and Multifunctionality. Int. J. Mol. Sci. 2016, 17, 599. [Google Scholar] [CrossRef]
- Gáspári, Z.; Ortutay, C.; Perczel, A. A simple fold with variations: The pacifastin inhibitor family. Bioinformatics 2004, 20, 448–451. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Y.; Yang, X.; Guo, H.; Lin, J.; Yang, J.; He, M.; Wang, J.; Liu, X.; Shi, T.; et al. Predicting the early risk of ophthalmopathy in Graves’ disease patients using TCR repertoire. Clin. Transl. Med. 2020, 10, e218. [Google Scholar] [CrossRef]
- Samsonova, E.V.; Krause, P.; Bäck, T.; Ijzerman, A.P. Characteristic amino acid combinations in olfactory G protein-coupled receptors. Proteins Struct. Funct. Bioinform. 2007, 67, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Posada, D.; Zardoya, R. MtArt: A New Model of Amino Acid Replacement for Arthropoda. Mol. Biol. Evol. 2006, 24, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Bywater, R.P. Prediction of Protein Structural Features from Sequence Data Based on Shannon Entropy and Kolmogorov Complexity. PLoS ONE 2015, 10, e0119306. [Google Scholar] [CrossRef] [PubMed]
- Min, S.; Lee, B.; Yoon, S. Deep learning in bioinformatics. Brief. Bioinform. 2016, 18, bbw068. [Google Scholar] [CrossRef] [PubMed]
- Stepniewska-Dziubinska, M.M.; Zielenkiewicz, P.; Siedlecki, P. Development and evaluation of a deep learning model for protein–ligand binding affinity prediction. Bioinformatics 2018, 34, 3666–3674. [Google Scholar] [CrossRef] [PubMed]
- Dodge, C.; Schneider, R.; Sander, C. The HSSP database of protein structure—Sequence alignments and family profiles. Nucleic Acids Res. 1998, 26, 313–315. [Google Scholar] [CrossRef]
- Sander, C.; Schneider, R. Database of homology-derived protein structures and the structural meaning of sequence alignment. Proteins Struct. Funct. Bioinform. 1991, 9, 56–68. [Google Scholar] [CrossRef]
- Goodfellow, I.; Bengio, Y.; Courville, A. Deep Learning; MIT Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Ioffe, S.; Szegedy, C. Batch Normalization: Accelerating Deep Network Training by Reducing Internal Covariate Shift. arXiv 2015, arXiv:1502.03167. [Google Scholar]
- Hinton, G.E.; Salakhutdinov, R.R. Reducing the Dimensionality of Data with Neural Networks. Science 2006, 313, 504–507. [Google Scholar] [CrossRef]
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization. arXiv 2017, arXiv:1412.6980. [Google Scholar]
- Paszke, A.; Gross, S.; Chintala, S.; Chanan, G.; Yang, E.; DeVito, Z.; Lin, Z.; Desmaison, A.; Antiga, L.; Lerer, A. Automatic Differentiation in PyTorch. October 2017. Available online: https://openreview.net/forum?id=BJJsrmfCZ (accessed on 28 July 2022).
- Crimella, C.; Arnoldi, A.; Crippa, F.; Mostacciuolo, M.L.; Boaretto, F.; Sironi, M.; D’Angelo, M.G.; Manzoni, S.; Piccinini, L.; Turconi, A.C.; et al. Point mutations and a large intragenic deletion in SPG11 in complicated spastic paraplegia without thin corpus callosum. J. Med. Genet. 2009, 46, 345–351. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rademaker, D.T.; Xue, L.C.; ‘t Hoen, P.A.C.; Vriend, G. Entropy and Variability: A Second Opinion by Deep Learning. Biomolecules 2022, 12, 1740. https://doi.org/10.3390/biom12121740
Rademaker DT, Xue LC, ‘t Hoen PAC, Vriend G. Entropy and Variability: A Second Opinion by Deep Learning. Biomolecules. 2022; 12(12):1740. https://doi.org/10.3390/biom12121740
Chicago/Turabian StyleRademaker, Daniel T., Li C. Xue, Peter A. C. ‘t Hoen, and Gert Vriend. 2022. "Entropy and Variability: A Second Opinion by Deep Learning" Biomolecules 12, no. 12: 1740. https://doi.org/10.3390/biom12121740
APA StyleRademaker, D. T., Xue, L. C., ‘t Hoen, P. A. C., & Vriend, G. (2022). Entropy and Variability: A Second Opinion by Deep Learning. Biomolecules, 12(12), 1740. https://doi.org/10.3390/biom12121740