
LRRK2 and Lipid Pathways: Implications for Parkinson’s Disease

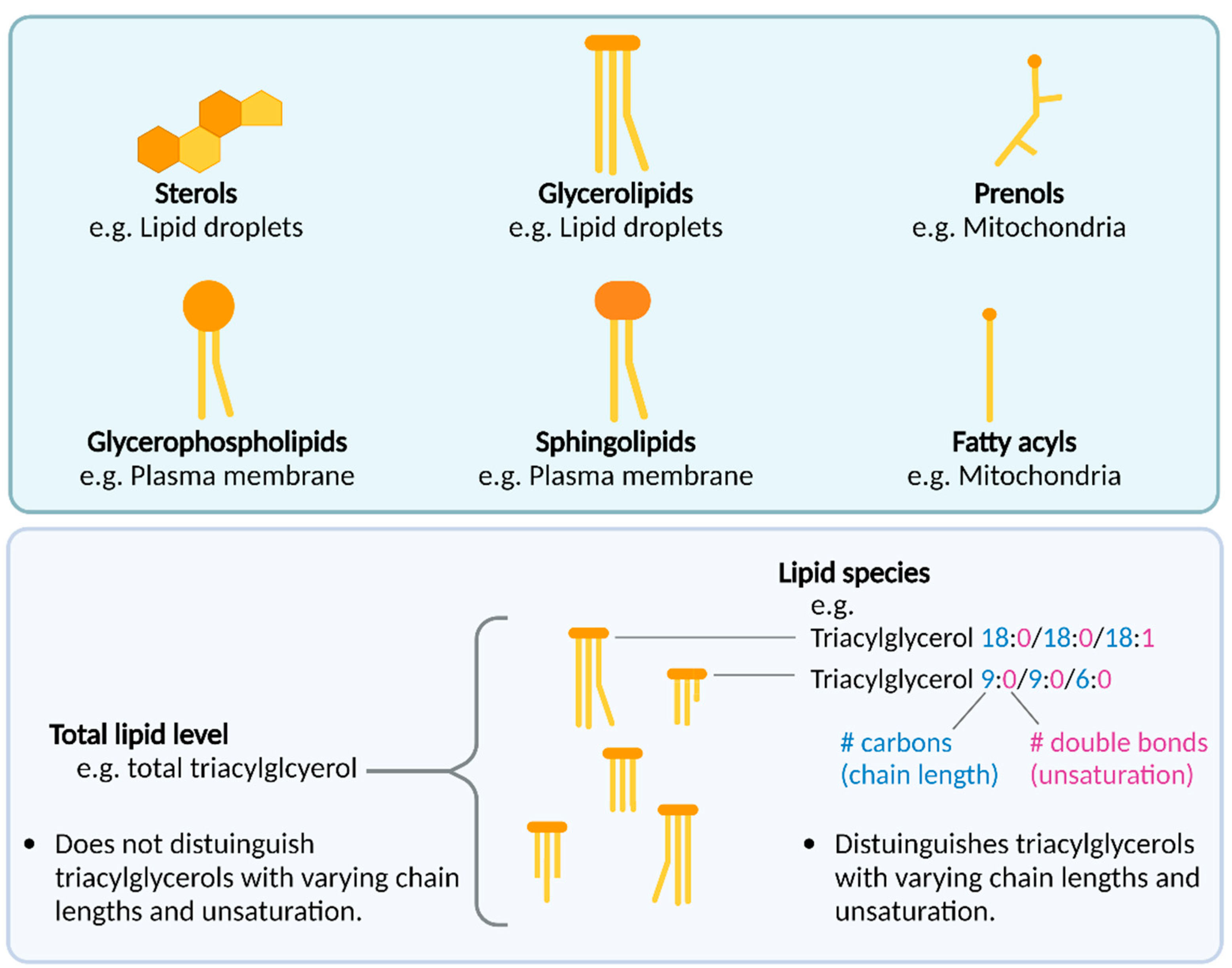{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Overview
1.2. An Introduction to Lipids
1.3. Lipid Alterations in LRRK2 Knockouts
1.4. Lipid Alterations in LRRK2 Mutation Carrier Humans

2. LRRK2 and Lysosomal Lipid Metabolism
3. LRRK2, Mitochondrial Energy and Glucose/Insulin Pathways
3.1. LRRK2 and Insulin/Glucose
3.2. LRRK2, Mitochondria and the Production and Storage of Energy
4. LRRK2 and Lipids in the Synaptic Vesicle Cycle
5. Lipid Biomarkers and Pharmacological LRRK2 Inhibition
6. Lipid Modulators in LRRK2 Models and Parkinson’s Disease
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Monfrini, E.; Di Fonzo, A. Leucine-Rich Repeat Kinase (LRRK2) Genetics and Parkinson’s Disease. Adv. Neurobiol. 2017, 14, 3–30. [Google Scholar] [CrossRef] [PubMed]
- Healy, D.G.; Falchi, M.; O’Sullivan, S.S.; Bonifati, V.; Durr, A.; Bressman, S.; Brice, A.; Aasly, J.; Zabetian, C.P.; Goldwurm, S.; et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol. 2008, 7, 583–590. [Google Scholar] [CrossRef] [Green Version]
- Gilks, W.P.; Abou-Sleiman, P.M.; Gandhi, S.; Jain, S.; Singleton, A.; Lees, A.J.; Shaw, K.; Bhatia, K.P.; Bonifati, V.; Quinn, N.P.; et al. A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet 2005, 365, 415–416. [Google Scholar] [CrossRef]
- Biskup, S.; Moore, D.J.; Celsi, F.; Higashi, S.; West, A.B.; Andrabi, S.A.; Kurkinen, K.; Yu, S.-W.; Savitt, J.M.; Waldvogel, H.J.; et al. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann. Neurol. 2006, 60, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.; Jeong, H.; Kwon, J.; Heo, H.Y.; Kwon, J.J.; Yun, H.J.; Kim, C.H.; Han, B.S.; Tong, Y.; Shen, J.; et al. LRRK2 regulates synaptic vesicle endocytosis. Exp. Cell Res. 2008, 314, 2055–2065. [Google Scholar] [CrossRef]
- Piccoli, G.; Condliffe, S.B.; Bauer, M.; Giesert, F.; Boldt, K.; De Astis, S.; Meixner, A.; Sarioglu, H.; Vogt-Weisenhorn, D.M.; Wurst, W.; et al. LRRK2 Controls Synaptic Vesicle Storage and Mobilization within the Recycling Pool. J. Neurosci. 2011, 31, 2225–2237. [Google Scholar] [CrossRef] [Green Version]
- Alegre-Abarrategui, J.; Christian, H.; Lufino, M.M.; Mutihac, R.; Venda, L.L.; Ansorge, O.; Wade-Martins, R. LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum. Mol. Genet. 2009, 18, 4022–4034. [Google Scholar] [CrossRef] [Green Version]
- Piomelli, D.; Astarita, G.; Rapaka, R. A neuroscientist’s guide to lipidomics. Nat. Rev. Neurosci. 2007, 8, 743–754. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.H.; Seo, Y.; Jo, Y.S.; Lee, S.; Cho, E.; Cazenave-Gassiot, A.; Shin, Y.-S.; Moon, M.H.; An, H.J.; Wenk, M.R.; et al. Brain lipidomics: From functional landscape to clinical significance. Sci. Adv. 2022, 8, eadc9317. [Google Scholar] [CrossRef]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell. Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef]
- Holthuis, J.C.M.; Menon, A.K. Lipid landscapes and pipelines in membrane homeostasis. Nature 2014, 510, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.; Shimizu, T.; Spener, F.; van Meer, G.; Wakelam, M.J.; Dennis, E.A. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 2009, 50, S9–S14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrazza, R.; Cogo, S.; Melrose, H.; Bubacco, L.; Greggio, E.; Guella, G.; Civiero, L.; Plotegher, N. LRRK2 deficiency impacts ceramide metabolism in brain. Biochem. Biophys. Res. Commun. 2016, 478, 1141–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuji, R.N.; Flagella, M.; Baca, M.; Baptista, M.A.S.; Brodbeck, J.; Chan, B.K.; Fiske, B.K.; Honigberg, L.; Jubb, A.M.; Katavolos, P.; et al. Effect of selective LRRK2 kinase inhibition on nonhuman primate lung. Sci. Transl. Med. 2015, 7, 273ra215. [Google Scholar] [CrossRef] [PubMed]
- Baptista, M.A.S.; Dave, K.D.; Frasier, M.A.; Sherer, T.B.; Greeley, M.; Beck, M.J.; Varsho, J.S.; Parker, G.A.; Moore, C.; Churchill, M.J.; et al. Loss of Leucine-Rich Repeat Kinase 2 (LRRK2) in Rats Leads to Progressive Abnormal Phenotypes in Peripheral Organs. PLoS ONE 2013, 8, e80705. [Google Scholar] [CrossRef] [Green Version]
- Ness, D.; Ren, Z.; Gardai, S.; Sharpnack, D.; Johnson, V.J.; Brennan, R.J.; Brigham, E.F.; Olaharski, A.J. Leucine-Rich Repeat Kinase 2 (LRRK2)-Deficient Rats Exhibit Renal Tubule Injury and Perturbations in Metabolic and Immunological Homeostasis. PLoS ONE 2013, 8, e66164. [Google Scholar] [CrossRef] [Green Version]
- Boddu, R.; Hull, T.D.; Bolisetty, S.; Hu, X.; Moehle, M.S.; Daher, J.P.L.; Kamal, A.I.; Joseph, R.; George, J.F.; Agarwal, A.; et al. Leucine-rich repeat kinase 2 deficiency is protective in rhabdomyolysis-induced kidney injury. Hum. Mol. Genet. 2015, 24, 4078–4093. [Google Scholar] [CrossRef]
- Abbott, S.K.; Li, H.; Muñoz, S.S.; Knoch, B.; Batterham, M.; Murphy, K.E.; Halliday, G.M.; Garner, B. Altered ceramide acyl chain length and ceramide synthase gene expression in Parkinson’s disease. Mov. Disord. 2014, 29, 518–526. [Google Scholar] [CrossRef] [Green Version]
- Bardien, S.; Lesage, S.; Brice, A.; Carr, J. Genetic characteristics of leucine-rich repeat kinase 2 (LRRK2) associated Parkinson’s disease. Park. Relat. Disord. 2011, 17, 501–508. [Google Scholar] [CrossRef]
- Yu, M.; Arshad, M.; Wang, W.; Zhao, D.; Xu, L.; Zhou, L. LRRK2 mediated Rab8a phosphorylation promotes lipid storage. Lipids Health Dis. 2018, 17, 34. [Google Scholar] [CrossRef]
- Whiffin, N.; Armean, I.M.; Kleinman, A.; Marshall, J.L.; Minikel, E.V.; Goodrich, J.K.; Quaife, N.M.; Cole, J.B.; Wang, Q.; Karczewski, K.J.; et al. The effect of LRRK2 loss-of-function variants in humans. Nat. Med. 2020, 26, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Yakhine-Diop, S.M.S.; Morales-García, J.A.; Niso-Santano, M.; González-Polo, R.A.; Uribe-Carretero, E.; Martinez-Chacon, G.; Durand, S.; Maiuri, M.C.; Aiastui, A.; Zulaica, M.; et al. Metabolic alterations in plasma from patients with familial and idiopathic Parkinson’s disease. Aging (Albany NY) 2020, 12, 16690–16708. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Alessi, D.R. Advances in elucidating the function of leucine-rich repeat protein kinase-2 in normal cells and Parkinson’s disease. Curr. Opin. Cell. Biol. 2020, 63, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Alcalay, R.N.; Hsieh, F.; Tengstrand, E.; Padmanabhan, S.; Baptista, M.; Kehoe, C.; Narayan, S.; Boehme, A.K.; Merchant, K. Higher Urine bis (Monoacylglycerol) Phosphate Levels in LRRK2 G2019S Mutation Carriers: Implications for Therapeutic Development. Mov. Disord. 2020, 35, 134–141. [Google Scholar] [CrossRef]
- Gomes, S.; Garrido, A.; Tonelli, F.; Obiang, D.; Tolosa, E.; Marti, M.J.; Ruiz-Martinez, J.; Aragon, A.V.; Hernandez-Eguiazu, H.; Croitoru, I.; et al. Elevated urine BMPs—bis (Monoacylglycerol) Phosphates—in LRRK2 G2019S, R1441G/C and VPS35 D620N mutation carriers with and without Parkinson’s disease. medRxiv 2022. [Google Scholar] [CrossRef]
- Galper, J.; Dean, N.J.; Pickford, R.; Lewis, S.J.G.; Halliday, G.M.; Kim, W.S.; Dzamko, N. Lipid pathway dysfunction is prevalent in patients with Parkinson’s disease. Brain J. Neurol. 2022. [Google Scholar] [CrossRef]
- Stiban, J.; Perera, M. Very long chain ceramides interfere with C16-ceramide-induced channel formation: A plausible mechanism for regulating the initiation of intrinsic apoptosis. Biochim. Biophys. Acta (BBA) Biomembr. 2015, 1848, 561–567. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, D.; Wegner, M.-S.; Wanger, R.A.; Ferreirós, N.; Schreiber, Y.; Lucks, J.; Schiffmann, S.; Geisslinger, G.; Grösch, S. The equilibrium between long and very long chain ceramides is important for the fate of the cell and can be influenced by co-expression of CerS. Int. J. Biochem. Cell Biol. 2013, 45, 1195–1203. [Google Scholar] [CrossRef]
- Abou-Ghali, M.; Stiban, J. Regulation of ceramide channel formation and disassembly: Insights on the initiation of apoptosis. Saudi J. Biol. Sci. 2015, 22, 760–772. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef]
- McGranaghan, P.; Kirwan, J.A.; Garcia-Rivera, M.A.; Pieske, B.; Edelmann, F.; Blaschke, F.; Appunni, S.; Saxena, A.; Rubens, M.; Veledar, E.; et al. Lipid Metabolite Biomarkers in Cardiovascular Disease: Discovery and Biomechanism Translation from Human Studies. Metabolites 2021, 11, 621. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Wang, Y.; He, X.; Li, H.; Liu, H.; Zhang, X. A systematic review and meta-analysis of serum cholesterol and triglyceride levels in patients with Parkinson’s disease. Lipids Health Dis. 2020, 19, 97. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Xu, X.; Gu, X.; Ou, R.; Luo, X.; Shang, H.; Song, W. Effects of Higher Serum Lipid Levels on the Risk of Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front. Neurol. 2020, 11, 597. [Google Scholar] [CrossRef] [PubMed]
- Thaler, A.; Shenhar-Tsarfaty, S.; Shaked, Y.; Gurevich, T.; Omer, N.; Bar-Shira, A.; Gana-Weisz, M.; Goldstein, O.; Kestenbaum, M.; Cedarbaum, J.M.; et al. Metabolic syndrome does not influence the phenotype of LRRK2 and GBA related Parkinson’s disease. Sci. Rep. 2020, 10, 9329. [Google Scholar] [CrossRef] [PubMed]
- Macías-García, D.; Periñán, M.T.; Muñoz-Delgado, L.; Jimenez-Jaraba, M.V.; Labrador-Espinosa, M.Á.; Jesús, S.; Buiza-Rueda, D.; Méndez-Del Barrio, C.; Adarmes-Gómez, A.; Gómez-Garre, P.; et al. Serum lipid profile among sporadic and familial forms of Parkinson’s disease. Npj Park. Dis. 2021, 7, 59. [Google Scholar] [CrossRef] [PubMed]
- Johansen, C.T.; Hegele, R.A. Genetic bases of hypertriglyceridemic phenotypes. Curr. Opin. Lipidol. 2011, 22, 247–253. [Google Scholar] [CrossRef]
- Li, L.-H.; Dutkiewicz, E.P.; Huang, Y.-C.; Zhou, H.-B.; Hsu, C.-C. Analytical methods for cholesterol quantification. J. Food Drug Anal. 2019, 27, 375–386. [Google Scholar] [CrossRef] [Green Version]
- Aviram, R.; Manella, G.; Kopelman, N.; Neufeld-Cohen, A.; Zwighaft, Z.; Elimelech, M.; Adamovich, Y.; Golik, M.; Wang, C.; Han, X.; et al. Lipidomics Analyses Reveal Temporal and Spatial Lipid Organization and Uncover Daily Oscillations in Intracellular Organelles. Mol. Cell 2016, 62, 636–648. [Google Scholar] [CrossRef]
- Vance, J.E. Dysregulation of cholesterol balance in the brain: Contribution to neurodegenerative diseases. Dis. Model. Mech. 2012, 5, 746–755. [Google Scholar] [CrossRef] [Green Version]
- Phan, K.; He, Y.; Pickford, R.; Bhatia, S.; Katzeff, J.S.; Hodges, J.R.; Piguet, O.; Halliday, G.M.; Kim, W.S. Uncovering pathophysiological changes in frontotemporal dementia using serum lipids. Sci. Rep. 2020, 10, 3640. [Google Scholar] [CrossRef]
- Roosen, D.A.; Cookson, M.R. LRRK2 at the interface of autophagosomes, endosomes and lysosomes. Mol. Neurodegener. 2016, 11, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, A.G.; Aghamohammadzadeh, S.; Samaroo, H.; Chen, Y.; Mou, K.; Needle, E.; Hirst, W.D. Pathogenic LRRK2 mutations, through increased kinase activity, produce enlarged lysosomes with reduced degradative capacity and increase ATP13A2 expression. Hum. Mol. Genet. 2015, 24, 6013–6028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, Y.; Yamaguchi, H.; Giaime, E.; Boyle, S.; Kopan, R.; Kelleher, R.J.; Shen, J. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of α-synuclein, and apoptotic cell death in aged mice. Proc. Natl. Acad. Sci. 2010, 107, 9879–9884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, Y.; Giaime, E.; Yamaguchi, H.; Ichimura, T.; Liu, Y.; Si, H.; Cai, H.; Bonventre, J.V.; Shen, J. Loss of leucine-rich repeat kinase 2 causes age-dependent bi-phasic alterations of the autophagy pathway. Mol. Neurodegener. 2012, 7, 2. [Google Scholar] [CrossRef] [Green Version]
- Keller, J.N.; Dimayuga, E.; Chen, Q.; Thorpe, J.; Gee, J.; Ding, Q. Autophagy, proteasomes, lipofuscin, and oxidative stress in the aging brain. Int. J. Biochem. Cell Biol. 2004, 36, 2376–2391. [Google Scholar] [CrossRef]
- Kobayashi, T.; Beuchat, M.-H.; Chevallier, J.; Makino, A.; Mayran, N.; Escola, J.-M.; Lebrand, C.; Cosson, P.; Kobayashi, T.; Gruenberg, J. Separation and Characterization of Late Endosomal Membrane Domains*. J. Biol. Chem. 2002, 277, 32157–32164. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Stang, E.; Fang, K.S.; de Moerloose, P.; Parton, R.G.; Gruenberg, J. A lipid associated with the antiphospholipid syndrome regulates endosome structure and function. Nature 1998, 392, 193–197. [Google Scholar] [CrossRef]
- Kobayashi, T.; Beuchat, M.-H.; Lindsay, M.; Frias, S.; Palmiter, R.D.; Sakuraba, H.; Parton, R.G.; Gruenberg, J. Late endosomal membranes rich in lysobisphosphatidic acid regulate cholesterol transport. Nat. Cell Biol. 1999, 1, 113–118. [Google Scholar] [CrossRef]
- Chevallier, J.; Chamoun, Z.; Jiang, G.; Prestwich, G.; Sakai, N.; Matile, S.; Parton, R.G.; Gruenberg, J. Lysobisphosphatidic Acid Controls Endosomal Cholesterol Levels*. J. Biol. Chem. 2008, 283, 27871–27880. [Google Scholar] [CrossRef] [Green Version]
- Ilnytska, O.; Jeziorek, M.; Lai, K.; Altan-Bonnet, N.; Dobrowolski, R.; Storch, J. Lysobisphosphatidic acid (LBPA) enrichment promotes cholesterol egress via exosomes in Niemann Pick type C1 deficient cells. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2021, 1866, 158916. [Google Scholar] [CrossRef]
- Ebner, M.; Koch, P.A.; Haucke, V. Phosphoinositides in the control of lysosome function and homeostasis. Biochem. Soc. Trans. 2019, 47, 1173–1185. [Google Scholar] [CrossRef] [PubMed]
- Albanese, F.; Mercatelli, D.; Finetti, L.; Lamonaca, G.; Pizzi, S.; Shimshek, D.R.; Bernacchia, G.; Morari, M. Constitutive silencing of LRRK2 kinase activity leads to early glucocerebrosidase deregulation and late impairment of autophagy in vivo. Neurobiol. Dis. 2021, 159, 105487. [Google Scholar] [CrossRef] [PubMed]
- Alcalay, R.N.; Levy, O.A.; Waters, C.H.; Fahn, S.; Ford, B.; Kuo, S.-H.; Mazzoni, P.; Pauciulo, M.W.; Nichols, W.C.; Gan-Or, Z.; et al. Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain J. Neurol. 2015, 138, 2648–2658. [Google Scholar] [CrossRef] [Green Version]
- Ysselstein, D.; Nguyen, M.; Young, T.J.; Severino, A.; Schwake, M.; Merchant, K.; Krainc, D. LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson’s disease patients. Nat. Commun. 2019, 10, 5570. [Google Scholar] [CrossRef] [Green Version]
- Kedariti, M.; Frattini, E.; Baden, P.; Cogo, S.; Civiero, L.; Ziviani, E.; Zilio, G.; Bertoli, F.; Aureli, M.; Kaganovich, A.; et al. LRRK2 kinase activity regulates GCase level and enzymatic activity differently depending on cell type in Parkinson’s disease. Npj Park. Dis. 2022, 8, 92. [Google Scholar] [CrossRef]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. New Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, L.; Xiong, J.; Zhang, X.; Chen, Z.; Yu, L.; Chen, C.; Huang, J.; Zhang, Z.; Mohmed, A.A.; et al. Glucocerebrosidase L444P mutation confers genetic risk for Parkinson’s disease in central China. Behav. Brain Funct. 2012, 8, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guimarães Bde, C.; Pereira, A.C.; Rodrigues Fda, C.; dos Santos, A.V.; Campos, M., Jr.; dos Santos, J.M.; dos Santos, F.L.; de Rosso, A.L.; Nicaretta, D.H.; Pereira, J.S.; et al. Glucocerebrosidase N370S and L444P mutations as risk factors for Parkinson’s disease in Brazilian patients. Park. Relat. Disord 2012, 18, 688–689. [Google Scholar] [CrossRef]
- Aharon-Peretz, J.; Rosenbaum, H.; Gershoni-Baruch, R. Mutations in the Glucocerebrosidase Gene and Parkinson’s Disease in Ashkenazi Jews. N. Engl. J. Med. 2004, 351, 1972–1977. [Google Scholar] [CrossRef]
- Vaccaro, A.M.; Muscillo, M.; Suzuki, K. Characterization of human glucosylsphingosine glucosyl hydrolase and comparison with glucosylceramidase. Eur. J. Biochem. 1985, 146, 315–321. [Google Scholar] [CrossRef]
- Smith, L.; Schapira, A.H.V. GBA Variants and Parkinson Disease: Mechanisms and Treatments. Cells 2022, 11, 1261. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Perera, G.; Takahashi-Fujigasaki, J.; Mash, D.C.; Vonsattel, J.P.G.; Uchino, A.; Hasegawa, K.; Jeremy Nichols, R.; Holton, J.L.; Murayama, S.; et al. Reduced LRRK2 in association with retromer dysfunction in post-mortem brain tissue from LRRK2 mutation carriers. Brain J. Neurol. 2017, 141, 486–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omer, N.; Giladi, N.; Gurevich, T.; Bar-Shira, A.; Gana-Weisz, M.; Glinka, T.; Goldstein, O.; Kestenbaum, M.; Cedarbaum, J.M.; Mabrouk, O.S.; et al. Glucocerebrosidase Activity is not Associated with Parkinson’s Disease Risk or Severity. Mov. Disord. 2022, 37, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Ortega, R.A.; Bodamer, O.; Peake, R.W.A.; Raymond, D.; Bressman, S.B.; Saunders-Pullman, R. Assessment of Glucocerebrosidase Enzyme Activity in Parkinson Disease Using Multiple Approaches. Mov. Disord. 2022, 37, 655–656. [Google Scholar] [CrossRef]
- Ysselstein, D.; Young, T.J.; Nguyen, M.; Padmanabhan, S.; Hirst, W.D.; Dzamko, N.; Krainc, D. Evaluation of Strategies for Measuring Lysosomal Glucocerebrosidase Activity. Mov. Disord. Off. J. Mov. Disord. Soc. 2021, 36, 2719–2730. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, W. Functional analysis of acid and neutral sphingomyelinases in vitro and in vivo. Chem. Phys. Lipids 1999, 102, 107–121. [Google Scholar] [CrossRef]
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; Kerchner, G.A.; Ayalon, G.; Bingol, B.; Sheng, M.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef] [Green Version]
- Gan-Or, Z.; Ozelius, L.J.; Bar-Shira, A.; Saunders-Pullman, R.; Mirelman, A.; Kornreich, R.; Gana-Weisz, M.; Raymond, D.; Rozenkrantz, L.; Deik, A.; et al. The p.L302P mutation in the lysosomal enzyme gene SMPD1 is a risk factor for Parkinson disease. Neurology 2013, 80, 1606–1610. [Google Scholar] [CrossRef] [Green Version]
- Parveen, F.; Bender, D.; Law, S.-H.; Mishra, V.K.; Chen, C.-C.; Ke, L.-Y. Role of Ceramidases in Sphingolipid Metabolism and Human Diseases. Cells 2019, 8, 1573. [Google Scholar] [CrossRef] [Green Version]
- Robak, L.A.; Jansen, I.E.; van Rooij, J.; Uitterlinden, A.G.; Kraaij, R.; Jankovic, J.; Heutink, P.; Shulman, J.M.; Consortium, I.P.s.D.G. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain J. Neurol. 2017, 140, 3191–3203. [Google Scholar] [CrossRef] [Green Version]
- Brunham, L.R.; Kruit, J.K.; Pape, T.D.; Timmins, J.M.; Reuwer, A.Q.; Vasanji, Z.; Marsh, B.J.; Rodrigues, B.; Johnson, J.D.; Parks, J.S.; et al. β-cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinedione treatment. Nat. Med. 2007, 13, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.; Head, W.S.; Gunawardana, S.C.; Hasty, A.H.; Piston, D.W. Direct Effect of Cholesterol on Insulin Secretion: A Novel Mechanism for Pancreatic β-Cell Dysfunction. Diabetes 2007, 56, 2328–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotronen, A.; Velagapudi, V.R.; Yetukuri, L.; Westerbacka, J.; Bergholm, R.; Ekroos, K.; Makkonen, J.; Taskinen, M.R.; Orešič, M.; Yki-Järvinen, H. Serum saturated fatty acids containing triacylglycerols are better markers of insulin resistance than total serum triacylglycerol concentrations. Diabetologia 2009, 52, 684–690. [Google Scholar] [CrossRef] [Green Version]
- Tessneer, K.L.; Jackson, R.M.; Griesel, B.A.; Olson, A.L. Rab5 Activity Regulates GLUT4 Sorting Into Insulin-Responsive and Non-Insulin-Responsive Endosomal Compartments: A Potential Mechanism for Development of Insulin Resistance. Endocrinology 2014, 155, 3315–3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaldin-Fincati, J.R.; Pavarotti, M.; Frendo-Cumbo, S.; Bilan, P.J.; Klip, A. Update on GLUT4 Vesicle Traffic: A Cornerstone of Insulin Action. Trends Endocrinol. Metab. 2017, 28, 597–611. [Google Scholar] [CrossRef]
- Sano, H.; Eguez, L.; Teruel, M.N.; Fukuda, M.; Chuang, T.D.; Chavez, J.A.; Lienhard, G.E.; McGraw, T.E. Rab10, a Target of the AS160 Rab GAP, Is Required for Insulin-Stimulated Translocation of GLUT4 to the Adipocyte Plasma Membrane. Cell Metab. 2007, 5, 293–303. [Google Scholar] [CrossRef] [Green Version]
- Randhawa, V.K.; Ishikura, S.; Talior-Volodarsky, I.; Cheng, A.W.P.; Patel, N.; Hartwig, J.H.; Klip, A. GLUT4 Vesicle Recruitment and Fusion Are Differentially Regulated by Rac, AS160, and Rab8A in Muscle Cells*. J. Biol. Chem. 2008, 283, 27208–27219. [Google Scholar] [CrossRef] [Green Version]
- Davey, J.R.; Humphrey, S.J.; Junutula, J.R.; Mishra, A.K.; Lambright, D.G.; James, D.E.; Stöckli, J. TBC1D13 is a RAB35 Specific GAP that Plays an Important Role in GLUT4 Trafficking in Adipocytes. Traffic 2012, 13, 1429–1441. [Google Scholar] [CrossRef] [Green Version]
- Funk, N.; Munz, M.; Ott, T.; Brockmann, K.; Wenninger-Weinzierl, A.; Kühn, R.; Vogt-Weisenhorn, D.; Giesert, F.; Wurst, W.; Gasser, T.; et al. The Parkinson’s disease-linked Leucine-rich repeat kinase 2 (LRRK2) is required for insulin-stimulated translocation of GLUT4. Sci. Rep. 2019, 9, 4515. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.S.; Nolte, H.; Jacob, W.; Ziegler, A.B.; Pütz, S.; Grosjean, Y.; Szczepanowska, K.; Trifunovic, A.; Braun, T.; Heumann, H.; et al. Human R1441C LRRK2 regulates the synaptic vesicle proteome and phosphoproteome in a Drosophila model of Parkinson’s disease. Hum. Mol. Genet. 2016, 25, 5365–5382. [Google Scholar] [CrossRef]
- Pan, P.-Y.; Li, X.; Wang, J.; Powell, J.; Wang, Q.; Zhang, Y.; Chen, Z.; Wicinski, B.; Hof, P.; Ryan, T.A.; et al. Parkinson’s Disease-Associated LRRK2 Hyperactive Kinase Mutant Disrupts Synaptic Vesicle Trafficking in Ventral Midbrain Neurons. J. Neurosci. 2017, 37, 11366–11376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krebs, C.E.; Karkheiran, S.; Powell, J.C.; Cao, M.; Makarov, V.; Darvish, H.; Di Paolo, G.; Walker, R.H.; Shahidi, G.A.; Buxbaum, J.D.; et al. The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum. Mutat. 2013, 34, 1200–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quadri, M.; Fang, M.; Picillo, M.; Olgiati, S.; Breedveld, G.J.; Graafland, J.; Wu, B.; Xu, F.; Erro, R.; Amboni, M.; et al. Mutation in the SYNJ1 Gene Associated with Autosomal Recessive, Early-Onset Parkinsonism. Hum. Mutat. 2013, 34, 1208–1215. [Google Scholar] [CrossRef]
- Berg, D.; Postuma, R.B.; Adler, C.H.; Bloem, B.R.; Chan, P.; Dubois, B.; Gasser, T.; Goetz, C.G.; Halliday, G.; Joseph, L.; et al. MDS research criteria for prodromal Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2015, 30, 1600–1611. [Google Scholar] [CrossRef] [Green Version]
- Choudhry, H.; Aggarwal, M.; Pan, P.-Y. Mini-review: Synaptojanin 1 and its implications in membrane trafficking. Neurosci. Lett. 2021, 765, 136288. [Google Scholar] [CrossRef]
- Pan, P.-Y.; Zhu, J.; Rizvi, A.; Zhu, X.; Tanaka, H.; Dreyfus, C.F. Synaptojanin1 deficiency upregulates basal autophagosome formation in astrocytes. J. Biol. Chem. 2021, 297, 100873. [Google Scholar] [CrossRef]
- Zou, L.; Tian, Y.; Zhang, Z. Dysfunction of Synaptic Vesicle Endocytosis in Parkinson’s Disease. Front. Integr. Neurosci. 2021, 15, 619160. [Google Scholar] [CrossRef] [PubMed]
- Shinde, S.R.; Maddika, S. PTEN Regulates Glucose Transporter Recycling by Impairing SNX27 Retromer Assembly. Cell Rep. 2017, 21, 1655–1666. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-W.; Peng, Y.-J.; Lin, Y.-Y.; Mersmann, H.J.; Ding, S.-T. LRRK2 Regulates CPT1A to Promote β-Oxidation in HepG2 Cells. Molecules 2020, 25, 4122. [Google Scholar] [CrossRef]
- Mejia, E.M.; Hatch, G.M. Mitochondrial phospholipids: Role in mitochondrial function. J. Bioenerg. Biomembr. 2016, 48, 99–112. [Google Scholar] [CrossRef]
- Hollie, N.I.; Cash, J.G.; Matlib, M.A.; Wortman, M.; Basford, J.E.; Abplanalp, W.; Hui, D.Y. Micromolar changes in lysophosphatidylcholine concentration cause minor effects on mitochondrial permeability but major alterations in function. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2014, 1841, 888–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pol, A.; Gross, S.P.; Parton, R.G. Biogenesis of the multifunctional lipid droplet: Lipids, proteins, and sites. J. Cell Biol. 2014, 204, 635–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Danés, A.; Richaud-Patin, Y.; Carballo-Carbajal, I.; Jiménez-Delgado, S.; Caig, C.; Mora, S.; Di Guglielmo, C.; Ezquerra, M.; Patel, B.; Giralt, A.; et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol. Med. 2012, 4, 380–395. [Google Scholar] [CrossRef]
- Wu, L.; Xu, D.; Zhou, L.; Xie, B.; Yu, L.; Yang, H.; Huang, L.; Ye, J.; Deng, H.; Yuan, Y.A.; et al. Rab8a-AS160-MSS4 Regulatory Circuit Controls Lipid Droplet Fusion and Growth. Dev. Cell 2014, 30, 378–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steger, M.; Tonelli, F.; Ito, G.; Davies, P.; Trost, M.; Vetter, M.; Wachter, S.; Lorentzen, E.; Duddy, G.; Wilson, S.; et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. eLife 2016, 5, e12813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steger, M.; Diez, F.; Dhekne, H.S.; Lis, P.; Nirujogi, R.S.; Karayel, O.; Tonelli, F.; Martinez, T.N.; Lorentzen, E.; Pfeffer, S.R.; et al. Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. eLife 2017, 6, e31012. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Schulze, R.J.; Weller, S.G.; Krueger, E.W.; Schott, M.B.; Zhang, X.; Casey, C.A.; Liu, J.; Stöckli, J.; James, D.E.; et al. A novel Rab10-EHBP1-EHD2 complex essential for the autophagic engulfment of lipid droplets. Sci. Adv. 2016, 2, e1601470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Weller, S.G.; Drizyte-Miller, K.; Chen, J.; Krueger, E.W.; Mehall, B.; Stöckli, J.; Casey, C.A.; Cao, H.; McNiven, M.A. Maturation of Lipophagic Organelles in Hepatocytes Is Dependent Upon a Rab10/Dynamin-2 Complex. Hepatology 2020, 72, 486–502. [Google Scholar] [CrossRef]
- Wauters, F.; Cornelissen, T.; Imberechts, D.; Martin, S.; Koentjoro, B.; Sue, C.; Vangheluwe, P.; Vandenberghe, W. LRRK2 mutations impair depolarization-induced mitophagy through inhibition of mitochondrial accumulation of RAB10. Autophagy 2020, 16, 203–222. [Google Scholar] [CrossRef]
- Liu, P.; Bartz, R.; Zehmer, J.K.; Ying, Y.-s.; Zhu, M.; Serrero, G.; Anderson, R.G.W. Rab-regulated interaction of early endosomes with lipid droplets. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2007, 1773, 784–793. [Google Scholar] [CrossRef]
- Kuhlmann, N.; Milnerwood, A.J. A Critical LRRK at the Synapse? The Neurobiological Function and Pathophysiological Dysfunction of LRRK2. Front. Mol. Neurosci. 2020, 13, 153. [Google Scholar] [CrossRef] [PubMed]
- Binotti, B.; Jahn, R.; Chua, J.J. Functions of Rab Proteins at Presynaptic Sites. Cells 2016, 5, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wucherpfennig, T.; Wilsch-Bräuninger, M.; González-Gaitán, M. Role of Drosophila Rab5 during endosomal trafficking at the synapse and evoked neurotransmitter release. J. Cell Biol. 2003, 161, 609–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer von Mollard, G.; Stahl, B.; Walch-Solimena, C.; Takei, K.; Daniels, L.; Khoklatchev, A.; De Camilli, P.; Südhof, T.C.; Jahn, R. Localization of Rab5 to synaptic vesicles identifies endosomal intermediate in synaptic vesicle recycling pathway. Eur. J. Cell Biol. 1994, 65, 319–326. [Google Scholar] [PubMed]
- de Hoop, M.J.; Huber, L.A.; Stenmark, H.; Williamson, E.; Zerial, M.; Parton, R.G.; Dotti, C.G. The involvement of the small GTP-binding protein Rab5a in neuronal endocytosis. Neuron 1994, 13, 11–22. [Google Scholar] [CrossRef]
- Christoforidis, S.; Miaczynska, M.; Ashman, K.; Wilm, M.; Zhao, L.; Yip, S.-C.; Waterfield, M.D.; Backer, J.M.; Zerial, M. Phosphatidylinositol-3-OH kinases are Rab5 effectors. Nat. Cell Biol. 1999, 1, 249–252. [Google Scholar] [CrossRef]
- Shin, H.-W.; Hayashi, M.; Christoforidis, S.; Lacas-Gervais, S.; Hoepfner, S.; Wenk, M.R.; Modregger, J.; Uttenweiler-Joseph, S.; Wilm, M.; Nystuen, A.; et al. An enzymatic cascade of Rab5 effectors regulates phosphoinositide turnover in the endocytic pathway. J. Cell Biol. 2005, 170, 607–618. [Google Scholar] [CrossRef]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef]
- Semerdjieva, S.; Shortt, B.; Maxwell, E.; Singh, S.; Fonarev, P.; Hansen, J.; Schiavo, G.; Grant, B.D.; Smythe, E. Coordinated regulation of AP2 uncoating from clathrin-coated vesicles by rab5 and hRME-6. J. Cell Biol. 2008, 183, 499–511. [Google Scholar] [CrossRef] [Green Version]
- Matta, S.; Van Kolen, K.; da Cunha, R.; van den Bogaart, G.; Mandemakers, W.; Miskiewicz, K.; De Bock, P.-J.; Morais, V.A.; Vilain, S.; Haddad, D.; et al. LRRK2 Controls an EndoA Phosphorylation Cycle in Synaptic Endocytosis. Neuron 2012, 75, 1008–1021. [Google Scholar] [CrossRef]
- Ambroso, M.R.; Hegde, B.G.; Langen, R. Endophilin A1 induces different membrane shapes using a conformational switch that is regulated by phosphorylation. Proc. Natl. Acad. Sci. 2014, 111, 6982–6987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mani, M.; Lee, S.Y.; Lucast, L.; Cremona, O.; Di Paolo, G.; De Camilli, P.; Ryan, T.A. The Dual Phosphatase Activity of Synaptojanin1 Is Required for Both Efficient Synaptic Vesicle Endocytosis and Reavailability at Nerve Terminals. Neuron 2007, 56, 1004–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haucke, V. Phosphoinositide regulation of clathrin-mediated endocytosis. Biochem. Soc. Trans. 2005, 33, 1285–1289. [Google Scholar] [CrossRef] [Green Version]
- Zoncu, R.; Perera, R.M.; Sebastian, R.; Nakatsu, F.; Chen, H.; Balla, T.; Ayala, G.; Toomre, D.; De Camilli, P.V. Loss of endocytic clathrin-coated pits upon acute depletion of phosphatidylinositol 4,5-bisphosphate. Proc. Natl. Acad. Sci. 2007, 104, 3793–3798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Y.; Neifert, S.; Karuppagounder, S.S.; Liu, Q.; Stankowski, J.N.; Lee, B.D.; Ko, H.S.; Lee, Y.; Grima, J.C.; Mao, X.; et al. Robust kinase- and age-dependent dopaminergic and norepinephrine neurodegeneration in LRRK2 G2019S transgenic mice. Proc. Natl. Acad. Sci. 2018, 115, 1635–1640. [Google Scholar] [CrossRef] [Green Version]
- Cao, M.; Wu, Y.; Ashrafi, G.; McCartney, A.J.; Wheeler, H.; Bushong, E.A.; Boassa, D.; Ellisman, M.H.; Ryan, T.A.; De Camilli, P. Parkinson Sac Domain Mutation in Synaptojanin 1 Impairs Clathrin Uncoating at Synapses and Triggers Dystrophic Changes in Dopaminergic Axons. Neuron 2017, 93, 882–896. [Google Scholar] [CrossRef] [Green Version]
- Puchkov, D.; Haucke, V. Greasing the synaptic vesicle cycle by membrane lipids. Trends Cell Biol. 2013, 23, 493–503. [Google Scholar] [CrossRef]
- Goldschmidt, H.L.; Tu-Sekine, B.; Volk, L.; Anggono, V.; Huganir, R.L.; Raben, D.M. DGKθ Catalytic Activity Is Required for Efficient Recycling of Presynaptic Vesicles at Excitatory Synapses. Cell Rep. 2016, 14, 200–207. [Google Scholar] [CrossRef] [Green Version]
- Bauer, C.S.; Woolley, R.J.; Teschemacher, A.G.; Seward, E.P. Potentiation of Exocytosis by Phospholipase C-Coupled G-Protein-Coupled Receptors Requires the Priming Protein Munc13-1. J. Neurosci. 2007, 27, 212–219. [Google Scholar] [CrossRef] [Green Version]
- Majewski, H.; Iannazzo, L. Protein kinase C: A physiological mediator of enhanced transmitter output. Prog. Neurobiol. 1998, 55, 463–475. [Google Scholar] [CrossRef]
- Gui, Y.X.; Xu, Z.P.; Wen, L.; Liu, H.M.; Zhao, J.J.; Hu, X.Y. Four novel rare mutations of PLA2G6 in Chinese population with Parkinson’s disease. Park. Relat Disord 2013, 19, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Hu, J.; Jiang, Y.; Zhao, S.; Lin, C.; Yin, X.; Yan, Y.; Pu, J.; Lai, H.-Y.; Zhang, B. Early-Onset Parkinson’s Disease Caused by PLA2G6 Compound Heterozygous Mutation, a Case Report and Literature Review. Front. Neurol. 2019, 10, 915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, J.Y.; Tang, B.S.; Shi, C.H.; Lv, Z.Y.; Li, K.; Yu, R.L.; Shen, L.; Yan, X.X.; Guo, J.F. Analysis of PLA2G6 gene mutation in sporadic early-onset parkinsonism patients from Chinese population. Neurosci. Lett. 2012, 514, 156–158. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, H.; Tomiyama, H.; Tachibana, N.; Ogaki, K.; Li, Y.; Funayama, M.; Hashimoto, T.; Takashima, S.; Hattori, N. Phenotypic spectrum of patients with PLA2G6 mutation and PARK14-linked parkinsonism. Neurology 2010, 75, 1356–1361. [Google Scholar] [CrossRef] [PubMed]
- Mori, A.; Hatano, T.; Inoshita, T.; Shiba-Fukushima, K.; Koinuma, T.; Meng, H.; Kubo, S.-I.; Spratt, S.; Cui, C.; Yamashita, C.; et al. Parkinson’s disease-associated iPLA2-VIA/PLA2G6 regulates neuronal functions and α-synuclein stability through membrane remodeling. Proc. Natl. Acad. Sci. 2019, 116, 20689–20699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauwers, E.; Goodchild, R.; Verstreken, P. Membrane Lipids in Presynaptic Function and Disease. Neuron 2016, 90, 11–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, H.; Kobayashi, S.; Hirabayashi, Y.; Murakami-Murofushi, K. Cholesterol glucosylation is catalyzed by transglucosylation reaction of β-glucosidase 1. Biochem. Biophys. Res. Commun. 2013, 441, 838–843. [Google Scholar] [CrossRef] [Green Version]
- Binotti, B.; Jahn, R.; Pérez-Lara, Á. An overview of the synaptic vesicle lipid composition. Arch. Biochem. Biophys. 2021, 709, 108966. [Google Scholar] [CrossRef]
- Yang, S.-T.; Kreutzberger, A.J.B.; Lee, J.; Kiessling, V.; Tamm, L.K. The role of cholesterol in membrane fusion. Chem. Phys. Lipids 2016, 199, 136–143. [Google Scholar] [CrossRef] [Green Version]
- Takamori, S.; Holt, M.; Stenius, K.; Lemke, E.A.; Grønborg, M.; Riedel, D.; Urlaub, H.; Schenck, S.; Brügger, B.; Ringler, P.; et al. Molecular Anatomy of a Trafficking Organelle. Cell 2006, 127, 831–846. [Google Scholar] [CrossRef]
- Deutsch, J.W.; Kelly, R.B. Lipids of synaptic vesicles: Relevance to the mechanism of membrane fusion. Biochemistry 1981, 20, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Baker, R.R.; Morris, S.J.; Whittaker, V.P. The preparation and characterization of synaptic vesicles of high purity. Brain Res. 1976, 109, 285–309. [Google Scholar] [CrossRef]
- Lang, T.; Bruns, D.; Wenzel, D.; Riedel, D.; Holroyd, P.; Thiele, C.; Jahn, R. SNAREs are concentrated in cholesterol-dependent clusters that define docking and fusion sites for exocytosis. EMBO J. 2001, 20, 2202–2213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamberlain, L.H.; Burgoyne, R.D.; Gould, G.W. SNARE proteins are highly enriched in lipid rafts in PC12 cells: Implications for the spatial control of exocytosis. Proc. Natl. Acad. Sci. 2001, 98, 5619–5624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belmonte, S.A.; López, C.I.; Roggero, C.M.; De Blas, G.A.; Tomes, C.N.; Mayorga, L.S. Cholesterol content regulates acrosomal exocytosis by enhancing Rab3A plasma membrane association. Dev. Biol. 2005, 285, 393–408. [Google Scholar] [CrossRef] [Green Version]
- Südhof, T.C. The Synaptic Vesicle Cycle. Annu. Rev. Neurosci. 2004, 27, 509–547. [Google Scholar] [CrossRef] [Green Version]
- Chandra, S.; Chen, X.; Rizo, J.; Jahn, R.; Südhof, T.C. A broken alpha -helix in folded alpha -Synuclein. J. Biol. Chem. 2003, 278, 15313–15318. [Google Scholar] [CrossRef] [Green Version]
- Weinreb, P.H.; Zhen, W.; Poon, A.W.; Conway, K.A.; Lansbury, P.T. NACP, A Protein Implicated in Alzheimer’s Disease and Learning, Is Natively Unfolded. Biochemistry 1996, 35, 13709–13715. [Google Scholar] [CrossRef]
- Kim, J. Evidence that the precursor protein of non-A beta component of Alzheimer’s disease amyloid (NACP) has an extended structure primarily composed of random-coil. Mol. Cells 1997, 7, 78–83. [Google Scholar]
- Fauvet, B.; Mbefo, M.K.; Fares, M.B.; Desobry, C.; Michael, S.; Ardah, M.T.; Tsika, E.; Coune, P.; Prudent, M.; Lion, N.; et al. α-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J. Biol. Chem. 2012, 287, 15345–15364. [Google Scholar] [CrossRef] [Green Version]
- Burré, J.; Vivona, S.; Diao, J.; Sharma, M.; Brunger, A.T.; Südhof, T.C. Properties of native brain α-synuclein. Nature 2013, 498, E4-6, discussion E6-7. [Google Scholar] [CrossRef] [PubMed]
- Bussell, R., Jr.; Eliezer, D. A structural and functional role for 11-mer repeats in alpha-synuclein and other exchangeable lipid binding proteins. J. Mol. Biol. 2003, 329, 763–778. [Google Scholar] [CrossRef]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 1998, 273, 9443–9449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eliezer, D.; Kutluay, E.; Bussell, R., Jr.; Browne, G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J. Mol. Biol. 2001, 307, 1061–1073. [Google Scholar] [CrossRef]
- Middleton, E.R.; Rhoades, E. Effects of curvature and composition on α-synuclein binding to lipid vesicles. Biophys. J. 2010, 99, 2279–2288. [Google Scholar] [CrossRef] [Green Version]
- Jo, E.; McLaurin, J.; Yip, C.M.; St George-Hyslop, P.; Fraser, P.E. alpha-Synuclein membrane interactions and lipid specificity. J. Biol. Chem. 2000, 275, 34328–34334. [Google Scholar] [CrossRef] [Green Version]
- Perrin, R.J.; Woods, W.S.; Clayton, D.F.; George, J.M. Interaction of human alpha-Synuclein and Parkinson’s disease variants with phospholipids. Structural analysis using site-directed mutagenesis. J. Biol. Chem. 2000, 275, 34393–34398. [Google Scholar] [CrossRef] [Green Version]
- Burré, J. The Synaptic Function of α-Synuclein. J. Park. Dis. 2015, 5, 699–713. [Google Scholar] [CrossRef] [Green Version]
- Fusco, G.; Pape, T.; Stephens, A.D.; Mahou, P.; Costa, A.R.; Kaminski, C.F.; Kaminski Schierle, G.S.; Vendruscolo, M.; Veglia, G.; Dobson, C.M.; et al. Structural basis of synaptic vesicle assembly promoted by α-synuclein. Nat. Commun. 2016, 7, 12563. [Google Scholar] [CrossRef] [Green Version]
- Gitler, A.D.; Bevis, B.J.; Shorter, J.; Strathearn, K.E.; Hamamichi, S.; Su, L.J.; Caldwell, K.A.; Caldwell, G.A.; Rochet, J.-C.; McCaffery, J.M.; et al. The Parkinson’s disease protein α-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. 2008, 105, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Diao, J.; Burré, J.; Vivona, S.; Cipriano, D.J.; Sharma, M.; Kyoung, M.; Südhof, T.C.; Brunger, A.T. Native α-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2. eLife 2013, 2, e00592. [Google Scholar] [CrossRef] [PubMed]
- Bodner, C.R.; Dobson, C.M.; Bax, A. Multiple Tight Phospholipid-Binding Modes of α-Synuclein Revealed by Solution NMR Spectroscopy. J. Mol. Biol. 2009, 390, 775–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soper, J.H.; Roy, S.; Stieber, A.; Lee, E.; Wilson, R.B.; Trojanowski, J.Q.; Burd, C.G.; Lee, V.M.-Y. α-Synuclein–induced Aggregation of Cytoplasmic Vesicles in Saccharomyces cerevisiae. Mol. Biol. Cell 2008, 19, 1093–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, W.K.; De Simone, A.; Barritt, J.D.; Vendruscolo, M.; Dobson, C.M.; Fusco, G. A Role of Cholesterol in Modulating the Binding of α-Synuclein to Synaptic-Like Vesicles. Front. Neurosci. 2020, 14, 18. [Google Scholar] [CrossRef] [Green Version]
- Bar-On, P.; Crews, L.; Koob, A.O.; Mizuno, H.; Adame, A.; Spencer, B.; Masliah, E. Statins reduce neuronal α-synuclein aggregation in in vitro models of Parkinson’s disease. J. Neurochem. 2008, 105, 1656–1667. [Google Scholar] [CrossRef] [Green Version]
- Fecchio, C.; Palazzi, L.; Polverino de Laureto, P. α-Synuclein and Polyunsaturated Fatty Acids: Molecular Basis of the Interaction and Implication in Neurodegeneration. Molecules 2018, 23, 1531. [Google Scholar] [CrossRef] [Green Version]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The Role of Lipids in Parkinson’s Disease. Cells 2019, 8, 27. [Google Scholar] [CrossRef] [Green Version]
- Jennings, D.; Huntwork-Rodriguez, S.; Henry, A.G.; Sasaki, J.C.; Meisner, R.; Diaz, D.; Solanoy, H.; Wang, X.; Negrou, E.; Bondar, V.V.; et al. Preclinical and clinical evaluation of the LRRK2 inhibitor DNL201 for Parkinson’s disease. Sci. Transl. Med. 2022, 14, eabj2658. [Google Scholar] [CrossRef]
- Lin, C.-H.; Lin, H.-I.; Chen, M.-L.; Lai, T.-T.; Cao, L.-P.; Farrer, M.J.; Wu, R.-M.; Chien, C.-T. Lovastatin protects neurite degeneration in LRRK2-G2019S parkinsonism through activating the Akt/Nrf pathway and inhibiting GSK3β activity. Hum. Mol. Genet. 2016, 25, 1965–1978. [Google Scholar] [CrossRef] [Green Version]
- Littarru, G.P.; Langsjoen, P. Coenzyme Q10 and statins: Biochemical and clinical implications. Mitochondrion 2007, 7, S168–S174. [Google Scholar] [CrossRef]
- Stancu, C.; Sima, A. Statins: Mechanism of action and effects. J. Cell. Mol. Med. 2001, 5, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Koob, A.O.; Ubhi, K.; Paulsson, J.F.; Kelly, J.; Rockenstein, E.; Mante, M.; Adame, A.; Masliah, E. Lovastatin ameliorates α-synuclein accumulation and oxidation in transgenic mouse models of α-synucleinopathies. Exp. Neurol. 2010, 221, 267–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.-H.; Chang, C.-H.; Tai, C.-H.; Cheng, M.-F.; Chen, Y.-C.; Chao, Y.-T.; Huang, T.-L.; Yen, R.-F.; Wu, R.-M. A Double-Blind, Randomized, Controlled Trial of Lovastatin in Early-Stage Parkinson’s Disease. Mov. Disord. 2021, 36, 1229–1237. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galper, J.; Kim, W.S.; Dzamko, N. LRRK2 and Lipid Pathways: Implications for Parkinson’s Disease. Biomolecules 2022, 12, 1597. https://doi.org/10.3390/biom12111597
Galper J, Kim WS, Dzamko N. LRRK2 and Lipid Pathways: Implications for Parkinson’s Disease. Biomolecules. 2022; 12(11):1597. https://doi.org/10.3390/biom12111597
Chicago/Turabian StyleGalper, Jasmin, Woojin S. Kim, and Nicolas Dzamko. 2022. "LRRK2 and Lipid Pathways: Implications for Parkinson’s Disease" Biomolecules 12, no. 11: 1597. https://doi.org/10.3390/biom12111597
APA StyleGalper, J., Kim, W. S., & Dzamko, N. (2022). LRRK2 and Lipid Pathways: Implications for Parkinson’s Disease. Biomolecules, 12(11), 1597. https://doi.org/10.3390/biom12111597