

Druggable Metabolic Vulnerabilities Are Exposed and Masked during Progression to Castration Resistant Prostate Cancer

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Metabolic Reprogramming during Progression to CRPC
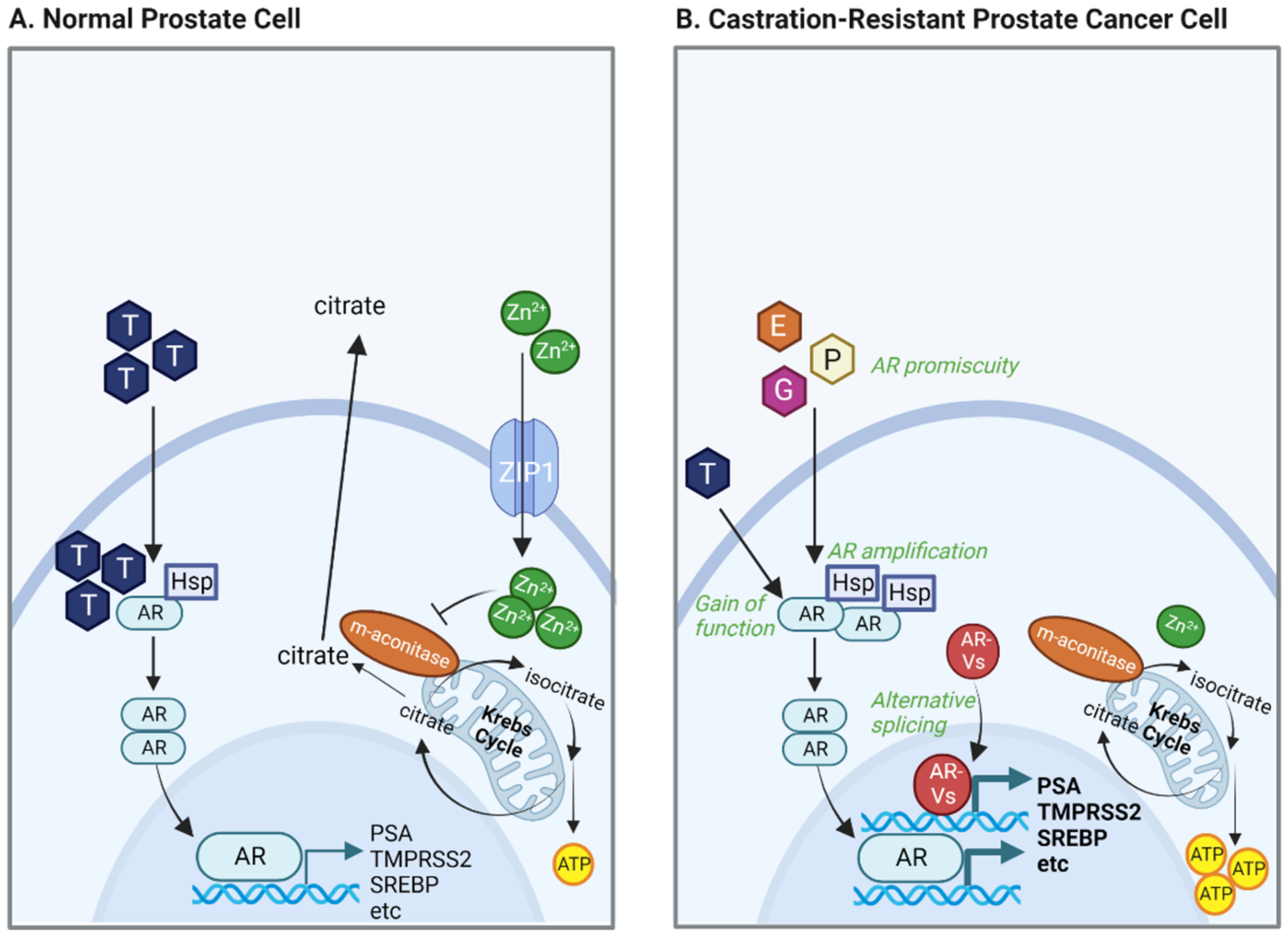
2.1. Zinc-Driven Metabolism
2.2. AR Alterations
3. Major Bioenergetic Sources
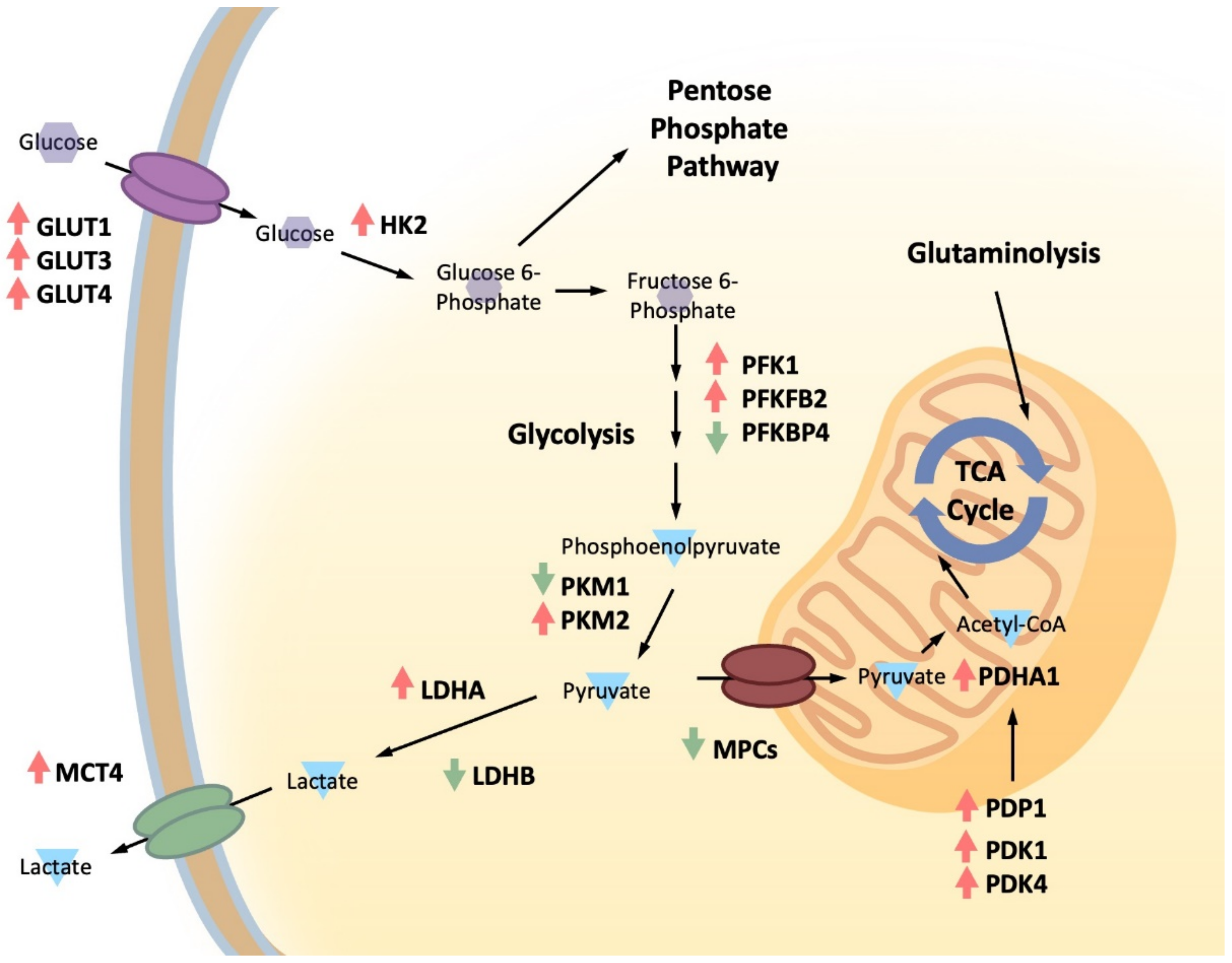
3.1. Glucose Metabolism
3.2. Fructose
3.3. Amino Acid Metabolism
3.3.1. Glutamine Metabolism
3.3.2. One-Carbon Metabolism
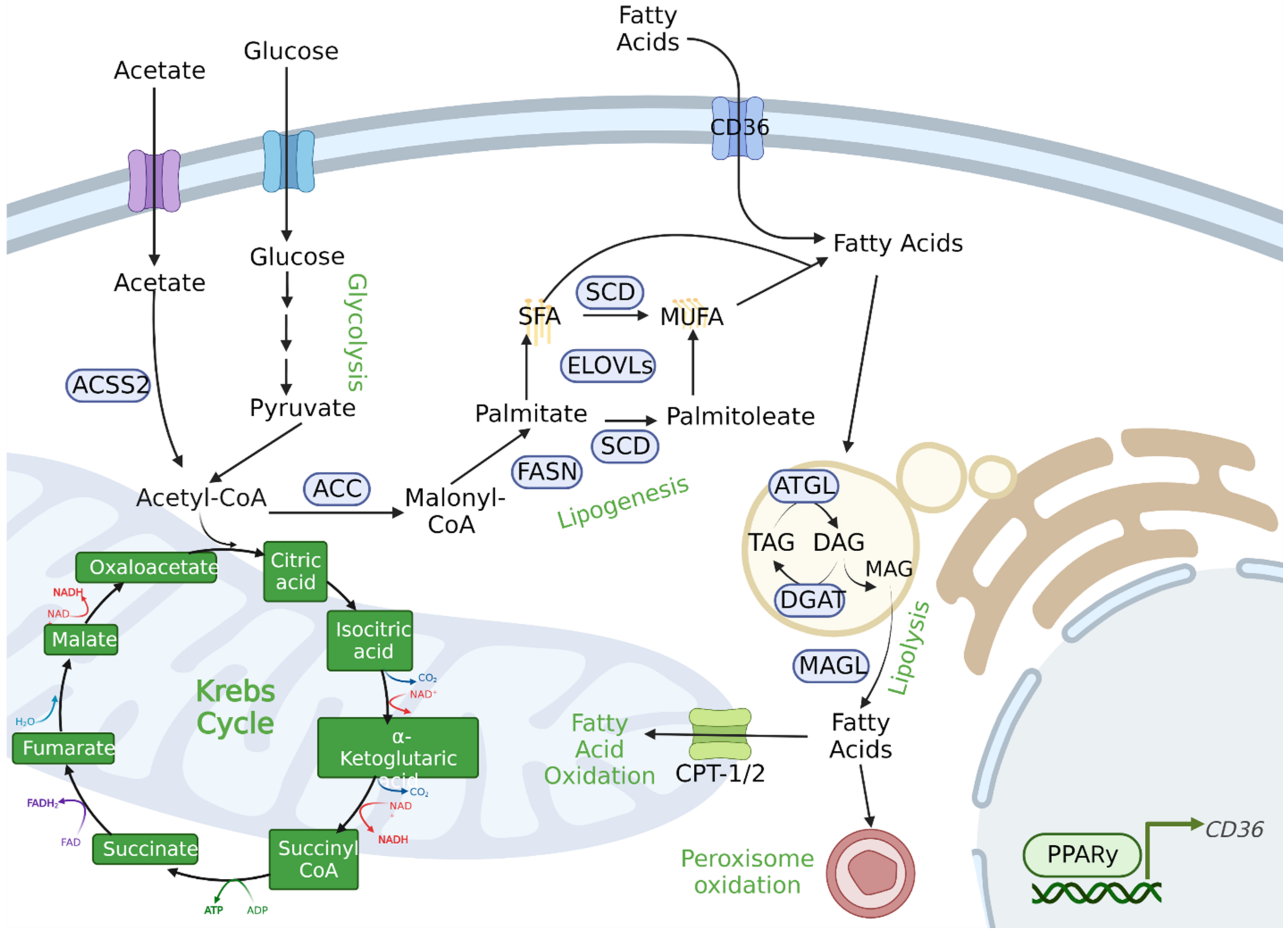
3.4. Lipid Metabolism
FA Storage and Utilization
4. Metabolites and Gene Regulation
4.1. Post-Translational Modifications
4.1.1. Lactylation
4.1.2. Methylation
5. Tumor Microenvironment (TME)
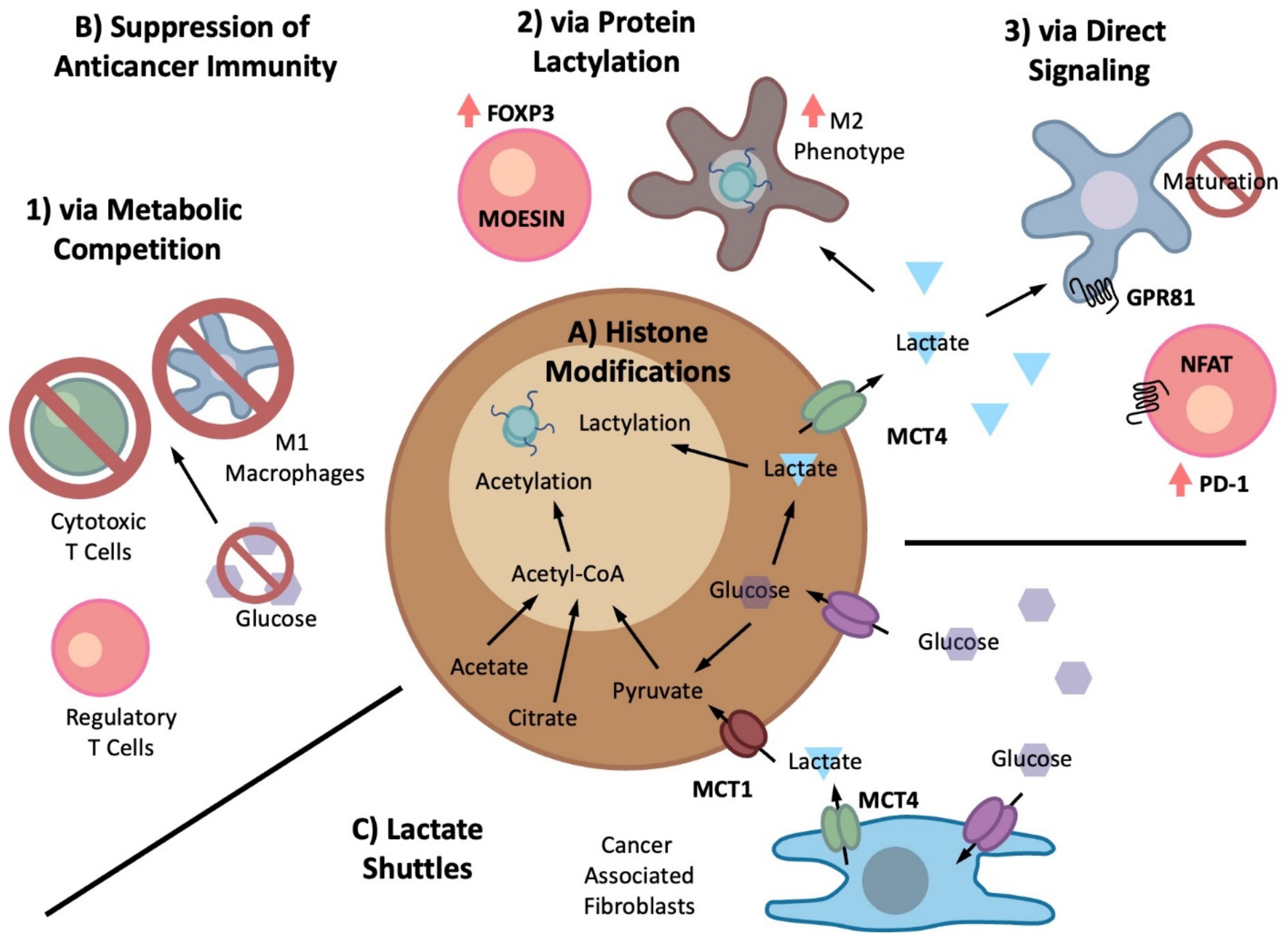
5.1. Metabolite-Driven Immunosuppression
Cancer-Generated Lactic Acid
5.2. Lactate Shuttle
5.3. Fatty Acid Signaling
6. Therapeutic Interventions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) | |||||
| Target | Agent | Modes of Action | Study Design and Results | Reference | |
| Glycolysis | |||||
| PKM2 | TEPP-46 | Small molecule pyruvate kinase activators have been identified that stabilize the PKM2 tetramer to promote an enzyme state similar to PKM1. | Activation of PKM2 led to tumor inhibition in mice with prostate specific deletion of PTEN. | [420] | |
| LDHA | GSK2837808A | A potent, selective and NADH-competitive inhibitor of LDH-A. | Combination therapy with radiation improved antitumoral T-cell response and reduced tumor progression in pancreatic cancer models. | [453,454] | |
| MCT1 | AR-C122982/SR13800 | Selective MCT1 inhibitor. | Antitumor effects on MYC-overexpressing breast cancer and neuroblastoma in preclinical models. | [455,456] | |
| Mitochondrial metabolism | |||||
| MPC | MSDC-0160 | Thiazolidinedione that reversibly binds to MPC1 and 2 heterodimer. | Inhibition of tumor growth in preclinical models of both castration-sensitive and resistant AR-driven prostate tumors. | [147,457] | |
| MPC | UK-5099 | Inhibits the MPC complex by binding to Cys54 of MPC2 in a covalent manner. | Inhibition of tumor growth in preclinical models of both castration-sensitive and resistant AR-driven prostate tumors. | [458] | |
| Complex V | Gboxin | Inhibits Complex V (F0F1-ATP synthase). | Antitumorigenic effects on in vivo and in vitro glioblastoma model. | [459] | |
| Amino acid metabolism | |||||
| ASCT2 | V-9302 | Competitive small molecule antagonist of ASCT2, leading to decrease in glutamine influx. | Antitumor effects on preclinical models including xenografts from triple negative breast cancer cell line HCC-1806. | [460] | |
| Improves antitumor T cell activity in triple-negative breast cancer. | [461] | ||||
| GLS1 (KGA and GAC) | CB-839/Telaglenastat | Orally available potent allosteric inhibitor for GLS1. | Antitumor effects on triple negative breast cancer models and lung tumor models. | [425,462,463] | |
| IDO1 | BMS-986205 | Inhibits apo-IDO1 binding to heme which is co-factor for IDO1 catalytic action. | Iinrodostat potently and specifically inhibits IDO1 to block an immunosuppressive mechanism that could be responsible for tumor escape from host immune surveillance with favorable PK/PD characteristics that support clinical development. | [464] | |
| Fatty acid metabolism | |||||
| SREBP | Fatostatin | Prevent nuclear translocation of SREBPs by inhibiting the SREBP cleavage-activating protein (SCAP) that facilitates the ER-Golgi translocation of SREBPs. | Inhibition of SREBP and AR transcription networks is associated with antitumorigenic effects on in vitro and in vivo PCa models. | [465,466] | |
| FASN | IPI-9119 | Inhibits the FASN thioesterase domain by promoting acylation of the catalytic serine. | Tumor inhibition is associated with altered fatty acid metabolism and reduced AR signaling. | [86] | |
| ACC1 and ACC2 | ND-654 | Allosteric inhibitor of the ACC (Acetyl-CoA carboxylase) enzymes that prevents dimerization of ACC. | Inhibition of de novo lipogenesis in liver and the development of hepatocellular carcinoma. | [467] | |
| ACC1 and ACC2 | ND-646 | Allosteric inhibitor of the ACC (Acetyl-CoA carboxylase) enzymes that prevents dimerization of ACC. | Suppression of lung tumor growth in the mouse models of non-small cell lung cancer. | [468] | |
| (B) | |||||
| Target | Agent | Modes of Action | Clinical Relevance (NCT Number, Study Phase, etc.) | Study Design | Reference |
| Glycolysis | |||||
| MCT1 and MCT2 | AZD3965 | A potent pyrimidine-derived inhibitor of MCT1 with activity against MCT2 but selectivity over MCT3 and MCT4. | NCT01791595 (Phase 1) | Trial of AZD3965 in patients with advanced cancer. | [469,470,471,472] |
| Mitochondrial metabolism | |||||
| PDH/alfa-KGDH | CPI-613/devimistat | Lipoate analog that inhibits two lipoate-dependent enzymes, ketoglutarate dehydrogenase (KGDH) and pyruvate dehydrogenase (PDH). | NCT03504423 (Phase 3) | Study evaluating efficacy and safety of thymidylate synthase inhibitor FFX versus combination of CPI-613 with mFFX (modified FFX) in patients with metastatic adenocarcinoma of the pancreas. | [473,474,475,476,477] |
| NCT03435289 (Phase 1) | CPI-613 with nucleoside metabolic inhibitor Gemcitabine and albumin-bound paclitaxel Nab-paclitaxel for patients with advanced or metastatic pancreatic cancer. | ||||
| mutant IDH2 | Enasidenib/AG-221 | Allosteric binding of AG-221 stabilizes inhibitory open conformation of the mutant IDH2. | FDA approval | Treatment of relapsed or refractory acute myeloid leukemia (AML). | [478] |
| Complex I, PEN2, polypharmacologic | Metformin | Inhibits complex I to decrease OXPHOS activity. Metformin binds to PEN2 (presenilin 2) to form a complex with ATP6AP1, a subunit of the v-ATPase8 and inhibit v-ATPase. | NCT02614859 (Phase 2) | Bicalutamide with or without metformin for biochemical recurrence in overweight or obese PCa patients (BIMET-1). | [479,480,481,482,483] |
| NCT02339168 (Phase 1) | Enzalutamide and metformin hydrochloride in treating patients with hormone-resistant PCa. | ||||
| NCT03291938 (Phase 1) | Test in advanced cancers. | ||||
| NCT02339168 (Phase 1) | Enzalutamide and metformin hydrochloride in treating patients with hormone-resistant PCa. | ||||
| Complex I | IACS-010759 | Binds to inhibit complex I (NADH ubiquinone oxidoreductase). | NCT02882321 (Phase 1) | Treating patients with relapsed or refractory acute myeloid leukemia (AML). | [484,485] |
| Complex I | IM156 | Orally available metformin derivative. | NCT03272256 (Phase 1) | Patients with advanced solid tumor and lymphoma. | [486,487,488] |
| Amino acid metabolism | |||||
| LAT1 | JPH203 | Selective L-type amino acid transporter 1 inhibitor. | UMIN000016546 (Clinical trial registration in Japan) | Exploratory analyses of biomarker using change of blood free amino-acid concentration related to JPH203 (LAT1 inhibitor). | [489,490,491] |
| GLS1 (KGA and GAC) | CB-839/Telaglenastat | Orally available potent allosteric inhibitor for GLS1. | NCT03875313 (Phase 1b/2) | Combination with PARP inhibitor Talazoparib in patients with solid tumors. | [425,462,492] |
| NCT04250545 (Phase 1) | To determine the safety and tolerability of CB-839 combination with mTORC1/2 inhibitor MLN0128 (sapanisertib) and determine the recommended phase 2 dose (RP2D) of the combination. | ||||
| GLS1 (KGA and GAC) | IPN60090 | Orally available potent allosteric inhibitor for GLS1. | NCT03894540 (Phase 1) | Dose escalation and dose expansion study of IPN60090 in patients with advanced solid tumors. | [493] |
| Glutamine metabolism | Sirpiglenastat/DRP-104 | Pro-drug form of a broad acting glutamine antagonist that irreversibly inhibits multiple enzymes involved in glutamine metabolism. Different from JHU083. | NCT04471415 (Phase1/2) | As single agent and in combination with checkpoint inhibitor atezolizumab in patients with advanced solid tumors. | [494] |
| IDO1 | Epacadostat/INCB024360 | Orally available reversible competitive potent IDO1 inhibitor. | NCT03516708 (Phase 1) | Epacadostat (INCB024360) added to preoperative chemoradiation in patients with locally advanced rectal cancer. | [495] |
| NCT03589651 (Phase 1) | Checkpoint inhibitor INCMGA00012 in combination with other therapies in patients with advanced solid tumors. | [494] | |||
| IDO1 | Indoximod | Orally available reversible competitive potent IDO1 inhibitor. | NCT02073123 (Phase 1/2) | IDO inhibitor in combination with checkpoint inhibitors for adult patients with metastatic melanoma. | [334,464] |
| NCT04049669 (Phase 2) | Indoximod with chemotherapy and radiation for relapsed brain tumors or newly diagnosed diffuse intrinsic pontine gliomas (DIPG). | ||||
| NCT01560923 (Phase 2) | Combined therapy with Sipuleucel-T and Indoximod for patients with refractory metastatic PCa. | [334] | |||
| MAT2A | AG-270 | Allosterically inhibits MAT2A (methionine adenosyltransferase 2A) activity by preventing product release, leading to decrease in intracellular SAM levels. | NCT03435250 (Phase 1) | Participants with advanced solid tumors or lymphoma With MTAP loss. | [496,497] |
| Fatty acid metabolism | |||||
| HMG-CoA reductase | statin, Atorvastatin | Competitive inhibitor for HMG-CoA reductase to decrease cholesterol level. | NCT04026230 (Phase 3) | Atorvastatin on PCa progression during ADT (ESTO2). | [390,391,498] |
| NCT02003924 (Phase 3), PROSPER | Safety and efficacy study of enzalutamide in patients with nonmetastatic CRPC. | ||||
| NCT01212991 (Phase 3), PREVAIL | Safety and efficacy study of oral MDV3100 in chemotherapy-naive patients with progressive metastatic PCa. | ||||
| NCT00974311 (Phase 3), AFFIRM | Safety and efficacy study of MDV3100 in patients with CRPC who have been previously treated with docetaxel-based chemotherapy. | ||||
| Retrospective analyses of all three trials pooled and AFFIRM + PREVAIL pooled revealed that statin but not metformin use was significantly associated with a reduced risk of death in enzalutamide-treated patients. | |||||
| FASN | TVB-2640 | Inhibits the β-ketoacyl-ACP reductase activity of FASN. | NCT03179904 (Phase 2) | TVB-2640 and Trastuzumab (herceptin) in combination with paclitaxel or endocrine therapy for the treatment of HER2 positive metastatic breast cancer. | [499,500] |
| NCT03808558 (Phase 2) | TVB-2640 in KRAS non-small cell lung carcinomas. | ||||
| ACC1 and ACC2 | GS-0976/ND-630/NDI 010976/Firsocostat | Allosteric inhibitor of the ACC (Acetyl-CoA carboxylase) enzymes that prevents dimerization of ACC. | NCT03449446 (Phase 1) | Study to evaluate the safety and efficacy of Selonsertib, Firsocostat, Cilofexor, and combinations in participants with bridging fibrosis or compensated cirrhosis due to nonalcoholic steatohepatitis (NASH). | [501,502] |
| Another metabolism | |||||
| mutant IDH1 | Ivosidenib/AG-120 | Allosteric binding of Ivosidenib stabilizes inhibitory open conformation of the mutant IDH1 active site. | FDA approval | Newly diagnosed acute myeloid leukemia (AML) with a susceptible IDH1 mutation. | [503] |
| Sirt1 | EX-527/Selisistat/SEN0014196 | Stabilize the closed enzyme conformation preventing product release. | NCT04184323 (Phase 2) terminated due to lack to funding | SIRT-1 antagonism for endometrial receptivity. | [504] |
| This compound has reached Phase 2 clinical trials for Huntington’s disease (HD) found to be safe and well tolerated in early HD patients at plasma levels within the therapeutic concentration range in preclinical HD models. | [505] | ||||
| DHODH | Leflunomide | Competitive inhibitor for mitochondrial enzyme DHODH. | NCT03709446 (Phase1/2) | Treating patients with previously treated metastatic triple negative breast cancer. | [506,507] |
| NCT04997993 (Phase 1) | Patients with PTEN-null advanced solid malignancies. | ||||
| ODC1 | 2-difluoromethyl ornithine (DFMO)/eflornithine | Competitive and irreversible inhibitor of ODC1. | NCT04301843 (Phase 2) | Eflornithine (DFMO) and etoposide for relapsed/refractory neuroblastoma. | [508,509] |
| NCT01349881 (Phase 3) | Adenoma and second primary prevention trial. | ||||
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Mostaghel, E.A.; Plymate, S.R.; Montgomery, B. Molecular pathways: Targeting resistance in the androgen receptor for therapeutic benefit. Clin. Cancer Res. 2014, 20, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Uo, T.; Plymate, S.R.; Sprenger, C.C. Allosteric alterations in the androgen receptor and activity in prostate cancer. Endocr. Relat. Cancer 2017, 24, R335–R348. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.D.; Welsbie, D.S.; Tran, C.; Baek, S.H.; Chen, R.; Vessella, R.; Rosenfeld, M.G.; Sawyers, C.L. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2004, 10, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef]
- Uo, T.; Plymate, S.R.; Sprenger, C.C. The potential of AR-V7 as a therapeutic target. Expert Opin. Ther. Targets 2018, 22, 201–216. [Google Scholar] [CrossRef]
- Ferraldeschi, R.; Welti, J.; Luo, J.; Attard, G.; de Bono, J.S. Targeting the androgen receptor pathway in castration-resistant prostate cancer: Progresses and prospects. Oncogene 2015, 34, 1745–1757. [Google Scholar] [CrossRef]
- Westaby, D.; Fenor de La Maza, M.L.D.; Paschalis, A.; Jimenez-Vacas, J.M.; Welti, J.; de Bono, J.; Sharp, A. A New Old Target: Androgen Receptor Signaling and Advanced Prostate Cancer. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 131–153. [Google Scholar] [CrossRef]
- Beltran, H.; Hruszkewycz, A.; Scher, H.I.; Hildesheim, J.; Isaacs, J.; Yu, E.Y.; Kelly, K.; Lin, D.; Dicker, A.; Arnold, J.; et al. The Role of Lineage Plasticity in Prostate Cancer Therapy Resistance. Clin. Cancer Res. 2019, 25, 6916–6924. [Google Scholar] [CrossRef]
- Beltran, H.; Tomlins, S.; Aparicio, A.; Arora, V.; Rickman, D.; Ayala, G.; Huang, J.; True, L.; Gleave, M.E.; Soule, H.; et al. Aggressive variants of castration-resistant prostate cancer. Clin. Cancer Res. 2014, 20, 2846–2850. [Google Scholar] [CrossRef]
- Bluemn, E.G.; Coleman, I.M.; Lucas, J.M.; Coleman, R.T.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.F.; Kaipainen, A.; Corella, A.N.; et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 2017, 32, 474–489.e476. [Google Scholar] [CrossRef]
- Myung, J.K.; Banuelos, C.A.; Fernandez, J.G.; Mawji, N.R.; Wang, J.; Tien, A.H. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J. Clin. Investig. 2013, 123, 2948–2960. [Google Scholar] [CrossRef]
- Monaghan, A.E.; McEwan, I.J. A sting in the tail: The N-terminal domain of the androgen receptor as a drug target. Asian J. 2016, 18, 687–694. [Google Scholar] [CrossRef]
- Asangani, I.A.; Dommeti, V.L.; Wang, X.; Malik, R.; Cieslik, M.; Yang, R.; Escara-Wilke, J.; Wilder-Romans, K.; Dhanireddy, S.; Engelke, C.; et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014, 510, 278–282. [Google Scholar] [CrossRef]
- Yang, Y.C.; Banuelos, C.A.; Mawji, N.R.; Wang, J.; Kato, M.; Haile, S.; McEwan, I.J.; Plymate, S.; Sadar, M.D. Targeting Androgen Receptor Activation Function-1 with EPI to Overcome Resistance Mechanisms in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 4466–4477. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Chandhasin, C.; Osbourne, E.; Luo, J.; Sadar, M.D.; Perabo, F. Targeting the N-Terminal Domain of the Androgen Receptor: A New Approach for the Treatment of Advanced Prostate Cancer. Oncologist 2016, 21, 1427–1435. [Google Scholar] [CrossRef]
- Hong, N.H.; Le Moigne, R.; Pearson, P.; Lauriault, V.; Banuelos, C.A.; Mawji, N.R.; Tam, T.; Wang, J.; Virsik, P.; Andersen, R.J.; et al. 181 Poster—The preclinical characterization of the N-terminal domain androgen receptor inhibitor, EPI-7386, for the treatment of prostate cancer. Eur. J. Cancer 2020, 138, S51. [Google Scholar] [CrossRef]
- Moigne, R.L.; Banuelos, C.A.; Mawji, N.R.; Tam, T.; Wang, J.; Jian, K.; Andersen, R.J.; Cesano, A.; Sadar, M.D.; Zhou, H.J.; et al. 503P—EPI-7386 is a novel N-terminal domain androgen receptor inhibitor for the treatment of prostate cancer. Ann. Oncol. 2019, 30, v189–v190. [Google Scholar] [CrossRef]
- Ma, B.; Fan, Y.; Zhang, D.; Wei, Y.; Jian, Y.; Liu, D.; Wang, Z.; Gao, Y.; Ma, J.; Chen, Y.; et al. De Novo Design of an Androgen Receptor DNA Binding Domain-Targeted peptide PROTAC for Prostate Cancer Therapy. Adv. Sci. 2022, 9, 2201859. [Google Scholar] [CrossRef]
- Hong, N.H.; Biannic, B.; Virsik, P.; Zhou, H.-J.; Moigne, R.L. Abstract 429: Androgen receptor (AR) N-terminal domain degraders can degrade AR full length and AR splice variants in CRPC preclinical models. Cancer Res. 2022, 82, 429. [Google Scholar] [CrossRef]
- Hofman, M.S.; Emmett, L.; Sandhu, S.; Iravani, A.; Joshua, A.M.; Goh, J.C.; Pattison, D.A.; Tan, T.H.; Kirkwood, I.D.; Francis, R.J.; et al. TheraP: 177Lu-PSMA-617 (LuPSMA) versus cabazitaxel in metastatic castration-resistant prostate cancer (mCRPC) progressing after docetaxel—Overall survival after median follow-up of 3 years (ANZUP 1603). J. Clin. Oncol. 2022, 40, 5000. [Google Scholar] [CrossRef]
- Hofman, M.S.; Emmett, L.; Sandhu, S.; Iravani, A.; Joshua, A.M.; Goh, J.C.; Pattison, D.A.; Tan, T.H.; Kirkwood, I.D.; Ng, S.; et al. [(177)Lu]Lu-PSMA-617 versus cabazitaxel in patients with metastatic castration-resistant prostate cancer (TheraP): A randomised, open-label, phase 2 trial. Lancet 2021, 397, 797–804. [Google Scholar] [CrossRef]
- Sartor, O.; de Bono, J.; Chi, K.N.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Nordquist, L.T.; Vaishampayan, N.; El-Haddad, G.; et al. Lutetium-177-PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2021, 385, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Niaz, M.O.; Nelson, A.; Skafida, M.; Niaz, M.J. Review of 177Lu-PSMA-617 in Patients With Metastatic Castration-Resistant Prostate Cancer. Cureus 2020, 12, e8921. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Schiewer, M.J.; Knudsen, K.E. DNA Damage Response in Prostate Cancer. Cold Spring Harb. Perspect. Med. 2019, 9, a030486. [Google Scholar] [CrossRef]
- Mollica, V.; Rizzo, A.; Rosellini, M.; Marchetti, A.; Ricci, A.D.; Cimadamore, A.; Scarpelli, M.; Bonucci, C.; Andrini, E.; Errani, C.; et al. Bone Targeting Agents in Patients with Metastatic Prostate Cancer: State of the Art. Cancers 2021, 13, 546. [Google Scholar] [CrossRef]
- Furesi, G.; Rauner, M.; Hofbauer, L.C. Emerging Players in Prostate Cancer-Bone Niche Communication. Trends Cancer 2021, 7, 112–121. [Google Scholar] [CrossRef]
- He, Y.; Xu, W.; Xiao, Y.T.; Huang, H.; Gu, D.; Ren, S. Targeting signaling pathways in prostate cancer: Mechanisms and clinical trials. Signal Transduct. Target. Ther. 2022, 7, 198. [Google Scholar] [CrossRef]
- Sandhu, S.; Moore, C.M.; Chiong, E.; Beltran, H.; Bristow, R.G.; Williams, S.G. Prostate cancer. Lancet 2021, 398, 1075–1090. [Google Scholar] [CrossRef]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Landau, B.R.; Laszlo, J.; Stengle, J.; Burk, D. Certain Metabolic and Pharmacologic Effects in Cancer Patients Given Infusions of 2-Deoxy-D-Glucose. JNCI J. Natl. Cancer Inst. 1958, 21, 485–494. [Google Scholar] [CrossRef]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2022, 21, 141–162. [Google Scholar] [CrossRef]
- Farber, S.; Diamond, L.K.; Mercer, R.D.; Sylvester, R.F.; Wolff, J.A. Temporary Remissions in Acute Leukemia in Children Produced by Folic Acid Antagonist, 4-Aminopteroyl-Glutamic Acid (Aminopterin). N. Engl. J. Med. 1948, 238, 787–793. [Google Scholar] [CrossRef]
- Miller, D.R. A tribute to Sidney Farber—The father of modern chemotherapy. Br. J. Haematol. 2006, 134, 20–26. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Dang, C.V. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a014217. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Keshari, K.R. Metabolic analysis as a driver for discovery, diagnosis, and therapy. Cell 2022, 185, 2678–2689. [Google Scholar] [CrossRef] [PubMed]
- Di Gregorio, E.; Miolo, G.; Saorin, A.; Steffan, A.; Corona, G. From Metabolism to Genetics and Vice Versa: The Rising Role of Oncometabolites in Cancer Development and Therapy. Int. J. Mol. Sci 2021, 22, 5574. [Google Scholar] [CrossRef]
- Yang, M.; Soga, T.; Pollard, P.J. Oncometabolites: Linking altered metabolism with cancer. J. Clin. Investig. 2013, 123, 3652–3658. [Google Scholar] [CrossRef]
- Luengo, A.; Gui, D.Y.; Vander Heiden, M.G. Targeting Metabolism for Cancer Therapy. Cell Chem. Biol. 2017, 24, 1161–1180. [Google Scholar] [CrossRef]
- Ku, S.Y.; Gleave, M.E.; Beltran, H. Towards precision oncology in advanced prostate cancer. Nat. Rev. Urol. 2019, 16, 645–654. [Google Scholar] [CrossRef]
- Mateo, J.; McKay, R.; Abida, W.; Aggarwal, R.; Alumkal, J.; Alva, A.; Feng, F.; Gao, X.; Graff, J.; Hussain, M.; et al. Accelerating precision medicine in metastatic prostate cancer. Nat. Cancer 2020, 1, 1041–1053. [Google Scholar] [CrossRef]
- Awad, D.; Pulliam, T.L.; Lin, C.; Wilkenfeld, S.R.; Frigo, D.E. Delineation of the androgen-regulated signaling pathways in prostate cancer facilitates the development of novel therapeutic approaches. Curr. Opin. Pharmacol. 2018, 41, 1–11. [Google Scholar] [CrossRef]
- Uo, T.; Sprenger, C.C.; Plymate, S.R. Androgen Receptor Signaling and Metabolic and Cellular Plasticity During Progression to Castration Resistant Prostate Cancer. Front. Oncol. 2020, 10, 580617. [Google Scholar] [CrossRef]
- Bader, D.A.; McGuire, S.E. Tumour metabolism and its unique properties in prostate adenocarcinoma. Nat. Rev. Urol. 2020, 17, 214–231. [Google Scholar] [CrossRef]
- Giunchi, F.; Fiorentino, M.; Loda, M. The Metabolic Landscape of Prostate Cancer. Eur. Urol. Oncol. 2019, 2, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Zadra, G.; Loda, M. Metabolic Vulnerabilities of Prostate Cancer: Diagnostic and Therapeutic Opportunities. Cold Spring Harb. Perspect. Med. 2018, 8, a030569. [Google Scholar] [CrossRef] [PubMed]
- Peitzsch, C.; Gorodetska, I.; Klusa, D.; Shi, Q.; Alves, T.C.; Pantel, K.; Dubrovska, A. Metabolic regulation of prostate cancer heterogeneity and plasticity. Semin. Cancer Biol. 2022, 82, 94–119. [Google Scholar] [CrossRef] [PubMed]
- Fidelito, G.; Watt, M.J.; Taylor, R.A. Personalized Medicine for Prostate Cancer: Is Targeting Metabolism a Reality? Front. Oncol. 2021, 11, 778761. [Google Scholar] [CrossRef] [PubMed]
- Kolenko, V.; Teper, E.; Kutikov, A.; Uzzo, R. Zinc and zinc transporters in prostate carcinogenesis. Nat. Rev. Urol. 2013, 10, 219–226. [Google Scholar] [CrossRef]
- Costello, L.C.; Franklin, R.B. A comprehensive review of the role of zinc in normal prostate function and metabolism; and its implications in prostate cancer. Arch. Biochem. Biophys. 2016, 611, 100–112. [Google Scholar] [CrossRef]
- Franklin, R.B.; Zou, J.; Yu, Z.; Costello, L.C. EAAC1 is expressed in rat and human prostate epithelial cells; functions as a high-affinity L-aspartate transporter; and is regulated by prolactin and testosterone. BMC Biochem. 2006, 7, 10. [Google Scholar] [CrossRef]
- Franklin, R.B.; Kukoyi, B.I.; Akuffo, V.; Costello, L.C. Testosterone stimulation of mitochondrial aspartate aminotransferase levels and biosynthesis in rat ventral prostate. J. Steroid Biochem. 1987, 28, 247–256. [Google Scholar] [CrossRef]
- Costello, L.C.; Liu, Y.; Zou, J.; Franklin, R.B. The pyruvate dehydrogenase E1 alpha gene is testosterone and prolactin regulated in prostate epithelial cells. Endocr. Res. 2000, 26, 23–39. [Google Scholar] [CrossRef]
- Franklin, R.B.; Feng, P.; Milon, B.; Desouki, M.M.; Singh, K.K.; Kajdacsy-Balla, A.; Bagasra, O.; Costello, L.C. hZIP1 zinc uptake transporter down regulation and zinc depletion in prostate cancer. Mol. Cancer 2005, 4, 32. [Google Scholar] [CrossRef]
- Costello, L.C.; Franklin, R.B. The clinical relevance of the metabolism of prostate cancer; zinc and tumor suppression: Connecting the dots. Mol. Cancer 2006, 5, 17. [Google Scholar] [CrossRef][Green Version]
- Jadvar, H. Is There Use for FDG-PET in Prostate Cancer? Semin. Nucl. Med. 2016, 46, 502–506. [Google Scholar] [CrossRef]
- Sahin, E.; Elboga, U.; Kalender, E.; Basibuyuk, M.; Demir, H.D.; Celen, Y.Z. Clinical significance of incidental FDG uptake in the prostate gland detected by PET/CT. Int. J. Clin. Exp. Med. 2015, 8, 10577–10585. [Google Scholar]
- Taplin, M.E.; Bubley, G.J.; Shuster, T.D.; Frantz, M.E.; Spooner, A.E.; Ogata, G.K.; Keer, H.N.; Balk, S.P. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N. Engl. J. Med. 1995, 332, 1393–1398. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Koivisto, P.; Kononen, J.; Palmberg, C.; Tammela, T.; Hyytinen, E.; Isola, J.; Trapman, J.; Cleutjens, K.; Noordzij, A.; Visakorpi, T.; et al. Androgen receptor gene amplification: A possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997, 57, 314–319. [Google Scholar] [PubMed]
- Buchanan, G.; Greenberg, N.M.; Scher, H.I.; Harris, J.M.; Marshall, V.R.; Tilley, W.D. Collocation of androgen receptor gene mutations in prostate cancer. Clin. Cancer Res. 2001, 7, 1273–1281. [Google Scholar] [PubMed]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef]
- Dehm, S.M.; Schmidt, L.J.; Heemers, H.V.; Vessella, R.L.; Tindall, D.J. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008, 68, 5469–5477. [Google Scholar] [CrossRef]
- Lu, C.; Luo, J. Decoding the androgen receptor splice variants. Transl. Androl. Urol. 2013, 2, 178–186. [Google Scholar] [CrossRef]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef]
- Clegg, N.J.; Wongvipat, J.; Joseph, J.D.; Tran, C.; Ouk, S.; Dilhas, A.; Chen, Y.; Grillot, K.; Bischoff, E.D.; Cai, L.; et al. ARN-509: A novel antiandrogen for prostate cancer treatment. Cancer Res. 2012, 72, 1494–1503. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef]
- Smith, M.R.; Saad, F.; Chowdhury, S.; Oudard, S.; Hadaschik, B.A.; Graff, J.N.; Olmos, D.; Mainwaring, P.N.; Lee, J.Y.; Uemura, H.; et al. Apalutamide Treatment and Metastasis-free Survival in Prostate Cancer. N. Engl. J. Med. 2018, 378, 1408–1418. [Google Scholar] [CrossRef]
- Berchuck, J.E.; Viscuse, P.V.; Beltran, H.; Aparicio, A. Clinical considerations for the management of androgen indifferent prostate cancer. Prostate Cancer Prostatic Dis. 2021, 24, 623–637. [Google Scholar] [CrossRef]
- Beltran, H.; Tagawa, S.T.; Park, K.; MacDonald, T.; Milowsky, M.I.; Mosquera, J.M.; Rubin, M.A.; Nanus, D.M. Challenges in recognizing treatment-related neuroendocrine prostate cancer. J. Clin. Oncol. 2012, 30, e386-9. [Google Scholar] [CrossRef]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef]
- Culine, S.; El Demery, M.; Lamy, P.J.; Iborra, F.; Avances, C.; Pinguet, F. Docetaxel and cisplatin in patients with metastatic androgen independent prostate cancer and circulating neuroendocrine markers. J. Urol. 2007, 178, 844–848, discussion 848. [Google Scholar] [CrossRef]
- Flechon, A.; Pouessel, D.; Ferlay, C.; Perol, D.; Beuzeboc, P.; Gravis, G.; Joly, F.; Oudard, S.; Deplanque, G.; Zanetta, S.; et al. Phase II study of carboplatin and etoposide in patients with anaplastic progressive metastatic castration-resistant prostate cancer (mCRPC) with or without neuroendocrine differentiation: Results of the French Genito-Urinary Tumor Group (GETUG) P01 trial. Ann. Oncol. 2011, 22, 2476–2481. [Google Scholar] [CrossRef]
- Saunders, L.R.; Bankovich, A.J.; Anderson, W.C.; Aujay, M.A.; Bheddah, S.; Black, K.; Desai, R.; Escarpe, P.A.; Hampl, J.; Laysang, A.; et al. A DLL3-targeted antibody-drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Sci. Transl. Med. 2015, 7, 302ra136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, B.; Wu, W.; Li, L.; Broom, B.M.; Basourakos, S.P.; Korentzelos, D.; Luan, Y.; Wang, J.; Yang, G.; et al. Targeting the MYCN-PARP-DNA Damage Response Pathway in Neuroendocrine Prostate Cancer. Clin. Cancer Res. 2018, 24, 696–707. [Google Scholar] [CrossRef] [PubMed]
- Shafi, A.A.; Putluri, V.; Arnold, J.M.; Tsouko, E.; Maity, S.; Roberts, J.M.; Coarfa, C.; Frigo, D.E.; Putluri, N.; Sreekumar, A.; et al. Differential regulation of metabolic pathways by androgen receptor (AR) and its constitutively active splice variant, AR-V7, in prostate cancer cells. Oncotarget 2015, 6, 31997–32012. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, J.V.; Heemers, H.; van de Sande, T.; de Schrijver, E.; Brusselmans, K.; Heyns, W.; Verhoeven, G. Androgens, lipogenesis and prostate cancer. J. Steroid Biochem. Mol. Biol. 2004, 92, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, S.L.; Sobel, R.; Whitmore, T.G.; Akbari, M.; Bradley, D.R.; Gleave, M.E.; Nelson, C.C. Dysregulation of sterol response element-binding proteins and downstream effectors in prostate cancer during progression to androgen independence. Cancer Res. 2004, 64, 2212–2221. [Google Scholar] [CrossRef]
- Zadra, G.; Ribeiro, C.F.; Chetta, P.; Ho, Y.; Cacciatore, S.; Gao, X.; Syamala, S.; Bango, C.; Photopoulos, C.; Huang, Y.; et al. Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 631–640. [Google Scholar] [CrossRef]
- Rossi, S.; Graner, E.; Febbo, P.; Weinstein, L.; Bhattacharya, N.; Onody, T.; Bubley, G.; Balk, S.; Loda, M. Fatty acid synthase expression defines distinct molecular signatures in prostate cancer. Mol. Cancer Res. 2003, 1, 707–715. [Google Scholar]
- Jadvar, H. Prostate cancer: PET with 18F-FDG, 18F- or 11C-acetate, and 18F- or 11C-choline. J. Nucl. Med. 2011, 52, 81–89. [Google Scholar] [CrossRef]
- Emonds, K.M.; Swinnen, J.V.; Lerut, E.; Koole, M.; Mortelmans, L.; Mottaghy, F.M. Evaluation of androgen-induced effects on the uptake of [18F]FDG, [11C]choline and [11C]acetate in an androgen-sensitive and androgen-independent prostate cancer xenograft model. EJNMMI Res. 2013, 3, 31. [Google Scholar] [CrossRef]
- Bakht, M.K.; Lovnicki, J.M.; Tubman, J.; Stringer, K.F.; Chiaramonte, J.; Reynolds, M.R.; Derecichei, I.; Ferraiuolo, R.M.; Fifield, B.A.; Lubanska, D.; et al. Differential Expression of Glucose Transporters and Hexokinases in Prostate Cancer with a Neuroendocrine Gene Signature: A Mechanistic Perspective for 18F-FDG Imaging of PSMA-Suppressed Tumors. J. Nucl. Med. 2020, 61, 904–910. [Google Scholar] [CrossRef]
- Lewis, D.Y.; Boren, J.; Shaw, G.L.; Bielik, R.; Ramos-Montoya, A.; Larkin, T.J.; Martins, C.P.; Neal, D.E.; Soloviev, D.; Brindle, K.M. Late Imaging with [1-11C]Acetate Improves Detection of Tumor Fatty Acid Synthesis with PET. J. Nucl. Med. 2014, 55, 1144–1149. [Google Scholar] [CrossRef]
- Yu, E.Y.; Muzi, M.; Hackenbracht, J.A.; Rezvani, B.B.; Link, J.M.; Montgomery, R.B.; Higano, C.S.; Eary, J.F.; Mankoff, D.A. C11-acetate and F-18 FDG PET for men with prostate cancer bone metastases: Relative findings and response to therapy. Clin. Nucl. Med. 2011, 36, 192–198. [Google Scholar] [CrossRef]
- Fox, J.J.; Gavane, S.C.; Blanc-Autran, E.; Nehmeh, S.; Gonen, M.; Beattie, B.; Vargas, H.A.; Schoder, H.; Humm, J.L.; Fine, S.W.; et al. Positron Emission Tomography/Computed Tomography-Based Assessments of Androgen Receptor Expression and Glycolytic Activity as a Prognostic Biomarker for Metastatic Castration-Resistant Prostate Cancer. JAMA Oncol. 2018, 4, 217–224. [Google Scholar] [CrossRef]
- Liu, Y. FDG PET-CT demonstration of metastatic neuroendocrine tumor of prostate. World J. Surg. Oncol. 2008, 6, 64. [Google Scholar] [CrossRef]
- Spratt, D.E.; Gavane, S.; Tarlinton, L.; Fareedy, S.B.; Doran, M.G.; Zelefsky, M.J.; Osborne, J.R. Utility of FDG-PET in clinical neuroendocrine prostate cancer. Prostate 2014, 74, 1153–1159. [Google Scholar] [CrossRef]
- Sung, J.; Espiritu, J.I.; Segall, G.M.; Terris, M.K. Fluorodeoxyglucose positron emission tomography studies in the diagnosis and staging of clinically advanced prostate cancer. BJU Int. 2003, 92, 24–27. [Google Scholar] [CrossRef]
- Momcilovic, M.; Jones, A.; Bailey, S.T.; Waldmann, C.M.; Li, R.; Lee, J.T.; Abdelhady, G.; Gomez, A.; Holloway, T.; Schmid, E.; et al. In vivo imaging of mitochondrial membrane potential in non-small-cell lung cancer. Nature 2019, 575, 380–384. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef]
- Warburg, O. The Chemical Constitution of Respiration Ferment. Science 1928, 68, 437–443. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Scroggins, B.T.; Matsuo, M.; White, A.O.; Saito, K.; Munasinghe, J.P.; Sourbier, C.; Yamamoto, K.; Diaz, V.; Takakusagi, Y.; Ichikawa, K.; et al. Hyperpolarized [1-13C]-Pyruvate Magnetic Resonance Spectroscopic Imaging of Prostate Cancer In Vivo Predicts Efficacy of Targeting the Warburg Effect. Clin. Cancer Res. 2018, 24, 3137–3148. [Google Scholar] [CrossRef]
- Javaeed, A.; Ghauri, S.K. MCT4 has a potential to be used as a prognostic biomarker—A systematic review and meta-analysis. Oncol. Rev. 2019, 13, 403. [Google Scholar] [CrossRef] [PubMed]
- Bovenzi, C.D.; Hamilton, J.; Tassone, P.; Johnson, J.; Cognetti, D.M.; Luginbuhl, A.; Keane, W.M.; Zhan, T.; Tuluc, M.; Bar-Ad, V.; et al. Prognostic Indications of Elevated MCT4 and CD147 across Cancer Types: A Meta-Analysis. BioMed Res. Int. 2015, 2015, 242437. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.; Xue, H.; Wu, R.; Fazli, L.; Lin, D.; Collins, C.C.; Gleave, M.E.; Gout, P.W.; Wang, Y. The MCT4 Gene: A Novel, Potential Target for Therapy of Advanced Prostate Cancer. Clin. Cancer Res. 2016, 22, 2721–2733. [Google Scholar] [CrossRef]
- Yu, M.; Yongzhi, H.; Chen, S.; Luo, X.; Lin, Y.; Zhou, Y.; Jin, H.; Hou, B.; Deng, Y.; Tu, L.; et al. The prognostic value of GLUT1 in cancers: A systematic review and meta-analysis. Oncotarget 2017, 8, 43356–43367. [Google Scholar] [CrossRef]
- Lv, J.; Zhou, Z.; Wang, J.; Yu, H.; Lu, H.; Yuan, B.; Han, J.; Zhou, R.; Zhang, X.; Yang, X.; et al. Prognostic Value of Lactate Dehydrogenase Expression in Different Cancers: A Meta-Analysis. Am. J. Med. Sci. 2019, 358, 412–421. [Google Scholar] [CrossRef]
- Fregeau-Proulx, L.; Lacouture, A.; Berthiaume, L.; Weidmann, C.; Harvey, M.; Gonthier, K.; Pelletier, J.F.; Neveu, B.; Jobin, C.; Bastien, D.; et al. Multiple metabolic pathways fuel the truncated tricarboxylic acid cycle of the prostate to sustain constant citrate production and secretion. Mol. Metab. 2022, 62, 101516. [Google Scholar] [CrossRef]
- Pliszka, M.; Szablewski, L. Glucose Transporters as a Target for Anticancer Therapy. Cancers 2021, 13, 4184. [Google Scholar] [CrossRef]
- Meng, Y.; Xu, X.; Luan, H.; Li, L.; Dai, W.; Li, Z.; Bian, J. The progress and development of GLUT1 inhibitors targeting cancer energy metabolism. Future Med. Chem. 2019, 11, 2333–2352. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, Y.; Zhang, W.; Bergmeier, S.; Qian, Y.; Akbar, H.; Colvin, R.; Ding, J.; Tong, L.; Wu, S.; et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol. Cancer 2012, 11, 1672–1682. [Google Scholar] [CrossRef]
- Meziou, S.; Ringuette Goulet, C.; Hovington, H.; Lefebvre, V.; Lavallee, E.; Bergeron, M.; Brisson, H.; Champagne, A.; Neveu, B.; Lacombe, D.; et al. GLUT1 expression in high-risk prostate cancer: Correlation with (18)F-FDG-PET/CT and clinical outcome. Prostate Cancer Prostatic Dis. 2020, 23, 441–448. [Google Scholar] [CrossRef]
- Wang, J.; Xu, W.; Wang, B.; Lin, G.; Wei, Y.; Abudurexiti, M.; Zhu, W.; Liu, C.; Qin, X.; Dai, B.; et al. GLUT1 is an AR target contributing to tumor growth and glycolysis in castration-resistant and enzalutamide-resistant prostate cancers. Cancer Lett. 2020, 485, 45–55. [Google Scholar] [CrossRef]
- Gonzalez-Menendez, P.; Hevia, D.; Mayo, J.C.; Sainz, R.M. The dark side of glucose transporters in prostate cancer: Are they a new feature to characterize carcinomas? Int. J. Cancer 2018, 142, 2414–2424. [Google Scholar] [CrossRef]
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef]
- Tahir, S.A.; Yang, G.; Goltsov, A.; Song, K.D.; Ren, C.; Wang, J.; Chang, W.; Thompson, T.C. Caveolin-1-LRP6 signaling module stimulates aerobic glycolysis in prostate cancer. Cancer Res. 2013, 73, 1900–1911. [Google Scholar] [CrossRef]
- Ryniawec, J.M.; Coope, M.R.; Loertscher, E.; Bageerathan, V.; de Oliveira Pessoa, D.; Warfel, N.A.; Cress, A.E.; Padi, M.; Rogers, G.C. GLUT3/SLC2A3 Is an Endogenous Marker of Hypoxia in Prostate Cancer Cell Lines and Patient-Derived Xenograft Tumors. Diagnostics 2022, 12, 676. [Google Scholar] [CrossRef]
- Hoshi, S.; Meguro, S.; Imai, H.; Matsuoka, Y.; Yoshida, Y.; Onagi, A.; Tanji, R.; Honda-Takinami, R.; Matsuoka, K.; Koguchi, T.; et al. Upregulation of glucocorticoid receptor-mediated glucose transporter 4 in enzalutamide-resistant prostate cancer. Cancer Sci. 2021, 112, 1899–1910. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, B.; Wang, Z.S.; Guo, Y.L.; Shen, H. Construction of Glycolytic Regulator Gene Signature to Predict the Prognosis and Tumor Immune Cell Infiltration Levels for Prostate Cancer. Comput. Math. Methods Med. 2022, 2022, 9273559. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Li, X.; Peng, S.; Li, J.; Yan, W.; Cui, Y.; Xiao, H.; Wen, X. Increased expression of glycolytic enzymes in prostate cancer tissues and association with Gleason scores. Int. J. Clin. Exp. Pathol. 2017, 10, 11080–11089. [Google Scholar]
- Choi, S.Y.C.; Ettinger, S.L.; Lin, D.; Xue, H.; Ci, X.; Nabavi, N.; Bell, R.H.; Mo, F.; Gout, P.W.; Fleshner, N.E.; et al. Targeting MCT4 to reduce lactic acid secretion and glycolysis for treatment of neuroendocrine prostate cancer. Cancer Med. 2018, 7, 3385–3392. [Google Scholar] [CrossRef] [PubMed]
- Fontana, F.; Anselmi, M.; Limonta, P. Exploiting the Metabolic Consequences of PTEN Loss and Akt/Hexokinase 2 Hyperactivation in Prostate Cancer: A New Role for delta-Tocotrienol. Int. J. Mol. Sci. 2022, 23, 5269. [Google Scholar] [CrossRef] [PubMed]
- Shangguan, X.; He, J.; Ma, Z.; Zhang, W.; Ji, Y.; Shen, K.; Yue, Z.; Li, W.; Xin, Z.; Zheng, Q.; et al. SUMOylation controls the binding of hexokinase 2 to mitochondria and protects against prostate cancer tumorigenesis. Nat. Commun. 2021, 12, 1812. [Google Scholar] [CrossRef]
- Lee, H.J.; Li, C.F.; Ruan, D.; He, J.; Montal, E.D.; Lorenz, S.; Girnun, G.D.; Chan, C.H. Non-proteolytic ubiquitination of Hexokinase 2 by HectH9 controls tumor metabolism and cancer stem cell expansion. Nat. Commun. 2019, 10, 2625. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.L.; Yin, J.J.; Seng, V.; Casey, O.; Corey, E.; Morrissey, C.; Simpson, R.M.; Kelly, K. Androgen deprivation leads to increased carbohydrate metabolism and hexokinase 2-mediated survival in Pten/Tp53-deficient prostate cancer. Oncogene 2017, 36, 525–533. [Google Scholar] [CrossRef]
- Wang, L.; Wang, J.; Xiong, H.; Wu, F.; Lan, T.; Zhang, Y.; Guo, X.; Wang, H.; Saleem, M.; Jiang, C.; et al. Co-targeting hexokinase 2-mediated Warburg effect and ULK1-dependent autophagy suppresses tumor growth of PTEN- and TP53-deficiency-driven castration-resistant prostate cancer. eBioMedicine 2016, 7, 50–61. [Google Scholar] [CrossRef]
- Wang, L.; Xiong, H.; Wu, F.; Zhang, Y.; Wang, J.; Zhao, L.; Guo, X.; Chang, L.J.; Zhang, Y.; You, M.J.; et al. Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell Rep. 2014, 8, 1461–1474. [Google Scholar] [CrossRef]
- Ros, S.; Schulze, A. Balancing glycolytic flux: The role of 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatases in cancer metabolism. Cancer Metab. 2013, 1, 8. [Google Scholar] [CrossRef]
- Mor, I.; Cheung, E.C.; Vousden, K.H. Control of glycolysis through regulation of PFK1: Old friends and recent additions. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 211–216. [Google Scholar] [CrossRef]
- Vaz, C.V.; Marques, R.; Alves, M.G.; Oliveira, P.F.; Cavaco, J.E.; Maia, C.J.; Socorro, S. Androgens enhance the glycolytic metabolism and lactate export in prostate cancer cells by modulating the expression of GLUT1, GLUT3, PFK, LDH and MCT4 genes. J. Cancer Res. Clin. Oncol. 2016, 142, 5–16. [Google Scholar] [CrossRef]
- Moon, J.S.; Jin, W.J.; Kwak, J.H.; Kim, H.J.; Yun, M.J.; Kim, J.W.; Park, S.W.; Kim, K.S. Androgen stimulates glycolysis for de novo lipid synthesis by increasing the activities of hexokinase 2 and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 2 in prostate cancer cells. Biochem. J. 2011, 433, 225–233. [Google Scholar] [CrossRef]
- Kuang, Q.; Liang, Y.; Zhuo, Y.; Cai, Z.; Jiang, F.; Xie, J.; Zheng, Y.; Zhong, W. The ALDOA Metabolism Pathway as a Potential Target for Regulation of Prostate Cancer Proliferation. OncoTargets Ther. 2021, 14, 3353–3366. [Google Scholar] [CrossRef]
- Dayton, T.L.; Jacks, T.; Vander Heiden, M.G. PKM2, cancer metabolism, and the road ahead. EMBO Rep. 2016, 17, 1721–1730. [Google Scholar] [CrossRef]
- Davidson, S.M.; Schmidt, D.R.; Heyman, J.E.; O’Brien, J.P.; Liu, A.C.; Israelsen, W.J.; Dayton, T.L.; Sehgal, R.; Bronson, R.T.; Freinkman, E.; et al. Pyruvate Kinase M1 Suppresses Development and Progression of Prostate Adenocarcinoma. Cancer Res. 2022, 82, 2403–2416. [Google Scholar] [CrossRef]
- Yoon, Y.J.; Kim, Y.H.; Jin, Y.; Chi, S.W.; Moon, J.H.; Han, D.C.; Kwon, B.M. 2’-hydroxycinnamaldehyde inhibits cancer cell proliferation and tumor growth by targeting the pyruvate kinase M2. Cancer Lett. 2018, 434, 42–55. [Google Scholar] [CrossRef]
- Zhu, S.; Guo, Y.; Zhang, X.; Liu, H.; Yin, M.; Chen, X.; Peng, C. Pyruvate kinase M2 (PKM2) in cancer and cancer therapeutics. Cancer Lett. 2021, 503, 240–248. [Google Scholar] [CrossRef]
- Lee, Y.B.; Min, J.K.; Kim, J.G.; Cap, K.C.; Islam, R.; Hossain, A.J.; Dogsom, O.; Hamza, A.; Mahmud, S.; Choi, D.R.; et al. Multiple functions of pyruvate kinase M2 in various cell types. J. Cell. Physiol. 2022, 237, 128–148. [Google Scholar] [CrossRef]
- Dylgjeri, E.; Kothari, V.; Shafi, A.A.; Semenova, G.; Gallagher, P.T.; Guan, Y.F.; Pang, A.; Goodwin, J.F.; Irani, S.; McCann, J.J.; et al. A Novel Role for DNA-PK in Metabolism by Regulating Glycolysis in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2022, 28, 1446–1459. [Google Scholar] [CrossRef]
- Guo, W.; Zhang, Z.; Li, G.; Lai, X.; Gu, R.; Xu, W.; Chen, H.; Xing, Z.; Chen, L.; Qian, J.; et al. Pyruvate Kinase M2 Promotes Prostate Cancer Metastasis Through Regulating ERK1/2-COX-2 Signaling. Front. Oncol. 2020, 10, 544288. [Google Scholar] [CrossRef]
- Giannoni, E.; Taddei, M.L.; Morandi, A.; Comito, G.; Calvani, M.; Bianchini, F.; Richichi, B.; Raugei, G.; Wong, N.; Tang, D.; et al. Targeting stromal-induced pyruvate kinase M2 nuclear translocation impairs oxphos and prostate cancer metastatic spread. Oncotarget 2015, 6, 24061–24074. [Google Scholar] [CrossRef]
- Dai, J.; Escara-Wilke, J.; Keller, J.M.; Jung, Y.; Taichman, R.S.; Pienta, K.J.; Keller, E.T. Primary prostate cancer educates bone stroma through exosomal pyruvate kinase M2 to promote bone metastasis. J. Exp. Med. 2019, 216, 2883–2899. [Google Scholar] [CrossRef] [PubMed]
- Hasan, D.; Gamen, E.; Abu Tarboush, N.; Ismail, Y.; Pak, O.; Azab, B. PKM2 and HIF-1alpha regulation in prostate cancer cell lines. PLoS ONE 2018, 13, e0203745. [Google Scholar] [CrossRef] [PubMed]
- Yasumizu, Y.; Hongo, H.; Kosaka, T.; Mikami, S.; Nishimoto, K.; Kikuchi, E.; Oya, M. PKM2 under hypoxic environment causes resistance to mTOR inhibitor in human castration resistant prostate cancer. Oncotarget 2018, 9, 27698–27707. [Google Scholar] [CrossRef] [PubMed][Green Version]
- McCommis, K.S.; Finck, B.N. Mitochondrial pyruvate transport: A historical perspective and future research directions. Biochem. J. 2015, 466, 443–454. [Google Scholar] [CrossRef]
- Bensard, C.L.; Wisidagama, D.R.; Olson, K.A.; Berg, J.A.; Krah, N.M.; Schell, J.C.; Nowinski, S.M.; Fogarty, S.; Bott, A.J.; Wei, P.; et al. Regulation of Tumor Initiation by the Mitochondrial Pyruvate Carrier. Cell Metab. 2020, 31, 284–300.e7. [Google Scholar] [CrossRef]
- Schell, J.C.; Olson, K.A.; Jiang, L.; Hawkins, A.J.; Van Vranken, J.G.; Xie, J.; Egnatchik, R.A.; Earl, E.G.; DeBerardinis, R.J.; Rutter, J. A role for the mitochondrial pyruvate carrier as a repressor of the Warburg effect and colon cancer cell growth. Mol. Cell 2014, 56, 400–413. [Google Scholar] [CrossRef]
- Bader, D.A.; Hartig, S.M.; Putluri, V.; Foley, C.; Hamilton, M.P.; Smith, E.A.; Saha, P.K.; Panigrahi, A.; Walker, C.; Zong, L.; et al. Mitochondrial pyruvate import is a metabolic vulnerability in androgen receptor-driven prostate cancer. Nat. Metab. 2019, 1, 70–85. [Google Scholar] [CrossRef]
- Yang, C.; Ko, B.; Hensley, C.T.; Jiang, L.; Wasti, A.T.; Kim, J.; Sudderth, J.; Calvaruso, M.A.; Lumata, L.; Mitsche, M.; et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol. Cell 2014, 56, 414–424. [Google Scholar] [CrossRef]
- Vacanti, N.M.; Divakaruni, A.S.; Green, C.R.; Parker, S.J.; Henry, R.R.; Ciaraldi, T.P.; Murphy, A.N.; Metallo, C.M. Regulation of substrate utilization by the mitochondrial pyruvate carrier. Mol. Cell 2014, 56, 425–435. [Google Scholar] [CrossRef]
- Xu, H.; Liu, Z.; Gao, D.; Li, P.; Shen, Y.; Sun, Y.; Xu, L.; Song, N.; Wang, Y.; Zhan, M.; et al. Reprogramming hormone-sensitive prostate cancer to a lethal neuroendocrine cancer lineage by mitochondrial pyruvate carrier (MPC). Mol. Metab. 2022, 59, 101466. [Google Scholar] [CrossRef]
- Roche, T.E.; Baker, J.C.; Yan, X.; Hiromasa, Y.; Gong, X.; Peng, T.; Dong, J.; Turkan, A.; Kasten, S.A. Distinct regulatory properties of pyruvate dehydrogenase kinase and phosphatase isoforms. Prog. Nucleic Acid Res. Mol. Biol. 2001, 70, 33–75. [Google Scholar] [CrossRef]
- Nunes-Xavier, C.E.; Mingo, J.; Emaldi, M.; Flem-Karlsen, K.; Maelandsmo, G.M.; Fodstad, O.; Llarena, R.; Lopez, J.I.; Pulido, R. Heterogeneous Expression and Subcellular Localization of Pyruvate Dehydrogenase Complex in Prostate Cancer. Front. Oncol. 2022, 12, 873516. [Google Scholar] [CrossRef]
- Atas, E.; Oberhuber, M.; Kenner, L. The Implications of PDK1-4 on Tumor Energy Metabolism, Aggressiveness and Therapy Resistance. Front. Oncol. 2020, 10, 583217. [Google Scholar] [CrossRef]
- Kikani, C.K.; Verona, E.V.; Ryu, J.; Shen, Y.; Ye, Q.; Zheng, L.; Qian, Z.; Sakaue, H.; Nakamura, K.; Du, J.; et al. Proliferative and antiapoptotic signaling stimulated by nuclear-localized PDK1 results in oncogenesis. Sci. Signal. 2012, 5, ra80. [Google Scholar] [CrossRef]
- Wang, L.Y.; Hung, C.L.; Chen, Y.R.; Yang, J.C.; Wang, J.; Campbell, M.; Izumiya, Y.; Chen, H.W.; Wang, W.C.; Ann, D.K.; et al. KDM4A Coactivates E2F1 to Regulate the PDK-Dependent Metabolic Switch between Mitochondrial Oxidation and Glycolysis. Cell Rep. 2016, 16, 3016–3027. [Google Scholar] [CrossRef]
- Chen, J.; Guccini, I.; Di Mitri, D.; Brina, D.; Revandkar, A.; Sarti, M.; Pasquini, E.; Alajati, A.; Pinton, S.; Losa, M.; et al. Compartmentalized activities of the pyruvate dehydrogenase complex sustain lipogenesis in prostate cancer. Nat. Genet. 2018, 50, 219–228. [Google Scholar] [CrossRef]
- Li, Y.; Li, X.; Li, X.; Zhong, Y.; Ji, Y.; Yu, D.; Zhang, M.; Wen, J.G.; Zhang, H.; Goscinski, M.A.; et al. PDHA1 gene knockout in prostate cancer cells results in metabolic reprogramming towards greater glutamine dependence. Oncotarget 2016, 7, 53837–53852. [Google Scholar] [CrossRef]
- Li, W.; Qian, L.; Lin, J.; Huang, G.; Hao, N.; Wei, X.; Wang, W.; Liang, J. CD44 regulates prostate cancer proliferation, invasion and migration via PDK1 and PFKFB4. Oncotarget 2017, 8, 65143–65151. [Google Scholar] [CrossRef]
- Sheng, W.; Xu, W.; Ding, J.; Li, L.; You, X.; Wu, Y.; He, Q. Curcumol inhibits the malignant progression of prostate cancer and regulates the PDK1/AKT/mTOR pathway by targeting miR9. Oncol. Rep. 2021, 46, 246. [Google Scholar] [CrossRef]
- Oberhuber, M.; Pecoraro, M.; Rusz, M.; Oberhuber, G.; Wieselberg, M.; Haslinger, P.; Gurnhofer, E.; Schlederer, M.; Limberger, T.; Lagger, S.; et al. STAT3-dependent analysis reveals PDK4 as independent predictor of recurrence in prostate cancer. Mol. Syst. Biol. 2020, 16, e9247. [Google Scholar] [CrossRef]
- Jiang, X.; Guo, S.; Wang, S.; Zhang, Y.; Chen, H.; Wang, Y.; Liu, R.; Niu, Y.; Xu, Y. EIF4A3-Induced circARHGAP29 Promotes Aerobic Glycolysis in Docetaxel-Resistant Prostate Cancer through IGF2BP2/c-Myc/LDHA Signaling. Cancer Res. 2022, 82, 831–845. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, H.; Sumitomo, M.; Morinaga, S.; Kajikawa, K.; Kobayashi, I.; Nishikawa, G.; Kato, Y.; Watanabe, M.; Zennami, K.; Kanao, K.; et al. Targeting lactate dehydrogenaseA promotes docetaxelinduced cytotoxicity predominantly in castrationresistant prostate cancer cells. Oncol. Rep. 2019, 42, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Wang, B.J.; Fu, Y.K.; Huo, C.; Wang, Y.P.; Chen, B.C.; Liu, W.Y.; Tseng, J.C.; Jiang, S.S.; Sie, Z.L.; et al. Inhibition of KDM4C/c-Myc/LDHA signalling axis suppresses prostate cancer metastasis via interference of glycolytic metabolism. Clin. Transl. Med. 2022, 12, e764. [Google Scholar] [CrossRef]
- Kwon, O.K.; Bang, I.H.; Choi, S.Y.; Jeon, J.M.; Na, A.Y.; Gao, Y.; Cho, S.S.; Ki, S.H.; Choe, Y.; Lee, J.N.; et al. SIRT5 Is the desuccinylase of LDHA as novel cancer metastatic stimulator in aggressive prostate cancer. Genom. Proteom. Bioinform. 2022; in press. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, G.; Liu, Z.; Liu, S.; Cai, Z.; You, P.; Ke, Y.; Lai, L.; Huang, Y.; Gao, H.; et al. Aberrant FGFR Tyrosine Kinase Signaling Enhances the Warburg Effect by Reprogramming LDH Isoform Expression and Activity in Prostate Cancer. Cancer Res. 2018, 78, 4459–4470. [Google Scholar] [CrossRef]
- Andersen, S.; Solstad, O.; Moi, L.; Donnem, T.; Eilertsen, M.; Nordby, Y.; Ness, N.; Richardsen, E.; Busund, L.T.; Bremnes, R.M. Organized metabolic crime in prostate cancer: The coexpression of MCT1 in tumor and MCT4 in stroma is an independent prognosticator for biochemical failure. Urol. Oncol. 2015, 33, 338.e9–338.e17. [Google Scholar] [CrossRef]
- Sushentsev, N.; McLean, M.A.; Warren, A.Y.; Benjamin, A.J.V.; Brodie, C.; Frary, A.; Gill, A.B.; Jones, J.; Kaggie, J.D.; Lamb, B.W.; et al. Hyperpolarised (13)C-MRI identifies the emergence of a glycolytic cell population within intermediate-risk human prostate cancer. Nat. Commun. 2022, 13, 466. [Google Scholar] [CrossRef]
- Choi, S.Y.; Collins, C.C.; Gout, P.W.; Wang, Y. Cancer-generated lactic acid: A regulatory, immunosuppressive metabolite? J. Pathol. 2013, 230, 350–355. [Google Scholar] [CrossRef]
- Wang, J.X.; Choi, S.Y.C.; Niu, X.; Kang, N.; Xue, H.; Killam, J.; Wang, Y. Lactic Acid and an Acidic Tumor Microenvironment suppress Anticancer Immunity. Int. J. Mol. Sci 2020, 21, 8363. [Google Scholar] [CrossRef]
- Carreno, D.; Corro, N.; Torres-Estay, V.; Veliz, L.P.; Jaimovich, R.; Cisternas, P.; San Francisco, I.F.; Sotomayor, P.C.; Tanasova, M.; Inestrosa, N.C.; et al. Fructose and prostate cancer: Toward an integrated view of cancer cell metabolism. Prostate Cancer Prostatic Dis. 2019, 22, 49–58. [Google Scholar] [CrossRef]
- Frenette, G.; Thabet, M.; Sullivan, R. Polyol pathway in human epididymis and semen. J. Androl. 2006, 27, 233–239. [Google Scholar] [CrossRef]
- Szabo, Z.; Hamalainen, J.; Loikkanen, I.; Moilanen, A.M.; Hirvikoski, P.; Vaisanen, T.; Paavonen, T.K.; Vaarala, M.H. Sorbitol dehydrogenase expression is regulated by androgens in the human prostate. Oncol. Rep. 2010, 23, 1233–1239. [Google Scholar] [CrossRef]
- Carreno, D.V.; Corro, N.B.; Cerda-Infante, J.F.; Echeverria, C.E.; Asencio-Barria, C.A.; Torres-Estay, V.A.; Mayorga-Weber, G.A.; Rojas, P.A.; Veliz, L.P.; Cisternas, P.A.; et al. Dietary Fructose Promotes Prostate Cancer Growth. Cancer Res. 2021, 81, 2824–2832. [Google Scholar] [CrossRef]
- Echeverria, C.; Nualart, F.; Ferrada, L.; Smith, G.J.; Godoy, A.S. Hexose Transporters in Cancer: From Multifunctionality to Diagnosis and Therapy. Trends Endocrinol. Metab. 2021, 32, 198–211. [Google Scholar] [CrossRef]
- Xu, L.; Zhao, B.; Butler, W.; Xu, H.; Song, N.; Chen, X.; Spencer Hauck, J.; Gao, X.; Zhang, H.; Groth, J.; et al. Targeting glutamine metabolism network for the treatment of therapy-resistant prostate cancer. Oncogene 2022, 41, 1140–1154. [Google Scholar] [CrossRef]
- Strmiska, V.; Michalek, P.; Eckschlager, T.; Stiborova, M.; Adam, V.; Krizkova, S.; Heger, Z. Prostate cancer-specific hallmarks of amino acids metabolism: Towards a paradigm of precision medicine. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 248–258. [Google Scholar] [CrossRef]
- Fu, Y.M.; Lin, H.; Liu, X.; Fang, W.; Meadows, G.G. Cell death of prostate cancer cells by specific amino acid restriction depends on alterations of glucose metabolism. J. Cell. Physiol. 2010, 224, 491–500. [Google Scholar] [CrossRef]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef]
- Cardoso, H.J.; Figueira, M.I.; Vaz, C.V.; Carvalho, T.M.A.; Bras, L.A.; Madureira, P.A.; Oliveira, P.J.; Sardao, V.A.; Socorro, S. Glutaminolysis is a metabolic route essential for survival and growth of prostate cancer cells and a target of 5alpha-dihydrotestosterone regulation. Cell. Oncol. 2021, 44, 385–403. [Google Scholar] [CrossRef]
- Xu, L.; Yin, Y.; Li, Y.; Chen, X.; Chang, Y.; Zhang, H.; Liu, J.; Beasley, J.; McCaw, P.; Zhang, H.; et al. A glutaminase isoform switch drives therapeutic resistance and disease progression of prostate cancer. Proc. Natl. Acad. Sci. USA 2021, 118, e2012748118. [Google Scholar] [CrossRef]
- Malviya, G.; Patel, R.; Salji, M.; Martinez, R.S.; Repiscak, P.; Mui, E.; Champion, S.; Mrowinska, A.; Johnson, E.; AlRasheedi, M.; et al. 18F-Fluciclovine PET metabolic imaging reveals prostate cancer tumour heterogeneity associated with disease resistance to androgen deprivation therapy. EJNMMI Res. 2020, 10, 143. [Google Scholar] [CrossRef] [PubMed]
- Martinez, R.S.; Salji, M.J.; Rushworth, L.; Ntala, C.; Rodriguez Blanco, G.; Hedley, A.; Clark, W.; Peixoto, P.; Hervouet, E.; Renaude, E.; et al. SLFN5 Regulates LAT1-Mediated mTOR Activation in Castration-Resistant Prostate Cancer. Cancer Res. 2021, 81, 3664–3678. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Sakamoto, S.; Matsushima, J.; Kimura, T.; Ueda, T.; Mizokami, A.; Kanai, Y.; Ichikawa, T. Up-Regulation of LAT1 during Antiandrogen Therapy Contributes to Progression in Prostate Cancer Cells. J. Urol. 2016, 195, 1588–1597. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hardie, R.A.; Hoy, A.J.; van Geldermalsen, M.; Gao, D.; Fazli, L.; Sadowski, M.C.; Balaban, S.; Schreuder, M.; Nagarajah, R.; et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J. Pathol. 2015, 236, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Katt, W.P.; Lukey, M.J.; Cerione, R.A. A tale of two glutaminases: Homologous enzymes with distinct roles in tumorigenesis. Future Med. Chem. 2017, 9, 223–243. [Google Scholar] [CrossRef]
- Dorai, T.; Dorai, B.; Pinto, J.T.; Grasso, M.; Cooper, A.J.L. High Levels of Glutaminase II Pathway Enzymes in Normal and Cancerous Prostate Suggest a Role in ‘Glutamine Addiction’. Biomolecules 2019, 10, 2. [Google Scholar] [CrossRef]
- He, Z.; Gao, Y.; Li, T.; Yu, C.; Ou, L.; Luo, C. HepaCAMPIK3CA axis regulates the reprogramming of glutamine metabolism to inhibit prostate cancer cell proliferation. Int. J. Oncol. 2022, 60, 37. [Google Scholar] [CrossRef]
- Shao, Y.; Ye, G.; Ren, S.; Piao, H.L.; Zhao, X.; Lu, X.; Wang, F.; Ma, W.; Li, J.; Yin, P.; et al. Metabolomics and transcriptomics profiles reveal the dysregulation of the tricarboxylic acid cycle and related mechanisms in prostate cancer. Int. J. Cancer 2018, 143, 396–407. [Google Scholar] [CrossRef]
- Liu, W.; Le, A.; Hancock, C.; Lane, A.N.; Dang, C.V.; Fan, T.W.; Phang, J.M. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc. Natl. Acad. Sci. USA 2012, 109, 8983–8988. [Google Scholar] [CrossRef]
- Mukha, A.; Kahya, U.; Linge, A.; Chen, O.; Lock, S.; Lukiyanchuk, V.; Richter, S.; Alves, T.C.; Peitzsch, M.; Telychko, V.; et al. GLS-driven glutamine catabolism contributes to prostate cancer radiosensitivity by regulating the redox state, stemness and ATG5-mediated autophagy. Theranostics 2021, 11, 7844–7868. [Google Scholar] [CrossRef]
- Zhang, J.; Mao, S.; Guo, Y.; Wu, Y.; Yao, X.; Huang, Y. Inhibition of GLS suppresses proliferation and promotes apoptosis in prostate cancer. Biosci. Rep. 2019, 39, BSR20181826. [Google Scholar] [CrossRef]
- Zacharias, N.M.; McCullough, C.; Shanmugavelandy, S.; Lee, J.; Lee, Y.; Dutta, P.; McHenry, J.; Nguyen, L.; Norton, W.; Jones, L.W.; et al. Metabolic Differences in Glutamine Utilization Lead to Metabolic Vulnerabilities in Prostate Cancer. Sci. Rep. 2017, 7, 16159. [Google Scholar] [CrossRef]
- Peng, H.; Wang, Y.; Luo, W. Multifaceted role of branched-chain amino acid metabolism in cancer. Oncogene 2020, 39, 6747–6756. [Google Scholar] [CrossRef]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011, 481, 385–388. [Google Scholar] [CrossRef]
- Sachdeva, A.; Hart, C.A.; Carey, C.D.; Vincent, A.E.; Greaves, L.C.; Heer, R.; Oliveira, P.; Brown, M.D.; Clarke, N.W.; Turnbull, D.M. Automated quantitative high-throughput multiplex immunofluorescence pipeline to evaluate OXPHOS defects in formalin-fixed human prostate tissue. Sci. Rep. 2022, 12, 6660. [Google Scholar] [CrossRef]
- Burke, L.; Guterman, I.; Palacios Gallego, R.; Britton, R.G.; Burschowsky, D.; Tufarelli, C.; Rufini, A. The Janus-like role of proline metabolism in cancer. Cell Death Discov. 2020, 6, 104. [Google Scholar] [CrossRef]
- Tanner, J.J.; Fendt, S.M.; Becker, D.F. The Proline Cycle As a Potential Cancer Therapy Target. Biochemistry 2018, 57, 3433–3444. [Google Scholar] [CrossRef]
- Liu, W.; Hancock, C.N.; Fischer, J.W.; Harman, M.; Phang, J.M. Proline biosynthesis augments tumor cell growth and aerobic glycolysis: Involvement of pyridine nucleotides. Sci. Rep. 2015, 5, 17206. [Google Scholar] [CrossRef]
- Lin, D.; Ettinger, S.L.; Qu, S.; Xue, H.; Nabavi, N.; Choi, S.Y.C.; Bell, R.H.; Mo, F.; Haegert, A.M.; Gout, P.W.; et al. Metabolic heterogeneity signature of primary treatment-naive prostate cancer. Oncotarget 2017, 8, 25928–25941. [Google Scholar] [CrossRef]
- Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Amelio, I.; Cutruzzola, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Ganini, C.; Amelio, I.; Bertolo, R.; Candi, E.; Cappello, A.; Cipriani, C.; Mauriello, A.; Marani, C.; Melino, G.; Montanaro, M.; et al. Serine and one-carbon metabolisms bring new therapeutic venues in prostate cancer. Discov. Oncol. 2021, 12, 45. [Google Scholar] [CrossRef] [PubMed]
- Labuschagne, C.F.; van den Broek, N.J.; Mackay, G.M.; Vousden, K.H.; Maddocks, O.D. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 2014, 7, 1248–1258. [Google Scholar] [CrossRef] [PubMed]
- Corbin, J.M.; Ruiz-Echevarria, M.J. One-Carbon Metabolism in Prostate Cancer: The Role of Androgen Signaling. Int. J. Mol. Sci. 2016, 17, 1208. [Google Scholar] [CrossRef]
- Vazquez, A.; Tedeschi, P.M.; Bertino, J.R. Overexpression of the mitochondrial folate and glycine-serine pathway: A new determinant of methotrexate selectivity in tumors. Cancer Res. 2013, 73, 478–482. [Google Scholar] [CrossRef]
- Pallmann, N.; Deng, K.; Livgard, M.; Tesikova, M.; Jin, Y.; Frengen, N.S.; Kahraman, N.; Mokhlis, H.M.; Ozpolat, B.; Kildal, W.; et al. Stress-Mediated Reprogramming of Prostate Cancer One-Carbon Cycle Drives Disease Progression. Cancer Res. 2021, 81, 4066–4078. [Google Scholar] [CrossRef]
- Reina-Campos, M.; Linares, J.F.; Duran, A.; Cordes, T.; L’Hermitte, A.; Badur, M.G.; Bhangoo, M.S.; Thorson, P.K.; Richards, A.; Rooslid, T.; et al. Increased Serine and One-Carbon Pathway Metabolism by PKClambda/iota Deficiency Promotes Neuroendocrine Prostate Cancer. Cancer Cell 2019, 35, 385–400.E9. [Google Scholar] [CrossRef]
- Sreekumar, A.; Poisson, L.M.; Rajendiran, T.M.; Khan, A.P.; Cao, Q.; Yu, J.; Laxman, B.; Mehra, R.; Lonigro, R.J.; Li, Y.; et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 2009, 457, 910–914. [Google Scholar] [CrossRef]
- Reina-Campos, M.; Diaz-Meco, M.T.; Moscat, J. The complexity of the serine glycine one-carbon pathway in cancer. J. Cell Biol. 2020, 219, e201907022. [Google Scholar] [CrossRef]
- Casero, R.A., Jr.; Murray Stewart, T.; Pegg, A.E. Polyamine metabolism and cancer: Treatments, challenges and opportunities. Nat. Rev. Cancer 2018, 18, 681–695. [Google Scholar] [CrossRef]
- Bai, G.; Kasper, S.; Matusik, R.J.; Rennie, P.S.; Moshier, J.A.; Krongrad, A. Androgen regulation of the human ornithine decarboxylase promoter in prostate cancer cells. J. Androl. 1998, 19, 127–135. [Google Scholar]
- Zabala-Letona, A.; Arruabarrena-Aristorena, A.; Martin-Martin, N.; Fernandez-Ruiz, S.; Sutherland, J.D.; Clasquin, M.; Tomas-Cortazar, J.; Jimenez, J.; Torres, I.; Quang, P.; et al. mTORC1-dependent AMD1 regulation sustains polyamine metabolism in prostate cancer. Nature 2017, 547, 109–113. [Google Scholar] [CrossRef]
- Affronti, H.C.; Rowsam, A.M.; Pellerite, A.J.; Rosario, S.R.; Long, M.D.; Jacobi, J.J.; Bianchi-Smiraglia, A.; Boerlin, C.S.; Gillard, B.M.; Karasik, E.; et al. Pharmacological polyamine catabolism upregulation with methionine salvage pathway inhibition as an effective prostate cancer therapy. Nat. Commun. 2020, 11, 52. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef]
- Nagarajan, S.R.; Butler, L.M.; Hoy, A.J. The diversity and breadth of cancer cell fatty acid metabolism. Cancer Metab. 2021, 9, 2. [Google Scholar] [CrossRef]
- Smith, S.; Witkowski, A.; Joshi, A.K. Structural and functional organization of the animal fatty acid synthase. Prog. Lipid Res. 2003, 42, 289–317. [Google Scholar] [CrossRef]
- Schug, Z.T.; Peck, B.; Jones, D.T.; Zhang, Q.; Grosskurth, S.; Alam, I.S.; Goodwin, L.M.; Smethurst, E.; Mason, S.; Blyth, K.; et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 2015, 27, 57–71. [Google Scholar] [CrossRef]
- Schug, Z.T.; Vande Voorde, J.; Gottlieb, E. The metabolic fate of acetate in cancer. Nat. Rev. Cancer 2016, 16, 708–717. [Google Scholar] [CrossRef]
- Vavere, A.L.; Kridel, S.J.; Wheeler, F.B.; Lewis, J.S. 1-11C-acetate as a PET radiopharmaceutical for imaging fatty acid synthase expression in prostate cancer. J. Nucl. Med. 2008, 49, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Semenkovich, C.F.; Coleman, T.; Fiedorek, F.T., Jr. Human fatty acid synthase mRNA: Tissue distribution, genetic mapping, and kinetics of decay after glucose deprivation. J. Lipid Res. 1995, 36, 1507–1521. [Google Scholar] [CrossRef]
- Weiss, L.; Hoffmann, G.E.; Schreiber, R.; Andres, H.; Fuchs, E.; Korber, E.; Kolb, H.J. Fatty-acid biosynthesis in man, a pathway of minor importance. Purification, optimal assay conditions, and organ distribution of fatty-acid synthase. Biol. Chem. Hoppe Seyler 1986, 367, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Zadra, G.; Photopoulos, C.; Loda, M. The fat side of prostate cancer. Biochim. Biophys. Acta 2013, 1831, 1518–1532. [Google Scholar] [CrossRef]
- Rashid, A.; Pizer, E.S.; Moga, M.; Milgraum, L.Z.; Zahurak, M.; Pasternack, G.R.; Kuhajda, F.P.; Hamilton, S.R. Elevated expression of fatty acid synthase and fatty acid synthetic activity in colorectal neoplasia. Am. J. Pathol. 1997, 150, 201–208. [Google Scholar] [PubMed]
- Pizer, E.S.; Lax, S.F.; Kuhajda, F.P.; Pasternack, G.R.; Kurman, R.J. Fatty acid synthase expression in endometrial carcinoma: Correlation with cell proliferation and hormone receptors. Cancer 1998, 83, 528–537. [Google Scholar] [CrossRef]
- Gelebart, P.; Zak, Z.; Anand, M.; Belch, A.; Lai, R. Blockade of fatty acid synthase triggers significant apoptosis in mantle cell lymphoma. PLoS ONE 2012, 7, e33738. [Google Scholar] [CrossRef]
- Bhatt, A.P.; Jacobs, S.R.; Freemerman, A.J.; Makowski, L.; Rathmell, J.C.; Dittmer, D.P.; Damania, B. Dysregulation of fatty acid synthesis and glycolysis in non-Hodgkin lymphoma. Proc. Natl. Acad. Sci. USA 2012, 109, 11818–11823. [Google Scholar] [CrossRef]
- Wang, W.Q.; Zhao, X.Y.; Wang, H.Y.; Liang, Y. Increased fatty acid synthase as a potential therapeutic target in multiple myeloma. J. Zhejiang Univ. Sci. B 2008, 9, 441–447. [Google Scholar] [CrossRef]
- Cai, Y.; Wang, J.; Zhang, L.; Wu, D.; Yu, D.; Tian, X.; Liu, J.; Jiang, X.; Shen, Y.; Zhang, L.; et al. Expressions of fatty acid synthase and HER2 are correlated with poor prognosis of ovarian cancer. Med. Oncol. 2015, 32, 391. [Google Scholar] [CrossRef]
- Alo, P.L.; Visca, P.; Marci, A.; Mangoni, A.; Botti, C.; Di Tondo, U. Expression of fatty acid synthase (FAS) as a predictor of recurrence in stage I breast carcinoma patients. Cancer 1996, 77, 474–482. [Google Scholar] [CrossRef]
- Swinnen, J.V.; Roskams, T.; Joniau, S.; Van Poppel, H.; Oyen, R.; Baert, L.; Heyns, W.; Verhoeven, G. Overexpression of fatty acid synthase is an early and common event in the development of prostate cancer. Int. J. Cancer 2002, 98, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Shurbaji, M.S.; Kalbfleisch, J.H.; Thurmond, T.S. Immunohistochemical detection of a fatty acid synthase (OA-519) as a predictor of progression of prostate cancer. Hum. Pathol. 1996, 27, 917–921. [Google Scholar] [CrossRef]
- Abumrad, N.A.; el-Maghrabi, M.R.; Amri, E.Z.; Lopez, E.; Grimaldi, P.A. Cloning of a rat adipocyte membrane protein implicated in binding or transport of long-chain fatty acids that is induced during preadipocyte differentiation. Homology with human CD36. J. Biol. Chem. 1993, 268, 17665–17668. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, M.; Huang, W.; Chen, W.; Zhao, Y.; Schulte, M.L.; Volberding, P.; Gerbec, Z.; Zimmermann, M.T.; Zeighami, A.; et al. Mitochondrial Metabolic Reprogramming by CD36 Signaling Drives Macrophage Inflammatory Responses. Circ. Res. 2019, 125, 1087–1102. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, S.O.; Lennon, D.J.; Febbraio, M.; Podrez, E.A.; Hazen, S.L.; Silverstein, R.L. A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab. 2006, 4, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Wu, N.; Xu, B.; Chu, Y.; Li, X.; Su, S.; Chen, D.; Li, W.; Shi, Y.; Gao, X.; et al. Fatty acid-induced CD36 expression via O-GlcNAcylation drives gastric cancer metastasis. Theranostics 2019, 9, 5359–5373. [Google Scholar] [CrossRef]
- Lim, H.J.; Lee, S.; Lee, K.S.; Park, J.H.; Jang, Y.; Lee, E.J.; Park, H.Y. PPARgamma activation induces CD36 expression and stimulates foam cell like changes in rVSMCs. Prostaglandins Other Lipid Mediat. 2006, 80, 165–174. [Google Scholar] [CrossRef]
- Tao, N.; Wagner, S.J.; Lublin, D.M. CD36 is palmitoylated on both N- and C-terminal cytoplasmic tails. J. Biol. Chem. 1996, 271, 22315–22320. [Google Scholar] [CrossRef]
- Zhang, Y.; Dong, D.; Xu, X.; He, H.; Zhu, Y.; Lei, T.; Ou, H. Oxidized high-density lipoprotein promotes CD36 palmitoylation and increases lipid uptake in macrophages. J. Biol. Chem. 2022, 298, 102000. [Google Scholar] [CrossRef]
- Ye, H.; Adane, B.; Khan, N.; Sullivan, T.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Balys, M.; Ashton, J.M.; et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell 2016, 19, 23–37. [Google Scholar] [CrossRef]
- Feng, W.W.; Wilkins, O.; Bang, S.; Ung, M.; Li, J.; An, J.; Del Genio, C.; Canfield, K.; DiRenzo, J.; Wells, W.; et al. CD36-Mediated Metabolic Rewiring of Breast Cancer Cells Promotes Resistance to HER2-Targeted Therapies. Cell Rep. 2019, 29, 3405–3420.E5. [Google Scholar] [CrossRef]
- Martini, C.; DeNichilo, M.; King, D.P.; Cockshell, M.P.; Ebert, B.; Dale, B.; Ebert, L.M.; Woods, A.; Bonder, C.S. CD36 promotes vasculogenic mimicry in melanoma by mediating adhesion to the extracellular matrix. BMC Cancer 2021, 21, 765. [Google Scholar] [CrossRef]
- Watt, M.J.; Clark, A.K.; Selth, L.A.; Haynes, V.R.; Lister, N.; Rebello, R.; Porter, L.H.; Niranjan, B.; Whitby, S.T.; Lo, J.; et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci. Transl. Med. 2019, 11, eaau5758. [Google Scholar] [CrossRef]
- Zhao, Y.; Peng, X.; Baldwin, H.; Zhang, C.; Liu, Z.; Lu, X. Anti-androgen therapy induces transcriptomic reprogramming in metastatic castration-resistant prostate cancer in a murine model. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166151. [Google Scholar] [CrossRef]
- Rysman, E.; Brusselmans, K.; Scheys, K.; Timmermans, L.; Derua, R.; Munck, S.; Van Veldhoven, P.P.; Waltregny, D.; Daniels, V.W.; Machiels, J.; et al. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 2010, 70, 8117–8126. [Google Scholar] [CrossRef]
- Rae, C.; Fragkoulis, G.I.; Chalmers, A.J. Cytotoxicity and Radiosensitizing Activity of the Fatty Acid Synthase Inhibitor C75 Is Enhanced by Blocking Fatty Acid Uptake in Prostate Cancer Cells. Adv. Radiat. Oncol. 2020, 5, 994–1005. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef]
- Getiye, Y.; Rice, T.A.; Phillips, B.D.; Carrillo, D.F.; He, G. Dysregulated lipolysis and lipophagy in lipid droplets of macrophages from high fat diet-fed obese mice. J. Cell. Mol. Med. 2022, 26, 4825–4836. [Google Scholar] [CrossRef]
- Nomura, D.K.; Long, J.Z.; Niessen, S.; Hoover, H.S.; Ng, S.W.; Cravatt, B.F. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 2010, 140, 49–61. [Google Scholar] [CrossRef]
- Talley, J.T.; Mohiuddin, S.S. Biochemistry, Fatty Acid Oxidation; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Zha, S.; Ferdinandusse, S.; Hicks, J.L.; Denis, S.; Dunn, T.A.; Wanders, R.J.; Luo, J.; De Marzo, A.M.; Isaacs, W.B. Peroxisomal branched chain fatty acid beta-oxidation pathway is upregulated in prostate cancer. Prostate 2005, 63, 316–323. [Google Scholar] [CrossRef]
- Jiang, Z.; Woda, B.A.; Rock, K.L.; Xu, Y.; Savas, L.; Khan, A.; Pihan, G.; Cai, F.; Babcook, J.S.; Rathanaswami, P.; et al. P504S: A new molecular marker for the detection of prostate carcinoma. Am. J. Surg. Pathol. 2001, 25, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Houten, S.M.; Wanders, R.J.A.; Ranea-Robles, P. Metabolic interactions between peroxisomes and mitochondria with a special focus on acylcarnitine metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165720. [Google Scholar] [CrossRef] [PubMed]
- Jacob, F.; Monod, J. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 1961, 3, 318–356. [Google Scholar] [CrossRef]
- Marte, B. A visionary pair. Nat. Rev. Mol. Cell Biol. 2005, 6, S6. [Google Scholar] [CrossRef]
- Van der Knaap, J.A.; Verrijzer, C.P. Undercover: Gene control by metabolites and metabolic enzymes. Genes Dev. 2016, 30, 2345–2369. [Google Scholar] [CrossRef]
- Shimano, H.; Sato, R. SREBP-regulated lipid metabolism: Convergent physiology—Divergent pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef]
- Sans, C.L.; Satterwhite, D.J.; Stoltzman, C.A.; Breen, K.T.; Ayer, D.E. MondoA-Mlx heterodimers are candidate sensors of cellular energy status: Mitochondrial localization and direct regulation of glycolysis. Mol. Cell. Biol. 2006, 26, 4863–4871. [Google Scholar] [CrossRef]
- Wilde, B.R.; Ye, Z.; Lim, T.Y.; Ayer, D.E. Cellular acidosis triggers human MondoA transcriptional activity by driving mitochondrial ATP production. eLife 2019, 8, e40199. [Google Scholar] [CrossRef]
- Chen, C.L.; Hsu, S.C.; Chung, T.Y.; Chu, C.Y.; Wang, H.J.; Hsiao, P.W.; Yeh, S.D.; Ann, D.K.; Yen, Y.; Kung, H.J. Arginine is an epigenetic regulator targeting TEAD4 to modulate OXPHOS in prostate cancer cells. Nat. Commun. 2021, 12, 2398. [Google Scholar] [CrossRef]
- Dai, Z.; Ramesh, V.; Locasale, J.W. The evolving metabolic landscape of chromatin biology and epigenetics. Nat. Rev. Genet. 2020, 21, 737–753. [Google Scholar] [CrossRef]
- Campbell, S.L.; Wellen, K.E. Metabolic Signaling to the Nucleus in Cancer. Mol. Cell 2018, 71, 398–408. [Google Scholar] [CrossRef]
- Boon, R. Metabolic Fuel for Epigenetic: Nuclear Production Meets Local Consumption. Front. Genet. 2021, 12, 768996. [Google Scholar] [CrossRef]
- Meier, J.L. Metabolic mechanisms of epigenetic regulation. ACS Chem. Biol. 2013, 8, 2607–2621. [Google Scholar] [CrossRef]
- Ramazi, S.; Zahiri, J. Post-translational modifications in proteins: Resources, tools and prediction methods. Database 2021, 2021, baab012. [Google Scholar] [CrossRef]
- Akella, N.M.; Ciraku, L.; Reginato, M.J. Fueling the fire: Emerging role of the hexosamine biosynthetic pathway in cancer. BMC Biol. 2019, 17, 52. [Google Scholar] [CrossRef]
- Kaushik, A.K.; Shojaie, A.; Panzitt, K.; Sonavane, R.; Venghatakrishnan, H.; Manikkam, M.; Zaslavsky, A.; Putluri, V.; Vasu, V.T.; Zhang, Y.; et al. Inhibition of the hexosamine biosynthetic pathway promotes castration-resistant prostate cancer. Nat. Commun. 2016, 7, 11612. [Google Scholar] [CrossRef]
- Itkonen, H.M.; Urbanucci, A.; Martin, S.E.; Khan, A.; Mathelier, A.; Thiede, B.; Walker, S.; Mills, I.G. High OGT activity is essential for MYC-driven proliferation of prostate cancer cells. Theranostics 2019, 9, 2183–2197. [Google Scholar] [CrossRef]
- Choudhary, C.; Weinert, B.T.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. [Google Scholar] [CrossRef]
- Sivanand, S.; Viney, I.; Wellen, K.E. Spatiotemporal Control of Acetyl-CoA Metabolism in Chromatin Regulation. Trends Biochem. Sci. 2018, 43, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Amjad, S.; Nisar, S.; Bhat, A.A.; Shah, A.R.; Frenneaux, M.P.; Fakhro, K.; Haris, M.; Reddy, R.; Patay, Z.; Baur, J.; et al. Role of NAD+ in regulating cellular and metabolic signaling pathways. Mol. Metab. 2021, 49, 101195. [Google Scholar] [CrossRef] [PubMed]
- Hurtado-Bages, S.; Knobloch, G.; Ladurner, A.G.; Buschbeck, M. The taming of PARP1 and its impact on NAD+ metabolism. Mol. Metab. 2020, 38, 100950. [Google Scholar] [CrossRef]
- Huang, S.B.; Thapa, D.; Munoz, A.R.; Hussain, S.S.; Yang, X.; Bedolla, R.G.; Osmulski, P.; Gaczynska, M.E.; Lai, Z.; Chiu, Y.C.; et al. Androgen deprivation-induced elevated nuclear SIRT1 promotes prostate tumor cell survival by reactivation of AR signaling. Cancer Lett. 2021, 505, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.A.; Cole, P.A. The Chemical Biology of Reversible Lysine Post-translational Modifications. Cell Chem. Biol. 2020, 27, 953–969. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D.; Zhao, Y. Metabolic Regulation by Lysine Malonylation, Succinylation, and Glutarylation. Mol. Cell. Proteom. 2015, 14, 2308–2315. [Google Scholar] [CrossRef]
- Wu, X.; Tao, W.A. Uncovering ubiquitous protein lactylation. Nat. Methods 2022, 19, 793–794. [Google Scholar] [CrossRef]
- Izzo, L.T.; Wellen, K.E. Histone lactylation links metabolism and gene regulation. Nature 2019, 574, 492–493. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Chen, A.N.; Luo, Y.; Yang, Y.H.; Fu, J.T.; Geng, X.M.; Shi, J.P.; Yang, J. Lactylation, a Novel Metabolic Reprogramming Code: Current Status and Prospects. Front. Immunol. 2021, 12, 688910. [Google Scholar] [CrossRef]
- Gu, J.; Zhou, J.; Chen, Q.; Xu, X.; Gao, J.; Li, X.; Shao, Q.; Zhou, B.; Zhou, H.; Wei, S.; et al. Tumor metabolite lactate promotes tumorigenesis by modulating MOESIN lactylation and enhancing TGF-beta signaling in regulatory T cells. Cell Rep. 2022, 39, 110986. [Google Scholar] [CrossRef]
- Xiong, J.; He, J.; Zhu, J.; Pan, J.; Liao, W.; Ye, H.; Wang, H.; Song, Y.; Du, Y.; Cui, B.; et al. Lactylation-driven METTL3-mediated RNA m(6)A modification promotes immunosuppression of tumor-infiltrating myeloid cells. Mol. Cell 2022, 82, 1660–1677.E10. [Google Scholar] [CrossRef]
- Wan, N.; Wang, N.; Yu, S.; Zhang, H.; Tang, S.; Wang, D.; Lu, W.; Li, H.; Delafield, D.G.; Kong, Y.; et al. Cyclic immonium ion of lactyllysine reveals widespread lactylation in the human proteome. Nat. Methods 2022, 19, 854–864. [Google Scholar] [CrossRef]
- Moreno-Yruela, C.; Zhang, D.; Wei, W.; Baek, M.; Liu, W.; Gao, J.; Dankova, D.; Nielsen, A.L.; Bolding, J.E.; Yang, L.; et al. Class I histone deacetylases (HDAC1-3) are histone lysine delactylases. Sci. Adv. 2022, 8, eabi6696. [Google Scholar] [CrossRef]
- Yang, D.; Yin, J.; Shan, L.; Yi, X.; Zhang, W.; Ding, Y. Identification of lysine-lactylated substrates in gastric cancer cells. iScience 2022, 25, 104630. [Google Scholar] [CrossRef]
- Dai, S.K.; Liu, P.P.; Li, X.; Jiao, L.F.; Teng, Z.Q.; Liu, C.M. Dynamic profiling and functional interpretation of histone lysine crotonylation and lactylation during neural development. Development 2022, 149, dev200049. [Google Scholar] [CrossRef]
- Nian, F.; Qian, Y.; Xu, F.; Yang, M.; Wang, H.; Zhang, Z. LDHA promotes osteoblast differentiation through histone lactylation. Biochem. Biophys. Res. Commun. 2022, 615, 31–35. [Google Scholar] [CrossRef]
- Yang, J.; Luo, L.; Zhao, C.; Li, X.; Wang, Z.; Zeng, Z.; Yang, X.; Zheng, X.; Jie, H.; Kang, L.; et al. A Positive Feedback Loop between Inactive VHL-Triggered Histone Lactylation and PDGFRbeta Signaling Drives Clear Cell Renal Cell Carcinoma Progression. Int. J. Biol. Sci. 2022, 18, 3470–3483. [Google Scholar] [CrossRef] [PubMed]
- Mentch, S.J.; Locasale, J.W. One-carbon metabolism and epigenetics: Understanding the specificity. Ann. N. Y. Acad. Sci. 2016, 1363, 91–98. [Google Scholar] [CrossRef]
- Hwang, J.W.; Cho, Y.; Bae, G.U.; Kim, S.N.; Kim, Y.K. Protein arginine methyltransferases: Promising targets for cancer therapy. Exp. Mol. Med. 2021, 53, 788–808. [Google Scholar] [CrossRef]
- Kooistra, S.M.; Helin, K. Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell Biol. 2012, 13, 297–311. [Google Scholar] [CrossRef]
- Wang, L.-Y.; Guo, W.; Kim, K.; Pochampalli, M.; Hung, C.-L.; Izumiya, Y.; Kung, H.-J. Histone Demethylases in Prostate Cancer. In Nuclear Signaling Pathways and Targeting Transcription in Cancer; Kumar, R., Ed.; Springer: New York, NY, USA, 2014; pp. 373–397. [Google Scholar] [CrossRef]
- Wang, H.J.; Pochampalli, M.; Wang, L.Y.; Zou, J.X.; Li, P.S.; Hsu, S.C.; Wang, B.J.; Huang, S.H.; Yang, P.; Yang, J.C.; et al. KDM8/JMJD5 as a dual coactivator of AR and PKM2 integrates AR/EZH2 network and tumor metabolism in CRPC. Oncogene 2019, 38, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.D.; Jin, F.; Shin, S.; Oh, S.; Lightfoot, S.A.; Grande, J.P.; Johnson, A.J.; van Deursen, J.M.; Wren, J.D.; Janknecht, R. Histone demethylase JMJD2A drives prostate tumorigenesis through transcription factor ETV1. J. Clin. Investig. 2016, 126, 706–720. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Peng, G.; Sahgal, N.; Fazli, L.; Gleave, M.; Zhang, Y.; Hussain, A.; Qi, J. Regulation of c-Myc expression by the histone demethylase JMJD1A is essential for prostate cancer cell growth and survival. Oncogene 2016, 35, 2441–2452. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Zhang, F.; Xu, S.; Cui, X.; Hussain, A.; Fazli, L.; Gleave, M.; Dong, X.; Qi, J. Histone demethylase JMJD1A promotes alternative splicing of AR variant 7 (AR-V7) in prostate cancer cells. Proc. Natl. Acad Sci. USA 2018, 115, E4584–E4593. [Google Scholar] [CrossRef]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA methylation landscape of cancer. Trends Genet. 2021, 37, 1012–1027. [Google Scholar] [CrossRef]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef]
- Massie, C.E.; Mills, I.G.; Lynch, A.G. The importance of DNA methylation in prostate cancer development. J. Steroid Biochem. Mol. Biol. 2017, 166, 1–15. [Google Scholar] [CrossRef]
- Millar, D.S.; Ow, K.K.; Paul, C.L.; Russell, P.J.; Molloy, P.L.; Clark, S.J. Detailed methylation analysis of the glutathione S-transferase pi (GSTP1) gene in prostate cancer. Oncogene 1999, 18, 1313–1324. [Google Scholar] [CrossRef]
- Zhao, S.G.; Chen, W.S.; Li, H.; Foye, A.; Zhang, M.; Sjostrom, M.; Aggarwal, R.; Playdle, D.; Liao, A.; Alumkal, J.J.; et al. The DNA methylation landscape of advanced prostate cancer. Nat. Genet. 2020, 52, 778–789. [Google Scholar] [CrossRef]
- Chen, X.Y.; Zhang, J.; Zhu, J.S. The role of m(6)A RNA methylation in human cancer. Mol. Cancer 2019, 18, 103. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, J.; Gao, W.Q.; Yang, R. METTL14 promotes prostate tumorigenesis by inhibiting THBS1 via an m6A-YTHDF2-dependent mechanism. Cell Death. Discov. 2022, 8, 143. [Google Scholar] [CrossRef]
- Cotter, K.A.; Gallon, J.; Uebersax, N.; Rubin, P.; Meyer, K.D.; Piscuoglio, S.; Jaffrey, S.R.; Rubin, M.A. Mapping of m(6)A and Its Regulatory Targets in Prostate Cancer Reveals a METTL3-Low Induction of Therapy Resistance. Mol. Cancer Res. 2021, 19, 1398–1411. [Google Scholar] [CrossRef]
- Lopez, J.; Anazco-Guenkova, A.M.; Monteagudo-Garcia, O.; Blanco, S. Epigenetic and Epitranscriptomic Control in Prostate Cancer. Genes 2022, 13, 378. [Google Scholar] [CrossRef]
- Network, C.G.A.R. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef]
- Yong, C.; Stewart, G.D.; Frezza, C. Oncometabolites in renal cancer. Nat. Rev. Nephrol. 2020, 16, 156–172. [Google Scholar] [CrossRef]
- Vander Linden, C.; Corbet, C. Reconciling environment-mediated metabolic heterogeneity with the oncogene-driven cancer paradigm in precision oncology. Semin. Cell Dev. Biol. 2020, 98, 202–210. [Google Scholar] [CrossRef]
- Reina-Campos, M.; Moscat, J.; Diaz-Meco, M. Metabolism shapes the tumor microenvironment. Curr. Opin. Cell Biol. 2017, 48, 47–53. [Google Scholar] [CrossRef]
- Kim, J.; DeBerardinis, R.J. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019, 30, 434–446. [Google Scholar] [CrossRef]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic Interactions in the Tumor Microenvironment. Trends Cell Biol. 2017, 27, 863–875. [Google Scholar] [CrossRef]
- Dey, P.; Kimmelman, A.C.; DePinho, R.A. Metabolic Codependencies in the Tumor Microenvironment. Cancer Discov. 2021, 11, 1067–1081. [Google Scholar] [CrossRef]
- Li, F.; Simon, M.C. Cancer Cells Don’t Live Alone: Metabolic Communication within Tumor Microenvironments. Dev. Cell 2020, 54, 183–195. [Google Scholar] [CrossRef]
- Elia, I.; Haigis, M.C. Metabolites and the tumour microenvironment: From cellular mechanisms to systemic metabolism. Nat. Metab. 2021, 3, 21–32. [Google Scholar] [CrossRef]
- Cassim, S.; Pouyssegur, J. Tumor Microenvironment: A Metabolic Player that Shapes the Immune Response. Int. J. Mol. Sci. 2019, 21, 157. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.C.; Liu, P.S. Metabolic communication in tumors: A new layer of immunoregulation for immune evasion. J. Immunother. Cancer 2016, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Ge, R.; Wang, Z.; Cheng, L. Tumor microenvironment heterogeneity an important mediator of prostate cancer progression and therapeutic resistance. npj Precis. Oncol. 2022, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.N.; Heiden, M.G.V. Metabolism in the Tumor Microenvironment. Annu. Rev. Cancer Biol. 2020, 4, 17–40. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. eLife 2016, 5, e10250. [Google Scholar] [CrossRef]
- Lemberg, K.M.; Gori, S.S.; Tsukamoto, T.; Rais, R.; Slusher, B.S. Clinical development of metabolic inhibitors for oncology. J. Clin. Investig. 2022, 132, e148550. [Google Scholar] [CrossRef]
- Bader, J.E.; Voss, K.; Rathmell, J.C. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol. Cell 2020, 78, 1019–1033. [Google Scholar] [CrossRef]
- Stultz, J.; Fong, L. How to turn up the heat on the cold immune microenvironment of metastatic prostate cancer. Prostate Cancer Prostatic Dis. 2021, 24, 697–717. [Google Scholar] [CrossRef]
- Martin, A.M.; Nirschl, T.R.; Nirschl, C.J.; Francica, B.J.; Kochel, C.M.; van Bokhoven, A.; Meeker, A.K.; Lucia, M.S.; Anders, R.A.; DeMarzo, A.M.; et al. Paucity of PD-L1 expression in prostate cancer: Innate and adaptive immune resistance. Prostate Cancer Prostatic Dis. 2015, 18, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Melo, C.M.; Vidotto, T.; Chaves, L.P.; Lautert-Dutra, W.; Reis, R.B.D.; Squire, J.A. The Role of Somatic Mutations on the Immune Response of the Tumor Microenvironment in Prostate Cancer. Int. J. Mol. Sci. 2021, 22, 9550. [Google Scholar] [CrossRef] [PubMed]
- Guerra, L.; Bonetti, L.; Brenner, D. Metabolic Modulation of Immunity: A New Concept in Cancer Immunotherapy. Cell Rep. 2020, 32, 107848. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.G.; Kaymak, I.; Williams, K.S.; Ma, E.H.; Jones, R.G. Immunometabolism in the Tumor Microenvironment. Annu. Rev. Cancer Biol. 2021, 5, 137–159. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sanchez-Rodriguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef]
- Opitz, C.A.; Somarribas Patterson, L.F.; Mohapatra, S.R.; Dewi, D.L.; Sadik, A.; Platten, M.; Trump, S. The therapeutic potential of targeting tryptophan catabolism in cancer. Br. J. Cancer 2020, 122, 30–44. [Google Scholar] [CrossRef]
- Li, F.; Zhao, Z.; Zhang, Z.; Zhang, Y.; Guan, W. Tryptophan metabolism induced by TDO2 promotes prostatic cancer chemotherapy resistance in a AhR/c-Myc dependent manner. BMC Cancer 2021, 21, 1112. [Google Scholar] [CrossRef]
- Cheong, J.E.; Sun, L. Targeting the IDO1/TDO2-KYN-AhR Pathway for Cancer Immunotherapy—Challenges and Opportunities. Trends Pharm. Sci. 2018, 39, 307–325. [Google Scholar] [CrossRef]
- Wang, X.F.; Wang, H.S.; Wang, H.; Zhang, F.; Wang, K.F.; Guo, Q.; Zhang, G.; Cai, S.H.; Du, J. The role of indoleamine 2,3-dioxygenase (IDO) in immune tolerance: Focus on macrophage polarization of THP-1 cells. Cell. Immunol. 2014, 289, 42–48. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Malachowski, W.P.; DuHadaway, J.B.; Muller, A.J. Discovery of IDO1 Inhibitors: From Bench to Bedside. Cancer Res. 2017, 77, 6795–6811. [Google Scholar] [CrossRef]
- Jha, G.G.; Gupta, S.; Tagawa, S.T.; Koopmeiners, J.S.; Vivek, S.; Dudek, A.Z.; Cooley, S.A.; Blazar, B.R.; Miller, J.S. A phase II randomized, double-blind study of sipuleucel-T followed by IDO pathway inhibitor, indoximod, or placebo in the treatment of patients with metastatic castration resistant prostate cancer (mCRPC). J. Clin. Oncol. 2017, 35, 3066. [Google Scholar] [CrossRef]
- Luo, M.; Brooks, M.; Wicha, M.S. Asparagine and Glutamine: Co-conspirators Fueling Metastasis. Cell Metab. 2018, 27, 947–949. [Google Scholar] [CrossRef]
- Vettore, L.; Westbrook, R.L.; Tennant, D.A. New aspects of amino acid metabolism in cancer. Br. J. Cancer 2020, 122, 150–156. [Google Scholar] [CrossRef]
- Wei, Z.; Liu, X.; Cheng, C.; Yu, W.; Yi, P. Metabolism of Amino Acids in Cancer. Front. Cell Dev. Biol. 2020, 8, 603837. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Hui, S.; Ghergurovich, J.M.; Fan, J.; Intlekofer, A.M.; White, R.M.; Rabinowitz, J.D.; Thompson, C.B.; Zhang, J. As Extracellular Glutamine Levels Decline, Asparagine Becomes an Essential Amino Acid. Cell Metab. 2018, 27, 428–438.E5. [Google Scholar] [CrossRef]
- Linares, J.F.; Cordes, T.; Duran, A.; Reina-Campos, M.; Valencia, T.; Ahn, C.S.; Castilla, E.A.; Moscat, J.; Metallo, C.M.; Diaz-Meco, M.T. ATF4-Induced Metabolic Reprograming Is a Synthetic Vulnerability of the p62-Deficient Tumor Stroma. Cell Metab. 2017, 26, 817–829.E6. [Google Scholar] [CrossRef]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef]
- Rawat, D.; Chhonker, S.K.; Naik, R.A.; Mehrotra, A.; Trigun, S.K.; Koiri, R.K. Lactate as a signaling molecule: Journey from dead end product of glycolysis to tumor survival. Front. Biosci. 2019, 24, 366–381. [Google Scholar] [CrossRef]
- Wang, Z.H.; Peng, W.B.; Zhang, P.; Yang, X.P.; Zhou, Q. Lactate in the tumour microenvironment: From immune modulation to therapy. eBioMedicine 2021, 73, 103627. [Google Scholar] [CrossRef]
- Hayes, C.; Donohoe, C.L.; Davern, M.; Donlon, N.E. The oncogenic and clinical implications of lactate induced immunosuppression in the tumour microenvironment. Cancer Lett. 2021, 500, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Bottcher, M.; Baur, R.; Stoll, A.; Mackensen, A.; Mougiakakos, D. Linking Immunoevasion and Metabolic Reprogramming in B-Cell-Derived Lymphomas. Front. Oncol. 2020, 10, 594782. [Google Scholar] [CrossRef] [PubMed]
- Lotscher, J.; Balmer, M.L. Sensing between reactions—How the metabolic microenvironment shapes immunity. Clin. Exp. Immunol. 2019, 197, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, I.; Yogev, O.; Mattsson, J.; Schurich, A. The Metabolic Profile of Tumor and Virally Infected Cells Shapes Their Microenvironment Counteracting T Cell Immunity. Front. Immunol. 2019, 10, 2309. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, S.; Koyama, S.; Itahashi, K.; Tanegashima, T.; Lin, Y.T.; Togashi, Y.; Kamada, T.; Irie, T.; Okumura, G.; Kono, H.; et al. Lactic acid promotes PD-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell 2022, 40, 201–218.E9. [Google Scholar] [CrossRef]
- Deng, H.; Kan, A.; Lyu, N.; He, M.; Huang, X.; Qiao, S.; Li, S.; Lu, W.; Xie, Q.; Chen, H.; et al. Tumor-derived lactate inhibit the efficacy of lenvatinib through regulating PD-L1 expression on neutrophil in hepatocellular carcinoma. J. Immunother. Cancer 2021, 9, e002305. [Google Scholar] [CrossRef]
- Watson, M.J.; Vignali, P.D.A.; Mullett, S.J.; Overacre-Delgoffe, A.E.; Peralta, R.M.; Grebinoski, S.; Menk, A.V.; Rittenhouse, N.L.; DePeaux, K.; Whetstone, R.D.; et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature 2021, 591, 645–651. [Google Scholar] [CrossRef]
- Angelin, A.; Gil-de-Gomez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J., 3rd; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293.E7. [Google Scholar] [CrossRef]
- Ho, P.C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef]
- Kedia-Mehta, N.; Finlay, D.K. Competition for nutrients and its role in controlling immune responses. Nat. Commun. 2019, 10, 2123. [Google Scholar] [CrossRef]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef] [PubMed]
- Comito, G.; Iscaro, A.; Bacci, M.; Morandi, A.; Ippolito, L.; Parri, M.; Montagnani, I.; Raspollini, M.R.; Serni, S.; Simeoni, L.; et al. Lactate modulates CD4(+) T-cell polarization and induces an immunosuppressive environment, which sustains prostate carcinoma progression via TLR8/miR21 axis. Oncogene 2019, 38, 3681–3695. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Filipazzi, P.; Grioni, M.; Iero, M.; De Milito, A.; Ricupito, A.; Cova, A.; Canese, R.; Jachetti, E.; Rossetti, M.; et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012, 72, 2746–2756. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, I.; Prasad, P.D.; Thangaraju, M.; Manicassamy, S. Lactate-Dependent Regulation of Immune Responses by Dendritic Cells and Macrophages. Front. Immunol. 2021, 12, 691134. [Google Scholar] [CrossRef]
- Brown, T.P.; Bhattacharjee, P.; Ramachandran, S.; Sivaprakasam, S.; Ristic, B.; Sikder, M.O.F.; Ganapathy, V. The lactate receptor GPR81 promotes breast cancer growth via a paracrine mechanism involving antigen-presenting cells in the tumor microenvironme.ent. Oncogene 2020, 39, 3292–3304. [Google Scholar] [CrossRef]
- Hoque, R.; Farooq, A.; Ghani, A.; Gorelick, F.; Mehal, W.Z. Lactate reduces liver and pancreatic injury in Toll-like receptor- and inflammasome-mediated inflammation via GPR81-mediated suppression of innate immunity. Gastroenterology 2014, 146, 1763–1774. [Google Scholar] [CrossRef]
- Errea, A.; Cayet, D.; Marchetti, P.; Tang, C.; Kluza, J.; Offermanns, S.; Sirard, J.C.; Rumbo, M. Lactate Inhibits the Pro-Inflammatory Response and Metabolic Reprogramming in Murine Macrophages in a GPR81-Independent Manner. PLoS ONE 2016, 11, e0163694. [Google Scholar] [CrossRef]
- Raychaudhuri, D.; Bhattacharya, R.; Sinha, B.P.; Liu, C.S.C.; Ghosh, A.R.; Rahaman, O.; Bandopadhyay, P.; Sarif, J.; D’Rozario, R.; Paul, S.; et al. Lactate Induces Pro-tumor Reprogramming in Intratumoral Plasmacytoid Dendritic Cells. Front. Immunol. 2019, 10, 1878. [Google Scholar] [CrossRef]
- Chen, P.; Zuo, H.; Xiong, H.; Kolar, M.J.; Chu, Q.; Saghatelian, A.; Siegwart, D.J.; Wan, Y. Gpr132 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proc. Natl. Acad Sci. USA 2017, 114, 580–585. [Google Scholar] [CrossRef]
- Gottfried, E.; Kunz-Schughart, L.A.; Ebner, S.; Mueller-Klieser, W.; Hoves, S.; Andreesen, R.; Mackensen, A.; Kreutz, M. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 2006, 107, 2013–2021. [Google Scholar] [CrossRef]
- Dietl, K.; Renner, K.; Dettmer, K.; Timischl, B.; Eberhart, K.; Dorn, C.; Hellerbrand, C.; Kastenberger, M.; Kunz-Schughart, L.A.; Oefner, P.J.; et al. Lactic acid and acidification inhibit TNF secretion and glycolysis of human monocytes. J. Immunol. 2010, 184, 1200–1209. [Google Scholar] [CrossRef]
- Zhang, L.; Li, S. Lactic acid promotes macrophage polarization through MCT-HIF1alpha signaling in gastric cancer. Exp. Cell Res. 2020, 388, 111846. [Google Scholar] [CrossRef]
- El-Kenawi, A.; Gatenbee, C.; Robertson-Tessi, M.; Bravo, R.; Dhillon, J.; Balagurunathan, Y.; Berglund, A.; Vishvakarma, N.; Ibrahim-Hashim, A.; Choi, J.; et al. Acidity promotes tumour progression by altering macrophage phenotype in prostate cancer. Br. J. Cancer 2019, 121, 556–566. [Google Scholar] [CrossRef]
- Sangsuwan, R.; Thuamsang, B.; Pacifici, N.; Allen, R.; Han, H.; Miakicheva, S.; Lewis, J.S. Lactate Exposure Promotes Immunosuppressive Phenotypes in Innate Immune Cells. Cell. Mol. Bioeng. 2020, 13, 541–557. [Google Scholar] [CrossRef]
- Caronni, N.; Simoncello, F.; Stafetta, F.; Guarnaccia, C.; Ruiz-Moreno, J.S.; Opitz, B.; Galli, T.; Proux-Gillardeaux, V.; Benvenuti, F. Downregulation of Membrane Trafficking Proteins and Lactate Conditioning Determine Loss of Dendritic Cell Function in Lung Cancer. Cancer Res. 2018, 78, 1685–1699. [Google Scholar] [CrossRef]
- Bae, S.; Park, P.S.U.; Lee, Y.; Mun, S.H.; Giannopoulou, E.; Fujii, T.; Lee, K.P.; Violante, S.N.; Cross, J.R.; Park-Min, K.H. MYC-mediated early glycolysis negatively regulates proinflammatory responses by controlling IRF4 in inflammatory macrophages. Cell Rep. 2021, 35, 109264. [Google Scholar] [CrossRef]
- Tu, C.E.; Hu, Y.; Zhou, P.; Guo, X.; Gu, C.; Zhang, Y.; Li, A.; Liu, S. Lactate and TGF-beta antagonistically regulate inflammasome activation in the tumor microenvironment. J. Cell. Physiol. 2021, 236, 4528–4537. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, G.; Xu, Z.G.; Tu, H.; Hu, F.; Dai, J.; Chang, Y.; Chen, Y.; Lu, Y.; Zeng, H.; et al. Lactate Is a Natural Suppressor of RLR Signaling by Targeting MAVS. Cell 2019, 178, 176–189.E15. [Google Scholar] [CrossRef]
- Wang, X.; Liu, H.; Ni, Y.; Shen, P.; Han, X. Lactate shuttle: From substance exchange to regulatory mechanism. Hum. Cell 2022, 35, 1–14. [Google Scholar] [CrossRef]
- Draoui, N.; Feron, O. Lactate shuttles at a glance: From physiological paradigms to anti-cancer treatments. Dis. Models Mech. 2011, 4, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Pertega-Gomes, N.; Vizcaino, J.R.; Attig, J.; Jurmeister, S.; Lopes, C.; Baltazar, F. A lactate shuttle system between tumour and stromal cells is associated with poor prognosis in prostate cancer. BMC Cancer 2014, 14, 352. [Google Scholar] [CrossRef] [PubMed]
- Sanita, P.; Capulli, M.; Teti, A.; Galatioto, G.P.; Vicentini, C.; Chiarugi, P.; Bologna, M.; Angelucci, A. Tumor-stroma metabolic relationship based on lactate shuttle can sustain prostate cancer progression. BMC Cancer 2014, 14, 154. [Google Scholar] [CrossRef] [PubMed]
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; De Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012, 72, 5130–5140. [Google Scholar] [CrossRef] [PubMed]
- De la Cruz-Lopez, K.G.; Castro-Munoz, L.J.; Reyes-Hernandez, D.O.; Garcia-Carranca, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [PubMed]
- Eheman, C.; Henley, S.J.; Ballard-Barbash, R.; Jacobs, E.J.; Schymura, M.J.; Noone, A.M.; Pan, L.; Anderson, R.N.; Fulton, J.E.; Kohler, B.A.; et al. Annual Report to the Nation on the status of cancer, 1975–2008, featuring cancers associated with excess weight and lack of sufficient physical activity. Cancer 2012, 118, 2338–2366. [Google Scholar] [CrossRef]
- Laurent, V.; Guerard, A.; Mazerolles, C.; Le Gonidec, S.; Toulet, A.; Nieto, L.; Zaidi, F.; Majed, B.; Garandeau, D.; Socrier, Y.; et al. Periprostatic adipocytes act as a driving force for prostate cancer progression in obesity. Nat. Commun. 2016, 7, 10230. [Google Scholar] [CrossRef]
- Cao, Y. Adipocyte and lipid metabolism in cancer drug resistance. J. Clin. Investig. 2019, 129, 3006–3017. [Google Scholar] [CrossRef]
- Zimmermann, R.; Lass, A.; Haemmerle, G.; Zechner, R. Fate of fat: The role of adipose triglyceride lipase in lipolysis. Biochim. Biophys. Acta 2009, 1791, 494–500. [Google Scholar] [CrossRef]
- Berndt, J.; Kovacs, P.; Ruschke, K.; Kloting, N.; Fasshauer, M.; Schon, M.R.; Korner, A.; Stumvoll, M.; Bluher, M. Fatty acid synthase gene expression in human adipose tissue: Association with obesity and type 2 diabetes. Diabetologia 2007, 50, 1472–1480. [Google Scholar] [CrossRef]
- Swinnen, J.V.; Esquenet, M.; Goossens, K.; Heyns, W.; Verhoeven, G. Androgens stimulate fatty acid synthase in the human prostate cancer cell line LNCaP. Cancer Res. 1997, 57, 1086–1090. [Google Scholar] [PubMed]
- Tousignant, K.D.; Rockstroh, A.; Poad, B.L.J.; Talebi, A.; Young, R.S.E.; Taherian Fard, A.; Gupta, R.; Zang, T.; Wang, C.; Lehman, M.L.; et al. Therapy-induced lipid uptake and remodeling underpin ferroptosis hypersensitivity in prostate cancer. Cancer Metab. 2020, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Lochner, M.; Berod, L.; Sparwasser, T. Fatty acid metabolism in the regulation of T cell function. Trends Immunol. 2015, 36, 81–91. [Google Scholar] [CrossRef]
- Harris, R.E. Cyclooxygenase-2 (cox-2) blockade in the chemoprevention of cancers of the colon, breast, prostate, and lung. Inflammopharmacology 2009, 17, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Pruthi, R.S.; Derksen, J.E.; Moore, D. A pilot study of use of the cyclooxygenase-2 inhibitor celecoxib in recurrent prostate cancer after definitive radiation therapy or radical prostatectomy. BJU Int. 2004, 93, 275–278. [Google Scholar] [CrossRef]
- Bottcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis e Sousa, C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.E14. [Google Scholar] [CrossRef]
- Gupta, S.; Srivastava, M.; Ahmad, N.; Sakamoto, K.; Bostwick, D.G.; Mukhtar, H. Lipoxygenase-5 is overexpressed in prostate adenocarcinoma. Cancer 2001, 91, 737–743. [Google Scholar] [CrossRef]
- Joshua, A.M.; Armstrong, A.; Crumbaker, M.; Scher, H.I.; de Bono, J.; Tombal, B.; Hussain, M.; Sternberg, C.N.; Gillessen, S.; Carles, J.; et al. Statin and metformin use and outcomes in patients with castration-resistant prostate cancer treated with enzalutamide: A meta-analysis of AFFIRM, PREVAIL and PROSPER. Eur. J. Cancer 2022, 170, 285–295. [Google Scholar] [CrossRef]
- Khan, S.; Chang, S.H.; Hicks, V.; Wang, M.; Grubb, R.L., 3rd; Drake, B.F. Improved survival with post-diagnostic metformin and statin use in a racially diverse cohort of US Veterans with advanced prostate cancer. Prostate Cancer Prostatic Dis. 2021; online ahead of print. [Google Scholar] [CrossRef]
- Swinney, D.C.; Anthony, J. How were new medicines discovered? Nat. Rev. Drug Discov. 2011, 10, 507–519. [Google Scholar] [CrossRef]
- Zheng, W.; Thorne, N.; McKew, J.C. Phenotypic screens as a renewed approach for drug discovery. Drug Discov. Today 2013, 18, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Schenone, M.; Dancik, V.; Wagner, B.K.; Clemons, P.A. Target identification and mechanism of action in chemical biology and drug discovery. Nat. Chem. Biol. 2013, 9, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Wiriyasermkul, P.; Jin, C.; Quan, L.; Ohgaki, R.; Okuda, S.; Kusakizako, T.; Nishizawa, T.; Oda, K.; Ishitani, R.; et al. Cryo-EM structure of the human L-type amino acid transporter 1 in complex with glycoprotein CD98hc. Nat. Struct. Mol. Biol. 2019, 26, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Jeckelmann, J.M.; Fotiadis, D. Sub-Nanometer Cryo-EM Density Map of the Human Heterodimeric Amino Acid Transporter 4F2hc-LAT2. Int. J. Mol. Sci. 2020, 21, 7094. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Jiang, X.; Zhang, S.; Zhu, A.; Yuan, Y.; Xu, H.; Lei, J.; Yan, C. Structural basis of human monocarboxylate transporter 1 inhibition by anti-cancer drug candidates. Cell 2021, 184, 370–383.E13. [Google Scholar] [CrossRef]
- Zhang, B.; Jin, Q.; Xu, L.; Li, N.; Meng, Y.; Chang, S.; Zheng, X.; Wang, J.; Chen, Y.; Neculai, D.; et al. Cooperative transport mechanism of human monocarboxylate transporter 2. Nat. Commun. 2020, 11, 2429. [Google Scholar] [CrossRef]
- Yuan, Y.; Kong, F.; Xu, H.; Zhu, A.; Yan, N.; Yan, C. Cryo-EM structure of human glucose transporter GLUT4. Nat. Commun. 2022, 13, 2671. [Google Scholar] [CrossRef]
- Ban, F.; Dalal, K.; Li, H.; LeBlanc, E.; Rennie, P.S.; Cherkasov, A. Best Practices of Computer-Aided Drug Discovery: Lessons Learned from the Development of a Preclinical Candidate for Prostate Cancer with a New Mechanism of Action. J. Chem. Inf. Model. 2017, 57, 1018–1028. [Google Scholar] [CrossRef]
- Ton, A.T.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol. Inf. 2020, 39, e2000028. [Google Scholar] [CrossRef]
- Ekins, S. The Next Era: Deep Learning in Pharmaceutical Research. Pharm. Res. 2016, 33, 2594–2603. [Google Scholar] [CrossRef]
- Fernandez, M.; Ban, F.; Woo, G.; Hsing, M.; Yamazaki, T.; LeBlanc, E.; Rennie, P.S.; Welch, W.J.; Cherkasov, A. Toxic Colors: The Use of Deep Learning for Predicting Toxicity of Compounds Merely from Their Graphic Images. J. Chem. Inf. Model. 2018, 58, 1533–1543. [Google Scholar] [CrossRef]
- Gentile, F.; Agrawal, V.; Hsing, M.; Ton, A.T.; Ban, F.; Norinder, U.; Gleave, M.E.; Cherkasov, A. Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery. ACS Cent. Sci. 2020, 6, 939–949. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Santos-Martins, D.; Solis-Vasquez, L.; Tillack, A.F.; Sanner, M.F.; Koch, A.; Forli, S. Accelerating AutoDock4 with GPUs and Gradient-Based Local Search. J. Chem. Theory Comput. 2021, 17, 1060–1073. [Google Scholar] [CrossRef]
- Wang, L.; Wu, Y.; Deng, Y.; Kim, B.; Pierce, L.; Krilov, G.; Lupyan, D.; Robinson, S.; Dahlgren, M.K.; Greenwood, J.; et al. Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field. J. Am. Chem. Soc. 2015, 137, 2695–2703. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Shi, R.; Pan, P.; Lv, R.; Ma, C.; Wu, E.; Guo, R.; Zhao, Z.; Song, H.; Zhou, J.; Liu, Y.; et al. High-throughput glycolytic inhibitor discovery targeting glioblastoma by graphite dots-assisted LDI mass spectrometry. Sci. Adv. 2022, 8, eabl4923. [Google Scholar] [CrossRef]
- Wagner, B.K.; Schreiber, S.L. The Power of Sophisticated Phenotypic Screening and Modern Mechanism-of-Action Methods. Cell Chem. Biol. 2016, 23, 3–9. [Google Scholar] [CrossRef]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef]
- Boonekamp, F.J.; Knibbe, E.; Vieira-Lara, M.A.; Wijsman, M.; Luttik, M.A.H.; van Eunen, K.; Ridder, M.D.; Bron, R.; Almonacid Suarez, A.M.; van Rijn, P.; et al. Full humanization of the glycolytic pathway in Saccharomyces cerevisiae. Cell Rep. 2022, 39, 111010. [Google Scholar] [CrossRef]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L.; et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Shan, W.; Zhou, Y.; Tam, K.Y. The development of small-molecule inhibitors targeting hexokinase 2. Drug Discov. Today 2022, 27, 2574–2585. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 2011, 208, 313–326. [Google Scholar] [CrossRef]
- Mehta, M.M.; Weinberg, S.E.; Steinert, E.M.; Chhiba, K.; Martinez, C.A.; Gao, P.; Perlman, H.R.; Bryce, P.; Hay, N.; Chandel, N.S. Hexokinase 2 is dispensable for T cell-dependent immunity. Cancer Metab. 2018, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Herschman, H.R. A Tumor Agnostic Therapeutic Strategy for Hexokinase 1-Null/Hexokinase 2-Positive Cancers. Cancer Res. 2019, 79, 5907–5914. [Google Scholar] [CrossRef]
- Xu, S.; Zhou, T.; Doh, H.M.; Trinh, K.R.; Catapang, A.; Lee, J.T.; Braas, D.; Bayley, N.A.; Yamada, R.E.; Vasuthasawat, A.; et al. An HK2 Antisense Oligonucleotide Induces Synthetic Lethality in HK1(-)HK2(+) Multiple Myeloma. Cancer Res. 2019, 79, 2748–2760. [Google Scholar] [CrossRef]
- Jiang, J.; Walsh, M.J.; Brimacombe, K.R.; Anastasiou, D.; Yu, Y.; Israelsen, W.J.; Hong, B.S.; Tempel, W.; Dimov, S.; Veith, H.; et al. ML265: A potent PKM2 activator induces tetramerization and reduces tumor formation and size in a mouse xenograft model. In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information: Bethesda, MD, USA, 2010. [Google Scholar]
- Anastasiou, D.; Yu, Y.; Israelsen, W.J.; Jiang, J.K.; Boxer, M.B.; Hong, B.S.; Tempel, W.; Dimov, S.; Shen, M.; Jha, A.; et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat. Chem. Biol. 2012, 8, 839–847. [Google Scholar] [CrossRef]
- Lin, Y.H.; Satani, N.; Hammoudi, N.; Yan, V.C.; Barekatain, Y.; Khadka, S.; Ackroyd, J.J.; Georgiou, D.K.; Pham, C.D.; Arthur, K.; et al. An enolase inhibitor for the targeted treatment of ENO1-deleted cancers. Nat. Metab. 2020, 2, 1413–1426. [Google Scholar] [CrossRef]
- Muller, F.L.; Colla, S.; Aquilanti, E.; Manzo, V.E.; Genovese, G.; Lee, J.; Eisenson, D.; Narurkar, R.; Deng, P.; Nezi, L.; et al. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature 2012, 488, 337–342. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer 2014, 13, 890–901. [Google Scholar] [CrossRef]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.M.; Oh, M.H.; Sun, I.H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J.; et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, 366, 1013–1021. [Google Scholar] [CrossRef]
- Kridel, S.J.; Axelrod, F.; Rozenkrantz, N.; Smith, J.W. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res. 2004, 64, 2070–2075. [Google Scholar] [CrossRef]
- Pizer, E.S.; Jackisch, C.; Wood, F.D.; Pasternack, G.R.; Davidson, N.E.; Kuhajda, F.P. Inhibition of fatty acid synthesis induces programmed cell death in human breast cancer cells. Cancer Res. 1996, 56, 2745–2747. [Google Scholar]
- Kuhajda, F.P.; Pizer, E.S.; Li, J.N.; Mani, N.S.; Frehywot, G.L.; Townsend, C.A. Synthesis and antitumor activity of an inhibitor of fatty acid synthase. Proc. Natl. Acad Sci. USA 2000, 97, 3450–3454. [Google Scholar] [CrossRef]
- Loftus, T.M.; Jaworsky, D.E.; Frehywot, G.L.; Townsend, C.A.; Ronnett, G.V.; Lane, M.D.; Kuhajda, F.P. Reduced food intake and body weight in mice treated with fatty acid synthase inhibitors. Science 2000, 288, 2379–2381. [Google Scholar] [CrossRef]
- Thupari, J.N.; Landree, L.E.; Ronnett, G.V.; Kuhajda, F.P. C75 increases peripheral energy utilization and fatty acid oxidation in diet-induced obesity. Proc. Natl. Acad Sci. USA 2002, 99, 9498–9502. [Google Scholar] [CrossRef]
- Hardwicke, M.A.; Rendina, A.R.; Williams, S.P.; Moore, M.L.; Wang, L.; Krueger, J.A.; Plant, R.N.; Totoritis, R.D.; Zhang, G.; Briand, J.; et al. A human fatty acid synthase inhibitor binds beta-ketoacyl reductase in the keto-substrate site. Nat. Chem. Biol. 2014, 10, 774–779. [Google Scholar] [CrossRef]
- Alwarawrah, Y.; Hughes, P.; Loiselle, D.; Carlson, D.A.; Darr, D.B.; Jordan, J.L.; Xiong, J.; Hunter, L.M.; Dubois, L.G.; Thompson, J.W.; et al. Fasnall, a Selective FASN Inhibitor, Shows Potent Anti-tumor Activity in the MMTV-Neu Model of HER2(+) Breast Cancer. Cell Chem. Biol. 2016, 23, 678–688. [Google Scholar] [CrossRef]
- Kley, J.T.; Mack, J.; Hamilton, B.; Scheuerer, S.; Redemann, N. Discovery of BI 99179, a potent and selective inhibitor of type I fatty acid synthase with central exposure. Bioorg. Med. Chem. Lett. 2011, 21, 5924–5927. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G.; Infante, J.; Arkenau, H.T.; Patel, M.R.; Dean, E.; Borazanci, E.; Brenner, A.; Cook, N.; Lopez, J.; Pant, S.; et al. First-in-human study of the safety, pharmacokinetics, and pharmacodynamics of first-in-class fatty acid synthase inhibitor TVB-2640 alone and with a taxane in advanced tumors. eClinicalMedicine 2021, 34, 100797. [Google Scholar] [CrossRef] [PubMed]
- Bollag, G.; Tsai, J.; Zhang, J.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Vemurafenib: The first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 2012, 11, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Cross, D.; Janne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef] [PubMed]
- Golub, D.; Iyengar, N.; Dogra, S.; Wong, T.; Bready, D.; Tang, K.; Modrek, A.S.; Placantonakis, D.G. Mutant Isocitrate Dehydrogenase Inhibitors as Targeted Cancer Therapeutics. Front. Oncol. 2019, 9, 417. [Google Scholar] [CrossRef]
- Andronesi, O.C. Precision oncology in the era of radiogenomics: The case of D-2HG as an imaging biomarker for mutant IDH gliomas. Neuro Oncol. 2018, 20, 865–867. [Google Scholar] [CrossRef]
- Cadoux-Hudson, T.; Schofield, C.J.; McCullagh, J.S.O. Isocitrate dehydrogenase gene variants in cancer and their clinical significance. Biochem. Soc. Trans. 2021, 49, 2561–2572. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Mullard, A. What’s next for the synthetic lethality drug discovery engine? Nat. Rev. Drug Discov. 2022, 21, 477–479. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Wilson, F.H.; Ruth, J.R.; Paulk, J.; Tsherniak, A.; Marlow, S.E.; Vazquez, F.; Weir, B.A.; Fitzgerald, M.E.; Tanaka, M.; et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 2016, 351, 1214–1218. [Google Scholar] [CrossRef]
- Mavrakis, K.J.; McDonald, E.R., 3rd; Schlabach, M.R.; Billy, E.; Hoffman, G.R.; deWeck, A.; Ruddy, D.A.; Venkatesan, K.; Yu, J.; McAllister, G.; et al. Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science 2016, 351, 1208–1213. [Google Scholar] [CrossRef]
- Ito, T.; Young, M.J.; Li, R.; Jain, S.; Wernitznig, A.; Krill-Burger, J.M.; Lemke, C.T.; Monducci, D.; Rodriguez, D.J.; Chang, L.; et al. Paralog knockout profiling identifies DUSP4 and DUSP6 as a digenic dependence in MAPK pathway-driven cancers. Nat. Genet. 2021, 53, 1664–1672. [Google Scholar] [CrossRef]
- Butler, M.; van der Meer, L.T.; van Leeuwen, F.N. Amino Acid Depletion Therapies: Starving Cancer Cells to Death. Trends Endocrinol. Metab. 2021, 32, 367–381. [Google Scholar] [CrossRef]
- Batool, T.; Makky, E.A.; Jalal, M.; Yusoff, M.M. A Comprehensive Review on L-Asparaginase and Its Applications. Appl. Biochem. Biotechnol. 2016, 178, 900–923. [Google Scholar] [CrossRef]
- Matos, A.; Carvalho, M.; Bicho, M.; Ribeiro, R. Arginine and Arginases Modulate Metabolism, Tumor Microenvironment and Prostate Cancer Progression. Nutrients 2021, 13, 4503. [Google Scholar] [CrossRef]
- Cramer, S.L.; Saha, A.; Liu, J.; Tadi, S.; Tiziani, S.; Yan, W.; Triplett, K.; Lamb, C.; Alters, S.E.; Rowlinson, S.; et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat. Med. 2017, 23, 120–127. [Google Scholar] [CrossRef]
- Kanarek, N.; Petrova, B.; Sabatini, D.M. Dietary modifications for enhanced cancer therapy. Nature 2020, 579, 507–517. [Google Scholar] [CrossRef]
- Maddocks, O.D.K.; Athineos, D.; Cheung, E.C.; Lee, P.; Zhang, T.; van den Broek, N.J.F.; Mackay, G.M.; Labuschagne, C.F.; Gay, D.; Kruiswijk, F.; et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 2017, 544, 372–376. [Google Scholar] [CrossRef]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Billiard, J.; Dennison, J.B.; Briand, J.; Annan, R.S.; Chai, D.; Colon, M.; Dodson, C.S.; Gilbert, S.A.; Greshock, J.; Jing, J.; et al. Quinoline 3-sulfonamides inhibit lactate dehydrogenase A and reverse aerobic glycolysis in cancer cells. Cancer Metab. 2013, 1, 19. [Google Scholar] [CrossRef]
- Yang, X.; Lu, Y.; Hang, J.; Zhang, J.; Zhang, T.; Huo, Y.; Liu, J.; Lai, S.; Luo, D.; Wang, L.; et al. Lactate-Modulated Immunosuppression of Myeloid-Derived Suppressor Cells Contributes to the Radioresistance of Pancreatic Cancer. Cancer Immunol. Res. 2020, 8, 1440–1451. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.R.; Yang, C.; Scott, K.E.; Cameron, M.D.; Fallahi, M.; Li, W.; Hall, M.A.; Amelio, A.L.; Mishra, J.K.; Li, F.; et al. Blocking lactate export by inhibiting the Myc target MCT1 Disables glycolysis and glutathione synthesis. Cancer Res. 2014, 74, 908–920. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Valli, E.; Lam, H.; Scott, D.A.; Murray, J.; Hanssen, K.M.; Eden, G.; Gamble, L.D.; Pandher, R.; Flemming, C.L.; et al. Targeting metabolic activity in high-risk neuroblastoma through Monocarboxylate Transporter 1 (MCT1) inhibition. Oncogene 2020, 39, 3555–3570. [Google Scholar] [CrossRef] [PubMed]
- Tanis, S.P.; Parker, T.T.; Colca, J.R.; Fisher, R.M.; Kletzein, R.F. Synthesis and biological activity of metabolites of the antidiabetic, antihyperglycemic agent pioglitazone. J. Med. Chem. 1996, 39, 5053–5063. [Google Scholar] [CrossRef] [PubMed]
- Wiemer, E.A.; Michels, P.A.; Opperdoes, F.R. The inhibition of pyruvate transport across the plasma membrane of the bloodstream form of Trypanosoma brucei and its metabolic implications. Biochem. J. 1995, 312 Pt 2, 479–484. [Google Scholar] [CrossRef][Green Version]
- Shi, Y.; Lim, S.K.; Liang, Q.; Iyer, S.V.; Wang, H.Y.; Wang, Z.; Xie, X.; Sun, D.; Chen, Y.J.; Tabar, V.; et al. Gboxin is an oxidative phosphorylation inhibitor that targets glioblastoma. Nature 2019, 567, 341–346. [Google Scholar] [CrossRef]
- Schulte, M.L.; Fu, A.; Zhao, P.; Li, J.; Geng, L.; Smith, S.T.; Kondo, J.; Coffey, R.J.; Johnson, M.O.; Rathmell, J.C.; et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat. Med. 2018, 24, 194–202. [Google Scholar] [CrossRef]
- Edwards, D.N.; Ngwa, V.M.; Raybuck, A.L.; Wang, S.; Hwang, Y.; Kim, L.C.; Cho, S.H.; Paik, Y.; Wang, Q.; Zhang, S.; et al. Selective glutamine metabolism inhibition in tumor cells improves antitumor T lymphocyte activity in triple-negative breast cancer. J. Clin. Investig. 2021, 131, e140100. [Google Scholar] [CrossRef]
- Wicker, C.A.; Hunt, B.G.; Krishnan, S.; Aziz, K.; Parajuli, S.; Palackdharry, S.; Elaban, W.R.; Wise-Draper, T.M.; Mills, G.B.; Waltz, S.E.; et al. Glutaminase inhibition with telaglenastat (CB-839) improves treatment response in combination with ionizing radiation in head and neck squamous cell carcinoma models. Cancer Lett. 2021, 502, 180–188. [Google Scholar] [CrossRef]
- Varghese, S.; Pramanik, S.; Williams, L.J.; Hodges, H.R.; Hudgens, C.W.; Fischer, G.M.; Luo, C.K.; Knighton, B.; Tan, L.; Lorenzi, P.L.; et al. The Glutaminase Inhibitor CB-839 (Telaglenastat) Enhances the Antimelanoma Activity of T-Cell-Mediated Immunotherapies. Mol. Cancer 2021, 20, 500–511. [Google Scholar] [CrossRef]
- Balog, A.; Lin, T.A.; Maley, D.; Gullo-Brown, J.; Kandoussi, E.H.; Zeng, J.; Hunt, J.T. Preclinical Characterization of Linrodostat Mesylate, a Novel, Potent, and Selective Oral Indoleamine 2,3-Dioxygenase 1 Inhibitor. Mol. Cancer 2021, 20, 467–476. [Google Scholar] [CrossRef]
- Li, X.; Chen, Y.T.; Hu, P.; Huang, W.C. Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol. Cancer 2014, 13, 855–866. [Google Scholar] [CrossRef]
- Kamisuki, S.; Mao, Q.; Abu-Elheiga, L.; Gu, Z.; Kugimiya, A.; Kwon, Y.; Shinohara, T.; Kawazoe, Y.; Sato, S.; Asakura, K.; et al. A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chem. Biol. 2009, 16, 882–892. [Google Scholar] [CrossRef]
- Lally, J.S.V.; Ghoshal, S.; DePeralta, D.K.; Moaven, O.; Wei, L.; Masia, R.; Erstad, D.J.; Fujiwara, N.; Leong, V.; Houde, V.P.; et al. Inhibition of Acetyl-CoA Carboxylase by Phosphorylation or the Inhibitor ND-654 Suppresses Lipogenesis and Hepatocellular Carcinoma. Cell Metab. 2019, 29, 174–182.e5. [Google Scholar] [CrossRef]
- Svensson, R.U.; Parker, S.J.; Eichner, L.J.; Kolar, M.J.; Wallace, M.; Brun, S.N.; Lombardo, P.S.; Van Nostrand, J.L.; Hutchins, A.; Vera, L.; et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat. Med. 2016, 22, 1108–1119. [Google Scholar] [CrossRef]
- Beloueche-Babari, M.; Casals Galobart, T.; Delgado-Goni, T.; Wantuch, S.; Parkes, H.G.; Tandy, D.; Harker, J.A.; Leach, M.O. Monocarboxylate transporter 1 blockade with AZD3965 inhibits lipid biosynthesis and increases tumour immune cell infiltration. Br. J. Cancer 2020, 122, 895–903. [Google Scholar] [CrossRef]
- McNeillis, R.; Greystoke, A.; Walton, J.; Bacon, C.; Keun, H.; Siskos, A.; Petrides, G.; Leech, N.; Jenkinson, F.; Bowron, A.; et al. A case of malignant hyperlactaemic acidosis appearing upon treatment with the mono-carboxylase transporter 1 inhibitor AZD3965. Br. J. Cancer 2020, 122, 1141–1145. [Google Scholar] [CrossRef]
- Silva, A.; Antunes, B.; Batista, A.; Pinto-Ribeiro, F.; Baltazar, F.; Afonso, J. In Vivo Anticancer Activity of AZD3965: A Systematic Review. Molecules 2021, 27, 181. [Google Scholar] [CrossRef]
- Halford, S.E.R.; Walter, H.; McKay, P.; Townsend, W.; Linton, K.; Heinzmann, K.; Dragoni, I.; Brotherton, L.; Veal, G.; Siskos, A.; et al. Phase I expansion study of the first-in-class monocarboxylate transporter 1 (MCT1) inhibitor AZD3965 in patients with diffuse large B-cell lymphoma (DLBCL) and Burkitt lymphoma (BL). J. Clin. Oncol. 2021, 39, 3115. [Google Scholar] [CrossRef]
- Zachar, Z.; Marecek, J.; Maturo, C.; Gupta, S.; Stuart, S.D.; Howell, K.; Schauble, A.; Lem, J.; Piramzadian, A.; Karnik, S.; et al. Non-redox-active lipoate derivates disrupt cancer cell mitochondrial metabolism and are potent anticancer agents in vivo. J. Mol. Med. 2011, 89, 1137–1148. [Google Scholar] [CrossRef]
- Pardee, T.S.; Anderson, R.G.; Pladna, K.M.; Isom, S.; Ghiraldeli, L.P.; Miller, L.D.; Chou, J.W.; Jin, G.; Zhang, W.; Ellis, L.R.; et al. A Phase I Study of CPI-613 in Combination with High-Dose Cytarabine and Mitoxantrone for Relapsed or Refractory Acute Myeloid Leukemia. Clin. Cancer Res. 2018, 24, 2060–2073. [Google Scholar] [CrossRef] [PubMed]
- Stuart, S.D.; Schauble, A.; Gupta, S.; Kennedy, A.D.; Keppler, B.R.; Bingham, P.M.; Zachar, Z. A strategically designed small molecule attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process. Cancer Metab. 2014, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.C.; Maturo, C.; Perera, C.N.; Luddy, J.; Rodriguez, R.; Shorr, R. Translational assessment of mitochondrial dysfunction of pancreatic cancer from in vitro gene microarray and animal efficacy studies, to early clinical studies, via the novel tumor-specific anti-mitochondrial agent, CPI-613. Ann. Transl. Med. 2014, 2, 91. [Google Scholar] [CrossRef]
- Philip, P.A.; Bahary, N.; Mahipal, A.; Kasi, A.; Lima, C.M.S.P.R.; Alistar, A.T.; Oberstein, P.E.; Golan, T.; Sahai, V.; Metges, J.P.; et al. Phase 3, multicenter, randomized study of CPI-613 with modified FOLFIRINOX (mFFX) versus FOLFIRINOX (FFX) as first-line therapy for patients with metastatic adenocarcinoma of the pancreas (AVENGER500). J. Clin. Oncol. 2022, 40, 4023. [Google Scholar] [CrossRef]
- Yen, K.; Travins, J.; Wang, F.; David, M.D.; Artin, E.; Straley, K.; Padyana, A.; Gross, S.; DeLaBarre, B.; Tobin, E.; et al. AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations. Cancer Discov. 2017, 7, 478–493. [Google Scholar] [CrossRef] [PubMed]
- Saengboonmee, C.; Sanlung, T.; Wongkham, S. Repurposing Metformin for Cancer Treatment: A Great Challenge of a Promising Drug. Anticancer Res. 2021, 41, 5913–5918. [Google Scholar] [CrossRef]
- Ahn, H.K.; Lee, Y.H.; Koo, K.C. Current Status and Application of Metformin for Prostate Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2020, 21, 8540. [Google Scholar] [CrossRef]
- Vial, G.; Detaille, D.; Guigas, B. Role of Mitochondria in the Mechanism(s) of Action of Metformin. Front. Endocrinol. 2019, 10, 294. [Google Scholar] [CrossRef]
- Bilusic, M.; Toney, N.J.; Donahue, R.N.; Wroblewski, S.; Zibelman, M.; Ghatalia, P.; Ross, E.A.; Karzai, F.; Madan, R.A.; Dahut, W.L.; et al. A randomized phase 2 study of bicalutamide with or without metformin for biochemical recurrence in overweight or obese prostate cancer patients (BIMET-1). Prostate Cancer Prostatic Dis. 2022; online ahead of print. [Google Scholar] [CrossRef]
- Ma, T.; Tian, X.; Zhang, B.; Li, M.; Wang, Y.; Yang, C.; Wu, J.; Wei, X.; Qu, Q.; Yu, Y.; et al. Low-dose metformin targets the lysosomal AMPK pathway through PEN2. Nature 2022, 603, 159–165. [Google Scholar] [CrossRef]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.A.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef]
- Yap, T.A.; Ahnert, J.R.; Piha-Paul, S.A.; Fu, S.; Janku, F.; Karp, D.D.; Naing, A.; Dumbrava, E.E.I.; Pant, S.; Subbiah, V.; et al. Phase I trial of IACS-010759 (IACS), a potent, selective inhibitor of complex I of the mitochondrial electron transport chain, in patients (pts) with advanced solid tumors. J. Clin. Oncol. 2019, 37, 3014. [Google Scholar] [CrossRef]
- Zhao, H.; Swanson, K.D.; Zheng, B. Therapeutic Repurposing of Biguanides in Cancer. Trends Cancer 2021, 7, 714–730. [Google Scholar] [CrossRef] [PubMed]
- Izreig, S.; Gariepy, A.; Kaymak, I.; Bridges, H.R.; Donayo, A.O.; Bridon, G.; DeCamp, L.M.; Kitchen-Goosen, S.M.; Avizonis, D.; Sheldon, R.D.; et al. Repression of LKB1 by miR-17 approximately 92 Sensitizes MYC-Dependent Lymphoma to Biguanide Treatment. Cell Rep. Med. 2020, 1, 100014. [Google Scholar] [CrossRef] [PubMed]
- Rha, S.Y.; Beom, S.-H.; Shin, Y.G.; Yim, D.-S.; Moon, Y.W.; Kim, T.W.; Kim, S.Y.; Kim, G.M.; Kim, H.S.; Cheong, J.-H.; et al. Phase I study of IM156, a novel potent biguanide oxidative phosphorylation (OXPHOS) inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2020, 38, 3590. [Google Scholar] [CrossRef]
- Okano, N.; Naruge, D.; Kawai, K.; Kobayashi, T.; Nagashima, F.; Endou, H.; Furuse, J. First-in-human phase I study of JPH203, an L-type amino acid transporter 1 inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2020, 38, 1495–1506. [Google Scholar] [CrossRef]
- Oda, K.; Hosoda, N.; Endo, H.; Saito, K.; Tsujihara, K.; Yamamura, M.; Sakata, T.; Anzai, N.; Wempe, M.F.; Kanai, Y.; et al. L-type amino acid transporter 1 inhibitors inhibit tumor cell growth. Cancer Sci. 2010, 101, 173–179. [Google Scholar] [CrossRef]
- Hafliger, P.; Graff, J.; Rubin, M.; Stooss, A.; Dettmer, M.S.; Altmann, K.H.; Gertsch, J.; Charles, R.P. The LAT1 inhibitor JPH203 reduces growth of thyroid carcinoma in a fully immunocompetent mouse model. J. Exp. Clin. Cancer Res. 2018, 37, 234. [Google Scholar] [CrossRef]
- Myint, Z.W.; Sun, R.C.; Hensley, P.J.; James, A.C.; Wang, P.; Strup, S.E.; McDonald, R.J.; Yan, D.; St Clair, W.H.; Allison, D.B. Evaluation of Glutaminase Expression in Prostate Adenocarcinoma and Correlation with Clinicopathologic Parameters. Cancers 2021, 13, 2157. [Google Scholar] [CrossRef]
- Soth, M.J.; Le, K.; Di Francesco, M.E.; Hamilton, M.M.; Liu, G.; Burke, J.P.; Carroll, C.L.; Kovacs, J.J.; Bardenhagen, J.P.; Bristow, C.A.; et al. Discovery of IPN60090, a Clinical Stage Selective Glutaminase-1 (GLS-1) Inhibitor with Excellent Pharmacokinetic and Physicochemical Properties. J. Med. Chem. 2020, 63, 12957–12977. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Wild, R.; Estok, T.M. Sirpiglenastat (DRP-104) Induces Anti-tumor Efficacy Through Direct, Broad Antagonism of Glutamine Metabolism and Stimulation of the Innate & Adaptive Immune Systems. Mol. Cancer Ther. 2022, 21, 1561–1572. [Google Scholar] [CrossRef]
- Komiya, T.; Huang, C.H. Updates in the Clinical Development of Epacadostat and Other Indoleamine 2,3-Dioxygenase 1 Inhibitors (IDO1) for Human Cancers. Front. Oncol. 2018, 8, 423. [Google Scholar] [CrossRef]
- Kalev, P.; Hyer, M.L.; Gross, S.; Konteatis, Z.; Chen, C.C.; Fletcher, M.; Lein, M.; Aguado-Fraile, E.; Frank, V.; Barnett, A.; et al. MAT2A Inhibition Blocks the Growth of MTAP-Deleted Cancer Cells by Reducing PRMT5-Dependent mRNA Splicing and Inducing DNA Damage. Cancer Cell 2021, 39, 209–224.E11. [Google Scholar] [CrossRef]
- Hung, M.H.; Lee, J.S.; Ma, C.; Diggs, L.P.; Heinrich, S.; Chang, C.W.; Ma, L.; Forgues, M.; Budhu, A.; Chaisaingmongkol, J.; et al. Tumor methionine metabolism drives T-cell exhaustion in hepatocellular carcinoma. Nat. Commun. 2021, 12, 1455. [Google Scholar] [CrossRef]
- Yoon, H.; Luo, S.; Sanfilippo, K.M.; Linneman, T.; Whitmer, A.; Schoen, M.W. Statin type and survival of patients with metastatic castrate-resistant prostate cancer receiving abiraterone and enzalutamide: A nationwide retrospective cohort study. J. Clin. Oncol. 2022, 40, 50. [Google Scholar] [CrossRef]
- Heuer, T.S.; Ventura, R.; Mordec, K.; Lai, J.; Fridlib, M.; Buckley, D.; Kemble, G. FASN Inhibition and Taxane Treatment Combine to Enhance Anti-tumor Efficacy in Diverse Xenograft Tumor Models through Disruption of Tubulin Palmitoylation and Microtubule Organization and FASN Inhibition-Mediated Effects on Oncogenic Signaling and Gene Expression. eBioMedicine 2017, 16, 51–62. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase (FASN) as a therapeutic target in breast cancer. Expert Opin. Ther. Targets 2017, 21, 1001–1016. [Google Scholar] [CrossRef]
- Harriman, G.; Greenwood, J.; Bhat, S.; Huang, X.; Wang, R.; Paul, D.; Tong, L.; Saha, A.K.; Westlin, W.F.; Kapeller, R.; et al. Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. Proc. Natl. Acad. Sci. USA 2016, 113, E1796–E1805. [Google Scholar] [CrossRef]
- Alkhouri, N.; Lawitz, E.; Noureddin, M.; DeFronzo, R.; Shulman, G.I. GS-0976 (Firsocostat): An investigational liver-directed acetyl-CoA carboxylase (ACC) inhibitor for the treatment of non-alcoholic steatohepatitis (NASH). Expert Opin. Investig. Drugs 2020, 29, 135–141. [Google Scholar] [CrossRef]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers. ACS. Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef]
- Sussmuth, S.D.; Haider, S.; Landwehrmeyer, G.B.; Farmer, R.; Frost, C.; Tripepi, G.; Andersen, C.A.; Di Bacco, M.; Lamanna, C.; Diodato, E.; et al. An exploratory double-blind, randomized clinical trial with selisistat, a SirT1 inhibitor, in patients with Huntington’s disease. Br. J. Clin. Pharmacol. 2015, 79, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.; Fischer, F.; Nguyen, G.T.; Lakshminarasimhan, M.; Schutkowski, M.; Weyand, M.; Steegborn, C. Ex-527 inhibits Sirtuins by exploiting their unique NAD+-dependent deacetylation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, E2772–E2781. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chu, M. Leflunomide: A promising drug with good antitumor potential. Biochem. Biophys. Res. Commun. 2018, 496, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, S.; Mathur, D.; Zhou, R.W.; Mulholland, D.; Parsons, R. Leflunomide triggers synthetic lethality in PTEN-deficient prostate cancer. Prostate Cancer Prostatic Dis. 2020, 23, 718–723. [Google Scholar] [CrossRef]
- Shukla-Dave, A.; Castillo-Martin, M.; Chen, M.; Lobo, J.; Gladoun, N.; Collazo-Lorduy, A.; Khan, F.M.; Ponomarev, V.; Yi, Z.; Zhang, W.; et al. Ornithine Decarboxylase Is Sufficient for Prostate Tumorigenesis via Androgen Receptor Signaling. Am. J. Pathol. 2016, 186, 3131–3145. [Google Scholar] [CrossRef]
- Alexiou, G.A.; Lianos, G.D.; Ragos, V.; Galani, V.; Kyritsis, A.P. Difluoromethylornithine in cancer: New advances. Future Oncol. 2017, 13, 809–819. [Google Scholar] [CrossRef]
- Randall, E.C.; Zadra, G.; Chetta, P.; Lopez, B.G.C.; Syamala, S.; Basu, S.S.; Agar, J.N.; Loda, M.; Tempany, C.M.; Fennessy, F.M.; et al. Molecular Characterization of Prostate Cancer with Associated Gleason Score Using Mass Spectrometry Imaging. Mol. Cancer Res. 2019, 17, 1155–1165. [Google Scholar] [CrossRef]
- Seydel, C. Single-cell metabolomics hits its stride. Nat. Methods 2021, 18, 1452–1456. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, S.Y.C.; Ribeiro, C.F.; Wang, Y.; Loda, M.; Plymate, S.R.; Uo, T. Druggable Metabolic Vulnerabilities Are Exposed and Masked during Progression to Castration Resistant Prostate Cancer. Biomolecules 2022, 12, 1590. https://doi.org/10.3390/biom12111590
Choi SYC, Ribeiro CF, Wang Y, Loda M, Plymate SR, Uo T. Druggable Metabolic Vulnerabilities Are Exposed and Masked during Progression to Castration Resistant Prostate Cancer. Biomolecules. 2022; 12(11):1590. https://doi.org/10.3390/biom12111590
Chicago/Turabian StyleChoi, Stephen Y. C., Caroline Fidalgo Ribeiro, Yuzhuo Wang, Massimo Loda, Stephen R. Plymate, and Takuma Uo. 2022. "Druggable Metabolic Vulnerabilities Are Exposed and Masked during Progression to Castration Resistant Prostate Cancer" Biomolecules 12, no. 11: 1590. https://doi.org/10.3390/biom12111590
APA StyleChoi, S. Y. C., Ribeiro, C. F., Wang, Y., Loda, M., Plymate, S. R., & Uo, T. (2022). Druggable Metabolic Vulnerabilities Are Exposed and Masked during Progression to Castration Resistant Prostate Cancer. Biomolecules, 12(11), 1590. https://doi.org/10.3390/biom12111590