
Neutrophil Extracellular Traps (NETs) in Ocular Diseases: An Update


Abstract
1. Introduction
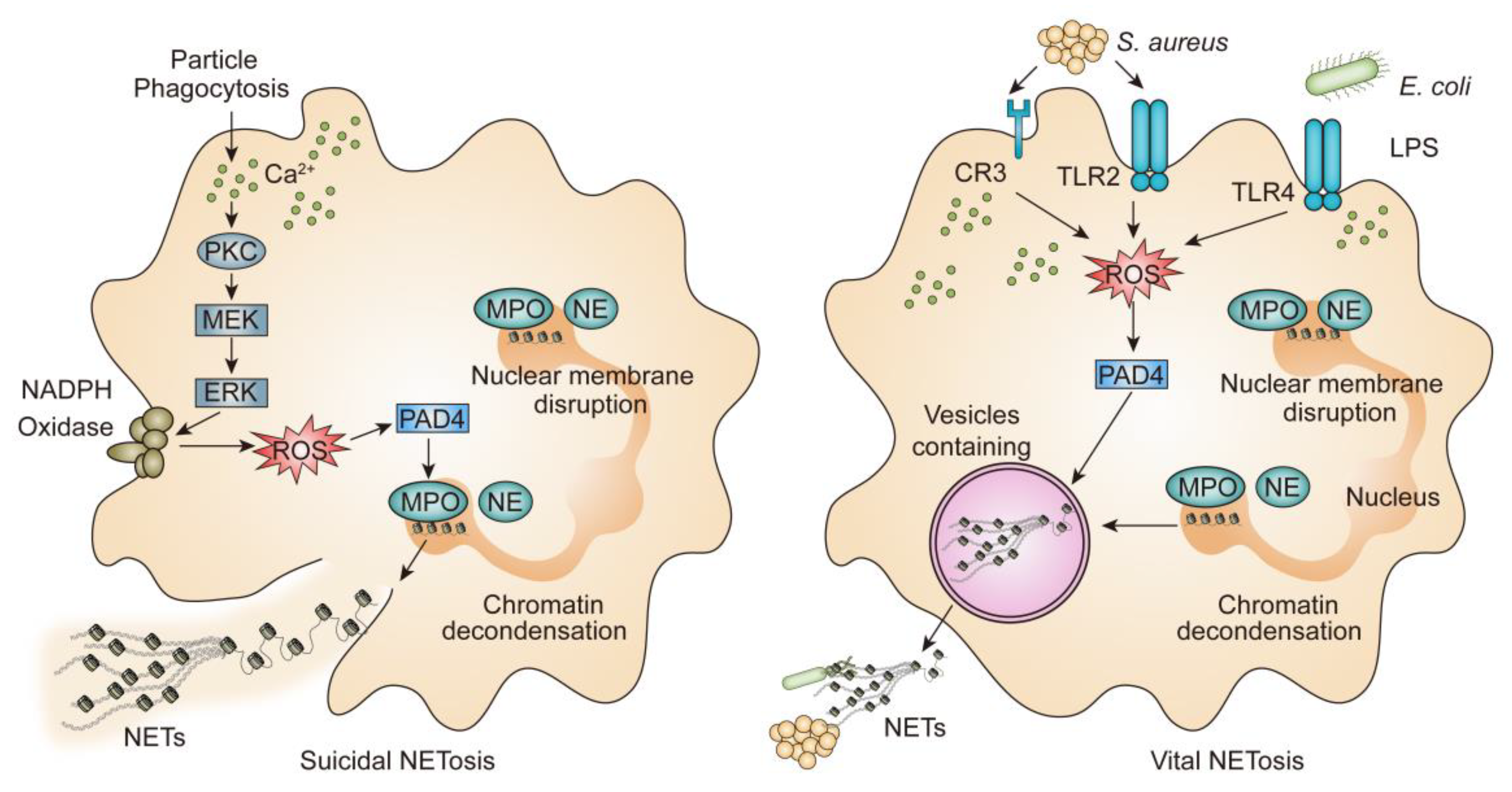
2. Mechanism of NETs
2.1. Lytic NETosis
2.2. Vital NETosis
3. Cornea and Ocular Surface
3.1. Dry Eye Disease (DED)
3.2. Infectious Keratitis
3.2.1. Bacteria Keratitis
3.2.2. Fungal Keratitis
3.3. Cornea Injury
4. Uveitis
5. Diabetic Retinopathy (DR)
6. Retinal Vein Occlusion (RVO)
7. Age-Related Macular Degeneration (AMD)
8. Conclusions and Perspective
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| IF | immunofluorescence |
| HE | hematoxylin eosin |
| MTS | 3-(4-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium |
| OSDI | ocular surface disease index |
| CGI | clinical global impression |
| SGA | subject global assessment |
| VBR | validated bulbar redness |
| WB | Western blot |
| ELISA | enzyme-linked immunosorbent assay |
| qRT-PCR | quantitative reverse transcription polymerase chain reaction |
| HCE | human corneal epithelial cell |
| hAM-MSC | human amniotic mesenchymal stem cell |
| IOP | intraocular pressure |
| OCT | optical coherence tomography |
References
- Segal, A.W. How neutrophils kill microbes. Annu. Rev. Immunol. 2005, 23, 197–223. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, H.O.; Roy, E.; Comerci, A.J.; van der Windt, D.J.; Zhang, H.; Huang, H.; Loughran, P.; Shiva, S.; Geller, D.A.; Bartlett, D.L.; et al. Neutrophil Extracellular Traps Drive Mitochondrial Homeostasis in Tumors to Augment Growth. Cancer Res. 2019, 79, 5626–5639. [Google Scholar] [CrossRef]
- Sivanandham, R.; Brocca-Cofano, E.; Krampe, N.; Falwell, E.; Venkatraman, S.M.K.; Ribeiro, R.M.; Apetrei, C.; Pandrea, I. Neutrophil extracellular trap production contributes to pathogenesis in SIV-infected nonhuman primates. J. Clin. Investig. 2018, 128, 5178–5183. [Google Scholar] [CrossRef] [PubMed]
- Urban, C.F.; Reichard, U.; Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol. 2006, 8, 668–676. [Google Scholar] [CrossRef]
- Estua-Acosta, G.A.; Zamora-Ortiz, R.; Buentello-Volante, B.; Garcia-Mejia, M.; Garfias, Y. Neutrophil Extracellular Traps: Current Perspectives in the Eye. Cells 2019, 8, 979. [Google Scholar] [CrossRef]
- Yu, H.C.; Lu, M.C. The roles of anti-citrullinated protein antibodies in the immunopathogenesis of rheumatoid arthritis. Ci Ji Yi Xue Za Zhi 2019, 31, 5–10. [Google Scholar]
- Chowdhury, C.S.; Giaglis, S.; Walker, U.A.; Buser, A.; Hahn, S.; Hasler, P. Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: Analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res. Ther. 2014, 16, R122. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2017, 18, 134–147. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Rohrbach, A.S.; Slade, D.J.; Thompson, P.R.; Mowen, K.A. Activation of PAD4 in NET formation. Front. Immunol. 2012, 3, 360. [Google Scholar] [CrossRef]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef]
- Wang, Y.; Li, M.; Stadler, S.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef] [PubMed]
- De Bont, C.M.; Koopman, W.J.H.; Boelens, W.C.; Pruijn, G.J.M. Stimulus-dependent chromatin dynamics, citrullination, calcium signalling and ROS production during NET formation. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1621–1629. [Google Scholar] [CrossRef]
- Hakkim, A.; Fuchs, T.A.; Martinez, N.E.; Hess, S.; Prinz, H.; Zychlinsky, A.; Waldmann, H. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat. Chem. Biol. 2011, 7, 75–77.e5. [Google Scholar] [CrossRef]
- Amulic, B.; Knackstedt, S.L.; Abed, U.A.; Deigendesch, N.; Harbort, C.J.; Caffrey, B.E.; Brinkmann, V.; Heppner, F.L.; Hinds, P.W.; Zychlinsky, A. Cell-Cycle Proteins Control Production of Neutrophil Extracellular Traps. Dev. Cell 2017, 43, 449–462. [Google Scholar] [CrossRef]
- Branzk, N.; Lubojemska, A.; Hardison, S.E.; Wang, Q.; Gutierrez, M.G.; Brown, G.D.; Papayannopoulos, V. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat. Immunol. 2014, 15, 1017–1025. [Google Scholar] [CrossRef]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.; Surette, M.G.; Sugai, M.; et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef] [PubMed]
- Speziale, P.; Pietrocola, G. Staphylococcus aureus induces neutrophil extracellular traps (NETs) and neutralizes their bactericidal potential. Comput. Struct. Biotechnol. J. 2021, 19, 3451–3457. [Google Scholar] [CrossRef]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Yipp, B.G.; Kubes, P. NETosis: How vital is it? Blood 2013, 122, 2784–2794. [Google Scholar] [CrossRef]
- Kumar, S.; Gupta, E.; Kaushik, S.; Jyoti, A. Neutrophil extracellular traps: Formation and involvement in disease progression, Iranian Journal of Allergy. Asthma Immunol. 2018, 17, 208–220. [Google Scholar]
- Yousefi, S.; Mihalache, C.; Kozlowski, E.; Schmid, I.; Simon, H.U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009, 16, 1438–1444. [Google Scholar] [CrossRef]
- Thanabalasuriar, A.; Kubes, P. Rise and shine: Open your eyes to produce anti-inflammatory NETs. J. Leukoc Biol. 2019, 105, 1083–1084. [Google Scholar] [CrossRef]
- Agrahari, V.; Mandal, A.; Agrahari, V.; Trinh, H.M.; Joseph, M.; Ray, A.; Hadji, H.; Mitra, R.; Pal, D.; Mitra, A.K. A comprehensive insight on ocular pharmacokinetics. Drug Deliv. Transl. Res. 2016, 6, 735–754. [Google Scholar] [CrossRef]
- Ueta, M.; Kinoshita, S. Innate immunity of the ocular surface. Brain Res. Bull 2010, 81, 219–228. [Google Scholar] [CrossRef]
- Taylor, A.W. Ocular immune privilege. Eye 2009, 23, 1885–1889. [Google Scholar] [CrossRef]
- Gomes, J.A.P.; Frizon, L.; Demeda, V.F. Ocular Surface Microbiome in Health and Disease. Asia Pac. J. Ophthalmol. 2020, 9, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Delbeke, H.; Younas, S.; Casteels, I.; Joossens, M. Current knowledge on the human eye microbiome: A systematic review of available amplicon and metagenomic sequencing data. Acta Ophthalmol. 2021, 99, 16–25. [Google Scholar] [CrossRef]
- Craig, J.P.; Nelson, J.D.; Azar, D.T.; Belmonte, C.; Bron, A.J.; Chauhan, S.K.; de Paiva, C.S.; Gomes, J.A.P.; Hammitt, K.M.; Jones, L.; et al. TFOS DEWS II Report Executive Summary. Ocul. Surf. 2017, 15, 802–812. [Google Scholar] [CrossRef]
- Koh, S.; Rhee, M.K. COVID-19 and Dry Eye. Eye Contact Lens. 2021, 47, 317–322. [Google Scholar] [CrossRef]
- Calonge, M.; Labetoulle, M.; Messmer, E.M.; Shah, S.; Akova, Y.A.; Boboridis, K.G.; Merayo-Lloves, J.; Aragona, P.; Benitez-Del-Castillo, J.; Geerling, G.; et al. Controlled Adverse Environment Chambers in Dry Eye Research. Curr. Eye Res. 2018, 43, 445–450. [Google Scholar] [CrossRef]
- Postnikoff, C.K.; Huisingh, C.; McGwin, G.; Nichols, K.K. Leukocyte Distribution in the Open Eye Tears of Normal and Dry Eye Subjects. Curr. Eye Res. 2018, 43, 1253–1259. [Google Scholar] [CrossRef]
- Rhee, M.K.; Mah, F.S. Inflammation in Dry Eye Disease: How Do We Break the Cycle? Ophthalmology 2017, 124, S14–S19. [Google Scholar] [CrossRef]
- Postnikoff, C.K.; Held, K.; Viswanath, V.; Nichols, K.K. Enhanced closed eye neutrophil degranulation in dry eye disease. Ocul. Surf. 2020, 18, 841–851. [Google Scholar] [CrossRef]
- Martinez-Alberquilla, I.; Gasull, X.; Perez-Luna, P.; Seco-Mera, R.; Ruiz-Alcocer, J.; Crooke, A. Neutrophils and neutrophil extracellular trap components: Emerging biomarkers and therapeutic targets for age-related eye diseases. Ageing Res. Rev. 2022, 74, 101553. [Google Scholar] [CrossRef]
- Ozcan, D.O.; Kurtul, B.E.; Ozcan, S.C.; Elbeyli, A. Increased Systemic Immune-Inflammation Index Levels in Patients with Dry Eye Disease. Ocul. Immunol. Inflamm. 2020, 30, 588–592. [Google Scholar] [CrossRef]
- Sekeryapan, B.; Uzun, F.; Buyuktarakci, S.; Bulut, A.; Oner, V. Neutrophil-to-lymphocyte ratio increases in patients with dry eye. Cornea 2016, 35, 983–986. [Google Scholar] [CrossRef]
- Tibrewal, S.; Ivanir, Y.; Sarkar, J.; Nayeb-Hashemi, N.; Bouchard, C.S.; Kim, E.; Jain, S. Hyperosmolar stress induces neutrophil extracellular trap formation: Implications for dry eye disease. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7961–7969. [Google Scholar] [CrossRef]
- An, S.; Raju, I.; Surenkhuu, B.; Kwon, J.E.; Gulati, S.; Karaman, M.; Pradeep, A.; Sinha, S.; Mun, C.; Jain, S. Neutrophil extracellular traps (NETs) contribute to pathological changes of ocular graft-vs.-host disease (oGVHD) dry eye: Implications for novel biomarkers and therapeutic strategies. Ocul. Surf. 2019, 17, 589–614. [Google Scholar] [CrossRef] [PubMed]
- Sonawane, S.; Khanolkar, V.; Namavari, A.; Chaudhary, S.; Gandhi, S.; Tibrewal, S.; Jassim, S.H.; Shaheen, B.; Hallak, J.; Horner, J.H.; et al. Ocular surface extracellular DNA and nuclease activity imbalance: A new paradigm for inflammation in dry eye disease. Invest. Ophthalmol. Vis. Sci. 2012, 53, 8253–8263. [Google Scholar] [CrossRef]
- Goto, E.; Monden, Y.; Takano, Y.; Mori, A.; Shimmura, S.; Shimazaki, J.; Tsubota, K. Treatment of non-inflamed obstructive meibomian gland dysfunction by an infrared warm compression device. Br. J. Ophthalmol. 2002, 86, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Mun, C.; Gulati, S.; Tibrewal, S.; Chen, Y.F.; An, S.; Surenkhuu, B.; Raju, I.; Buwick, M.; Ahn, A.; Kwon, J.E.; et al. A Phase I/II Placebo-Controlled Randomized Pilot Clinical Trial of Recombinant Deoxyribonuclease (DNase) Eye Drops Use in Patients With Dry Eye Disease. Transl. Vis. Sci. Technol. 2019, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.D.; Shimazaki, J.; Benitez-del-Castillo, J.M.; Craig, J.P.; McCulley, J.P.; Den, S.; Foulks, G.N. The international workshop on meibomian gland dysfunction: Report of the definition and classification subcommittee. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1930–1937. [Google Scholar] [CrossRef]
- Nicolas-Avila, J.A.; Adrover, J.M.; Hidalgo, A. Neutrophils in Homeostasis, Immunity, and Cancer. Immunity 2017, 46, 15–28. [Google Scholar] [CrossRef]
- Mahajan, A.; Hasikova, L.; Hampel, U.; Gruneboom, A.; Shan, X.; Herrmann, I.; Garreis, F.; Bock, F.; Knopf, J.; Singh, J.; et al. Aggregated neutrophil extracellular traps occlude Meibomian glands during ocular surface inflammation. Ocul. Surf. 2021, 20, 1–12. [Google Scholar] [CrossRef]
- Samejima, K.; Earnshaw, W.C. Trashing the genome: The role of nucleases during apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6, 677–688. [Google Scholar] [CrossRef]
- Zeiser, R.; Polverelli, N.; Ram, R.; Hashmi, S.K.; Chakraverty, R.; Middeke, J.M.; Musso, M.; Giebel, S.; Uzay, A.; Langmuir, P.; et al. Ruxolitinib for Glucocorticoid-Refractory Chronic Graft-versus-Host Disease. N. Engl. J. Med. 2021, 385, 228–238. [Google Scholar] [CrossRef]
- Nair, S.; Vanathi, M.; Mukhija, R.; Tandon, R.; Jain, S.; Ogawa, Y. Update on ocular graft-versus-host disease. Indian J. Ophthalmol. 2021, 69, 1038–1050. [Google Scholar] [PubMed]
- Napirei, M.; Ludwig, S.; Mezrhab, J.; Klockl, T.; Mannherz, H.G. Murine serum nucleases--contrasting effects of plasmin and heparin on the activities of DNase1 and DNase1-like 3 (DNase1l3). FEBS J. 2009, 276, 1059–1073. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [PubMed]
- Iniguez-Gutierrez, L.; Godinez-Mendez, L.A.; Fafutis-Morris, M.; Padilla-Arellano, J.R.; Corona-Rivera, A.; Bueno-Topete, M.R.; Rojas-Rejon, O.A.; Delgado-Rizo, V. Physiological concentrations of short-chain fatty acids induce the formation of neutrophil extracellular traps in vitro. Int. J. Immunopathol. Pharmacol. 2020, 34, 2058738420958949. [Google Scholar] [CrossRef] [PubMed]
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid. Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef]
- Ratajczak, W.; Ryl, A.; Mizerski, A.; Walczakiewicz, K.; Sipak, O.; Laszczynska, M. Immunomodulatory potential of gut microbiome-derived short-chain fatty acids (SCFAs). Acta Biochim. Pol. 2019, 66, 1–12. [Google Scholar] [CrossRef]
- Portincasa, P.; Bonfrate, L.; Vacca, M.; de Angelis, M.; Farella, I.; Lanza, E.; Khalil, M.; Wang, D.Q.; Sperandio, M.; di Ciaula, A. Gut Microbiota and Short Chain Fatty Acids: Implications in Glucose Homeostasis. Int. J. Mol. Sci. 2022, 23, 1105. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Gong, Y.; Chen, S.; Li, S.; Zhang, Y.; Zhong, H.; Wang, Z.; Chen, Y.; Deng, Q.; Jiang, Y.; et al. Comparative portrayal of ocular surface microbe with and without dry eye. J. Microbiol. 2019, 57, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Padayatty, S.J.; Katz, A.; Wang, Y.; Eck, P.; Kwon, O.; Lee, J.-H.; Chen, S.; Corpe, C.; Dutta, A.; Dutta, S.K. Vitamin C as an antioxidant: Evaluation of its role in disease prevention. J. Am. Coll. Nutr. 2003, 22, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Vissers, M.C.; Wilkie, R.P. Ascorbate deficiency results in impaired neutrophil apoptosis and clearance and is associated with up-regulation of hypoxia-inducible factor 1α. J. Leukoc. Biol. 2007, 81, 1236–1244. [Google Scholar] [CrossRef] [PubMed]
- Remijsen, Q.; Berghe, T.V.; Wirawan, E.; Asselbergh, B.; Parthoens, E.; de Rycke, R.; Noppen, S.; Delforge, M.; Willems, J.; Vandenabeele, P. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011, 21, 290–304. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, B.M.; Fisher, B.J.; Kraskauskas, D.; Farkas, D.; Brophy, D.F.; Fowler, A.A., 3rd; Natarajan, R. Vitamin C: A novel regulator of neutrophil extracellular trap formation. Nutrients 2013, 5, 3131–3151. [Google Scholar] [CrossRef]
- Kirchner, T.; Hermann, E.; Moller, S.; Klinger, M.; Solbach, W.; Laskay, T.; Behnen, M. Flavonoids and 5-aminosalicylic acid inhibit the formation of neutrophil extracellular traps. Mediat. Inflamm. 2013, 2013, 710239. [Google Scholar] [CrossRef]
- Thanabalasuriar, A.; Scott, B.N.V.; Peiseler, M.; Willson, M.E.; Zeng, Z.; Warrener, P.; Keller, A.E.; Surewaard, B.G.J.; Dozier, E.A.; Korhonen, J.T.; et al. Neutrophil Extracellular Traps Confine Pseudomonas aeruginosa Ocular Biofilms and Restrict Brain Invasion. Cell Host. Microbe 2019, 25, 526–536.e4. [Google Scholar] [CrossRef]
- Xing, Y.; Ye, Y.; Zuo, H.; Li, Y. Progress on the Function and Application of Thymosin beta4. Front. Endocrinol. 2021, 12, 767785. [Google Scholar] [CrossRef]
- Wang, Y.; Carion, T.W.; Ebrahim, A.S.; Sosne, G.; Berger, E.A. Adjunctive Thymosin Beta-4 Treatment Influences PMN Effector Cell Function during Pseudomonas aeruginosa-Induced Corneal Infection. Cells 2021, 10, 3579. [Google Scholar] [CrossRef]
- Tesfaye, T.; Beyene, G.; Gelaw, Y.; Bekele, S.; Saravanan, M. Bacterial profile and antimicrobial susceptibility pattern of external ocular infections in Jimma University specialized hospital, Southwest Ethiopia. Am. J. Infect. Dis. Microbiol. 2013, 1, 13–20. [Google Scholar] [CrossRef]
- Shrestha, G.S.; Vijay, A.K.; Stapleton, F.; Henriquez, F.L.; Carnt, N. Understanding clinical and immunological features associated with Pseudomonas and Staphylococcus keratitis. Cont. Lens. Anterior. Eye 2021, 44, 3–13. [Google Scholar] [CrossRef]
- O’Callaghan, R.J.; Callegan, M.C.; Moreau, J.M.; Green, L.C.; Foster, T.J.; Hartford, O.M.; Engel, L.S.; Hill, J.M. Specific roles of alpha-toxin and beta-toxin during Staphylococcus aureus corneal infection. Infect. Immun. 1997, 65, 1571–1578. [Google Scholar] [CrossRef]
- Jin, X.; Zhao, Y.; Zhang, F.; Wan, T.; Fan, F.; Xie, X.; Lin, Z. Neutrophil extracellular traps involvement in corneal fungal infection. Mol. Vis. 2016, 22, 944. [Google Scholar]
- Khames, A.; Khaleel, M.A.; El-Badawy, M.F.; El-Nezhawy, A.O.H. Natamycin solid lipid nanoparticles—Sustained ocular delivery system of higher corneal penetration against deep fungal keratitis: Preparation and optimization. Int. J. Nanomed. 2019, 14, 2515–2531. [Google Scholar] [CrossRef]
- Yang, S.S.; Hung, C.T.; Li, S.F.; Lee, H.M.; Chung, Y.C.; Chen, H.H.; Chang, S.C. Hepatitis B virus-related mortality in rheumatoid arthritis patients undergoing long-term low-dose glucocorticoid treatment: A population-based study. J. Formos. Med. Assoc. 2018, 117, 566–571. [Google Scholar] [CrossRef]
- Fan, F.; Huang, X.; Yuan, K.; Zhu, B.; Zhao, Y.; Hu, R.; Wan, T.; Zhu, L.; Jin, X. Glucocorticoids May Exacerbate Fungal Keratitis by Increasing Fungal Aggressivity and Inhibiting the Formation of Neutrophil Extracellular Traps. Curr. Eye Res. 2020, 45, 124–133. [Google Scholar] [CrossRef]
- Itakura, A.; McCarty, O.J. Pivotal role for the mTOR pathway in the formation of neutrophil extracellular traps via regulation of autophagy. Am. J. Physiol. Cell Physiol. 2013, 305, C348–C354. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef]
- Yost, C.C.; Denis, M.M.; Lindemann, S.; Rubner, F.J.; Marathe, G.K.; Buerke, M.; McIntyre, T.M.; Weyrich, A.S.; Zimmerman, G.A. Activated polymorphonuclear leukocytes rapidly synthesize retinoic acid receptor-alpha: A mechanism for translational control of transcriptional events. J. Exp. Med. 2004, 200, 671–680. [Google Scholar] [CrossRef]
- Yuan, K.; Zheng, J.; Huang, X.; Zhang, Y.; Han, Y.; Hu, R.; Jin, X. Neutrophil extracellular traps promote corneal neovascularization-induced by alkali burn. Int. Immunopharmacol. 2020, 88, 106902. [Google Scholar] [CrossRef]
- Wan, T.; Zhang, Y.; Yuan, K.; Min, J.; Mou, Y.; Jin, X. Acetylsalicylic Acid Promotes Corneal Epithelium Migration by Regulating Neutrophil Extracellular Traps in Alkali Burn. Front. Immunol. 2020, 11, 551057. [Google Scholar] [CrossRef] [PubMed]
- De Souza, C.N.; Breda, L.C.D.; Khan, M.A.; de Almeida, S.R.; Camara, N.O.S.; Sweezey, N.; Palaniyar, N. Alkaline pH Promotes NADPH Oxidase-Independent Neutrophil Extracellular Trap Formation: A Matter of Mitochondrial Reactive Oxygen Species Generation and Citrullination and Cleavage of Histone. Front. Immunol. 2017, 8, 1849. [Google Scholar] [CrossRef] [PubMed]
- Noyan, K.; Nguyen, S.; Betts, M.R.; Sonnerborg, A.; Buggert, M. Human Immunodeficiency Virus Type-1 Elite Controllers Maintain Low Co-Expression of Inhibitory Receptors on CD4+ T Cells. Front. Immunol. 2018, 9, 19. [Google Scholar] [CrossRef]
- Krishna, U.; Ajanaku, D.; Denniston, A.K.; Gkika, T. Uveitis: A sight-threatening disease which can impact all systems. Postgrad. Med. J. 2017, 93, 766–773. [Google Scholar] [CrossRef]
- Li, H.; Tan, H.; Liu, Z.; Pan, S.; Tan, S.; Zhu, Y.; Wang, Q.; Su, G.; Zhou, C.; Cao, Q.; et al. Succinic acid exacerbates experimental autoimmune uveitis by stimulating neutrophil extracellular traps formation via SUCNR1 receptor. Br. J. Ophthalmol. 2022, 320880. [Google Scholar] [CrossRef]
- Luger, D.; Silver, P.B.; Tang, J.; Cua, D.; Chen, Z.; Iwakura, Y.; Bowman, E.P.; Sgambellone, N.M.; Chan, C.C.; Caspi, R.R. Either a Th17 or a Th1 effector response can drive autoimmunity: Conditions of disease induction affect dominant effector category. J. Exp. Med. 2008, 205, 799–810. [Google Scholar] [CrossRef]
- Wakefield, D.; Li, Q.; McCluskey, P.; Nussenblatt, R.B.; Chan, C.-C. Immunohistochemical localization of T lymphocytes and macrophages and expression of interferon gamma and defensin in uveitis. Ocul. Immunol. Inflamm. 1994, 2, 153–159. [Google Scholar] [CrossRef]
- Chao, J.; Guo, Y.; Chao, L. Protective Role of Endogenous Kallistatin in Vascular Injury and Senescence by Inhibiting Oxidative Stress and Inflammation. Oxid. Med. Cell Longev. 2018, 2018, 4138560. [Google Scholar] [CrossRef]
- Chen, N.; Chen, S.; Zhang, Z.; Cui, X.; Wu, L.; Guo, K.; Shao, H.; Ma, J.X.; Zhang, X. Overexpressing Kallistatin Aggravates Experimental Autoimmune Uveitis Through Promoting Th17 Differentiation. Front. Immunol. 2021, 12, 756423. [Google Scholar] [CrossRef]
- Pan, S.; Tan, H.; Chang, R.; Wang, Q.; Zhu, Y.; Chen, L.; Li, H.; Su, G.; Zhou, C.; Cao, Q.; et al. High Ambient Temperature Aggravates Experimental Autoimmune Uveitis Symptoms. Front. Cell Dev. Biol. 2021, 9, 629306. [Google Scholar] [CrossRef]
- Zaslona, Z.; O’Neill, L.A.J. Cytokine-like Roles for Metabolites in Immunity. Mol. Cell 2020, 78, 814–823. [Google Scholar] [CrossRef]
- Mills, E.; O’Neill, L.A. Succinate: A metabolic signal in inflammation. Trends Cell Biol. 2014, 24, 313–320. [Google Scholar] [CrossRef]
- Malalana, F.; Stylianides, A.; McGowan, C. Equine recurrent uveitis: Human and equine perspectives. Vet. J. 2015, 206, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Deeg, C.A.; Hauck, S.M.; Amann, B.; Pompetzki, D.; Altmann, F.; Raith, A.; Schmalzl, T.; Stangassinger, M.; Ueffing, M. Equine recurrent uveitis—A spontaneous horse model of uveitis. Ophthalmic. Res. 2008, 40, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Fingerhut, L.; Yucel, L.; Strutzberg-Minder, K.; von Kockritz-Blickwede, M.; Ohnesorge, B.; de Buhr, N. Ex Vivo and In Vitro Analysis Identify a Detrimental Impact of Neutrophil Extracellular Traps on Eye Structures in Equine Recurrent Uveitis. Front. Immunol. 2022, 13, 830871. [Google Scholar] [CrossRef] [PubMed]
- Fingerhut, L.; Ohnesorge, B.; von Borstel, M.; Schumski, A.; Strutzberg-Minder, K.; Morgelin, M.; Deeg, C.A.; Haagsman, H.P.; Beineke, A.; von Kockritz-Blickwede, M.; et al. Neutrophil Extracellular Traps in the Pathogenesis of Equine Recurrent Uveitis (ERU). Cells 2019, 8, 1528. [Google Scholar] [CrossRef]
- Kulbrock, M.; Lehner, S.; Metzger, J.; Ohnesorge, B.; Distl, O. A genome-wide association study identifies risk loci to equine recurrent uveitis in German warmblood horses. PLoS ONE 2013, 8, e71619. [Google Scholar] [CrossRef]
- Tirosh-Levy, S.; Baum, M.; Schvartz, G.; Kalir, B.; Pe’er, O.; Shnaiderman-Torban, A.; Bernstein, M.; Blum, S.E.; Steinman, A. Seroprevalence of Leptospira spp. in horses in Israel. Pathogens 2021, 10, 408. [Google Scholar] [CrossRef]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef]
- Cheung, N.; Mitchell, P.; Wong, T.Y. Diabetic retinopathy. Lancet 2010, 376, 124–136. [Google Scholar] [CrossRef]
- Baena-Diez, J.M.; Penafiel, J.; Subirana, I.; Ramos, R.; Elosua, R.; Marin-Ibanez, A.; Guembe, M.J.; Rigo, F.; Tormo-Diaz, M.J.; Moreno-Iribas, C.; et al. Risk of Cause-Specific Death in Individuals with Diabetes: A Competing Risks Analysis. Diabetes Care 2016, 39, 1987–1995. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, X.; Yin, Y.; Mai, Y.; Wang, D.; Zhang, X. Hyperglycemia Induces Neutrophil Extracellular Traps Formation through an NADPH Oxidase-Dependent Pathway in Diabetic Retinopathy. Front. Immunol. 2018, 9, 3076. [Google Scholar] [CrossRef]
- Wong, T.Y.; Cheung, C.M.; Larsen, M.; Sharma, S.; Simo, R. Diabetic retinopathy. Nat. Rev. Dis. Prim. 2016, 2, 16012. [Google Scholar] [CrossRef] [PubMed]
- Binet, F.; Cagnone, G.; Crespo-Garcia, S.; Hata, M.; Neault, M.; Dejda, A.; Wilson, A.M.; Buscarlet, M.; Mawambo, G.T.; Howard, J.P.; et al. Neutrophil extracellular traps target senescent vasculature for tissue remodeling in retinopathy. Science 2020, 369, eaay5356. [Google Scholar] [CrossRef]
- Tang, J.; Kern, T.S. Inflammation in diabetic retinopathy. Prog. Retin. Eye Res. 2011, 30, 343–358. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, S.; McGuire, P.G.; Das, A. Diabetic retinopathy and inflammation: Novel therapeutic targets. Middle East Afr. J. Ophthalmol. 2012, 19, 52–59. [Google Scholar]
- Kastelan, S.; Oreskovic, I.; Biscan, F.; Kastelan, H.; Antunica, A.G. Inflammatory and angiogenic biomarkers in diabetic retinopathy. Biochem. Med. 2020, 30, 030502. [Google Scholar] [CrossRef]
- Shurtz-Swirski, R.; Sela, S.; Herskovits, A.T.; Shasha, S.M.; Shapiro, G.; Nasser, L.; Kristal, B. Involvement of peripheral polymorphonuclear leukocytes in oxidative stress and inflammation in type 2 diabetic patients. Diabetes Care 2001, 24, 104–110. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, J.E.; Gu, J.Y.; Yoo, H.J.; Park, S.H.; Kim, Y.I.; Nam-Goong, I.S.; Kim, E.S.; Kim, H.K. Evaluation of Circulating Markers of Neutrophil Extracellular Trap (NET) Formation as Risk Factors for Diabetic Retinopathy in a Case-Control Association Study. Exp. Clin. Endocrinol. Diabetes 2016, 124, 557–561. [Google Scholar] [CrossRef]
- Gupta, A.K.; Joshi, M.B.; Philippova, M.; Erne, P.; Hasler, P.; Hahn, S.; Resink, T.J. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 2010, 584, 3193–3197. [Google Scholar] [CrossRef]
- Schmaier, A.H. The contact activation and kallikrein/kinin systems: Pathophysiologic and physiologic activities. J. Thromb. Haemost. 2016, 14, 28–39. [Google Scholar] [CrossRef]
- Song, D.Y.; Gu, J.Y.; Yoo, H.J.; Kim, Y.I.; Nam-Goong, I.S.; Kim, E.S.; Kim, H.K. Activation of Factor XII and Kallikrein-Kinin System Combined with Neutrophil Extracellular Trap Formation in Diabetic Retinopathy. Exp. Clin. Endocrinol. Diabetes 2021, 129, 560–565. [Google Scholar] [CrossRef]
- Zhang, J.; Dai, Y.; Wei, C.; Zhao, X.; Zhou, Q.; Xie, L. DNase I improves corneal epithelial and nerve regeneration in diabetic mice. J. Cell Mol. Med. 2020, 24, 4547–4556. [Google Scholar] [CrossRef]
- Jaulim, A.; Ahmed, B.; Khanam, T.; Chatziralli, I.P. Branch retinal vein occlusion: Epidemiology, pathogenesis, risk factors, clinical features, diagnosis, and complications. An update of the literature. Retina 2013, 33, 901–910. [Google Scholar] [CrossRef]
- Kesler, A.; Shalev, V.; Rogowski, O.; Shimron, O.; Shainberg, B.; Sela, B.-A.; Shapira, I.; Salomon, O.; Berliner, S. Comparative analysis of homocysteine concentrations in patients with retinal vein occlusion versus thrombotic and atherosclerotic disorders. Blood Coagul. Fibrinolysis 2008, 19, 259–262. [Google Scholar] [CrossRef]
- Ulu, S.M.; Dogan, M.; Ahsen, A.; Altug, A.; Demir, K.; Acarturk, G.; Inan, S. Neutrophil-to-lymphocyte ratio as a quick and reliable predictive marker to diagnose the severity of diabetic retinopathy. Diabetes Technol. Ther. 2013, 15, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Ilhan, N.; Daglioglu, M.C.; Ilhan, O.; Coskun, M.; Tuzcu, E.A.; Kahraman, H.; Keskin, U. Assessment of Neutrophil/Lymphocyte Ratio in Patients with Age-related Macular Degeneration. Ocul. Immunol. Inflamm. 2015, 23, 287–290. [Google Scholar] [CrossRef]
- Liu, Z.; Perry, L.A.; Penny-Dimri, J.C.; Raveendran, D.; Hu, M.L.; Arslan, J.; Britten-Jones, A.C.; O’Hare, F.; Ayton, L.N.; Edwards, T.L. The association of neutrophil–lymphocyte ratio and platelet–lymphocyte ratio with retinal vein occlusion: A systematic review and meta-analysis. Acta Ophthalmol. 2021, 100, e635–e647. [Google Scholar] [CrossRef]
- Wan, W.; Liu, H.; Long, Y.; Wan, W.; Li, Q.; Zhu, W.; Wu, Y. The association between circulating neutrophil extracellular trap related biomarkers and retinal vein occlusion incidence: A case-control pilot study. Exp. Eye Res. 2021, 210, 108702. [Google Scholar] [CrossRef]
- Al-Zamil, W.M.; Yassin, S.A. Recent developments in age-related macular degeneration: A review. Clin. Interv. Aging 2017, 12, 1313–1330. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.S.; Mitchell, P.; Seddon, J.M.; Holz, F.G.; Wong, T.Y. Age-related macular degeneration. Lancet 2012, 379, 1728–1738. [Google Scholar] [CrossRef]
- Jetten, N.; Verbruggen, S.; Gijbels, M.J.; Post, M.J.; de Winther, M.P.; Donners, M.M. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis 2014, 17, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Niazi, S.; Nielsen, M.K.; Sorensen, T.L.; Subhi, Y. Neutrophil-to-lymphocyte ratio in age-related macular degeneration: A systematic review and meta-analysis. Acta Ophthalmol. 2019, 97, 558–566. [Google Scholar] [CrossRef]
- Lechner, J.; Chen, M.; Hogg, R.E.; Toth, L.; Silvestri, G.; Chakravarthy, U.; Xu, H. Alterations in Circulating Immune Cells in Neovascular Age-Related Macular Degeneration. Sci. Rep. 2015, 5, 16754. [Google Scholar] [CrossRef] [PubMed]
- An, Z.; Li, J.; Yu, J.; Wang, X.; Gao, H.; Zhang, W.; Wei, Z.; Zhang, J.; Zhang, Y.; Zhao, J.; et al. Neutrophil extracellular traps induced by IL-8 aggravate atherosclerosis via activation NF-kappaB signaling in macrophages. Cell Cycle 2019, 18, 2928–2938. [Google Scholar] [CrossRef] [PubMed]
- Tibrewal, S.; Sarkar, J.; Jassim, S.H.; Gandhi, S.; Sonawane, S.; Chaudhary, S.; Byun, Y.S.; Ivanir, Y.; Hallak, J.; Horner, J.H.; et al. Tear fluid extracellular DNA: Diagnostic and therapeutic implications in dry eye disease. Investig. Ophthalmol. Vis. Sci. 2013, 54, 8051–8061. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Zhang, L.; Yuan, K.; Huang, X.; Hu, R.; Jin, X. Neutrophil extracellular traps may have a dual role in Pseudomonas aeruginosa keratitis. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Navas, A.; Magana-Guerrero, F.S.; Dominguez-Lopez, A.; Chavez-Garcia, C.; Partido, G.; Graue-Hernandez, E.O.; Sanchez-Garcia, F.J.; Garfias, Y. Anti-Inflammatory and Anti-Fibrotic Effects of Human Amniotic Membrane Mesenchymal Stem Cells and Their Potential in Corneal Repair. Stem Cells Transl. Med. 2018, 7, 906–917. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Ocular Disease | Cell and Animal Model or Patients | Treatment | Detection Methods | References |
|---|---|---|---|---|
| Dry eye (oGVHD) | Thy1-YFP mice | Heparin (100 IU/mL) | Heparin dismantled NETs, IF, HE staining, MTS assay, kinetic assay. | [42] |
| Dry eye patients | 0.1% DNase | OSDI; CGI; SGA; corneal staining, conjunctival staining; mucoid debris/strands; VBR grading scale. | [45,124] | |
| Type 1 diabetic mice model | DNase I | Corneal epithelial wound healing, IF, WB, ELISA, qRT-PCR; corneal reactive oxygen species; measurement of corneal mechanical sensitivity; corneal whole-mount staining. | [111] | |
| Pseudomonas aeruginosa keratitis | P. aeruginosa keratitis mice model | DXM, tobramycin (Tobrex). Combination treatment | Ulcer area; density of opacity; corneal surface regularity; IF; scanning electron microscopy. | [125] |
| Ocular alkali burns | NaOH-stimulated neutrophils | Acetylsalicylic acid | Neutrophil-HCE adhesion assay; HCE proliferation and migration assay. | [79] |
| Rabbit corneal alkali burn model | hAM-MSC | IOP determination; assessment of corneal thickness using ultrahigh-resolution OCT. | [126] | |
| DR | Type 2 diabetic rat model | Anti-VEGF | ELISA; IF; qRT-PCR. | [100] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, J.; Wu, M.; Zhou, Y.; Zhu, M.; Liu, X. Neutrophil Extracellular Traps (NETs) in Ocular Diseases: An Update. Biomolecules 2022, 12, 1440. https://doi.org/10.3390/biom12101440
Zeng J, Wu M, Zhou Y, Zhu M, Liu X. Neutrophil Extracellular Traps (NETs) in Ocular Diseases: An Update. Biomolecules. 2022; 12(10):1440. https://doi.org/10.3390/biom12101440
Chicago/Turabian StyleZeng, Jia, Min Wu, Yamei Zhou, Manhui Zhu, and Xiaojuan Liu. 2022. "Neutrophil Extracellular Traps (NETs) in Ocular Diseases: An Update" Biomolecules 12, no. 10: 1440. https://doi.org/10.3390/biom12101440
APA StyleZeng, J., Wu, M., Zhou, Y., Zhu, M., & Liu, X. (2022). Neutrophil Extracellular Traps (NETs) in Ocular Diseases: An Update. Biomolecules, 12(10), 1440. https://doi.org/10.3390/biom12101440