
Computational Dissection of the Role of Trp305 in the Regulation of the Death-Associated Protein Kinase–Calmodulin Interaction

Abstract
1. Introduction
2. Materials and Methods
2.1. Construction of Simulation Systems
2.2. MD Simulations
2.3. Principal Component Analysis (PCA)
2.4. Cross-Correlation Analysis
2.5. MM/GBSA Binding Free Energy Calculations
3. Results and Discussion
3.1. Overview of the Structural Complex of DAPK1–CaM Interactions
3.2. System Stability
3.3. W305D Mutation Increases the Inter- and Intra-Domain Correlation Motions
3.4. W305D Mutation Increases the Dynamics of DAPK1–CaM Complex
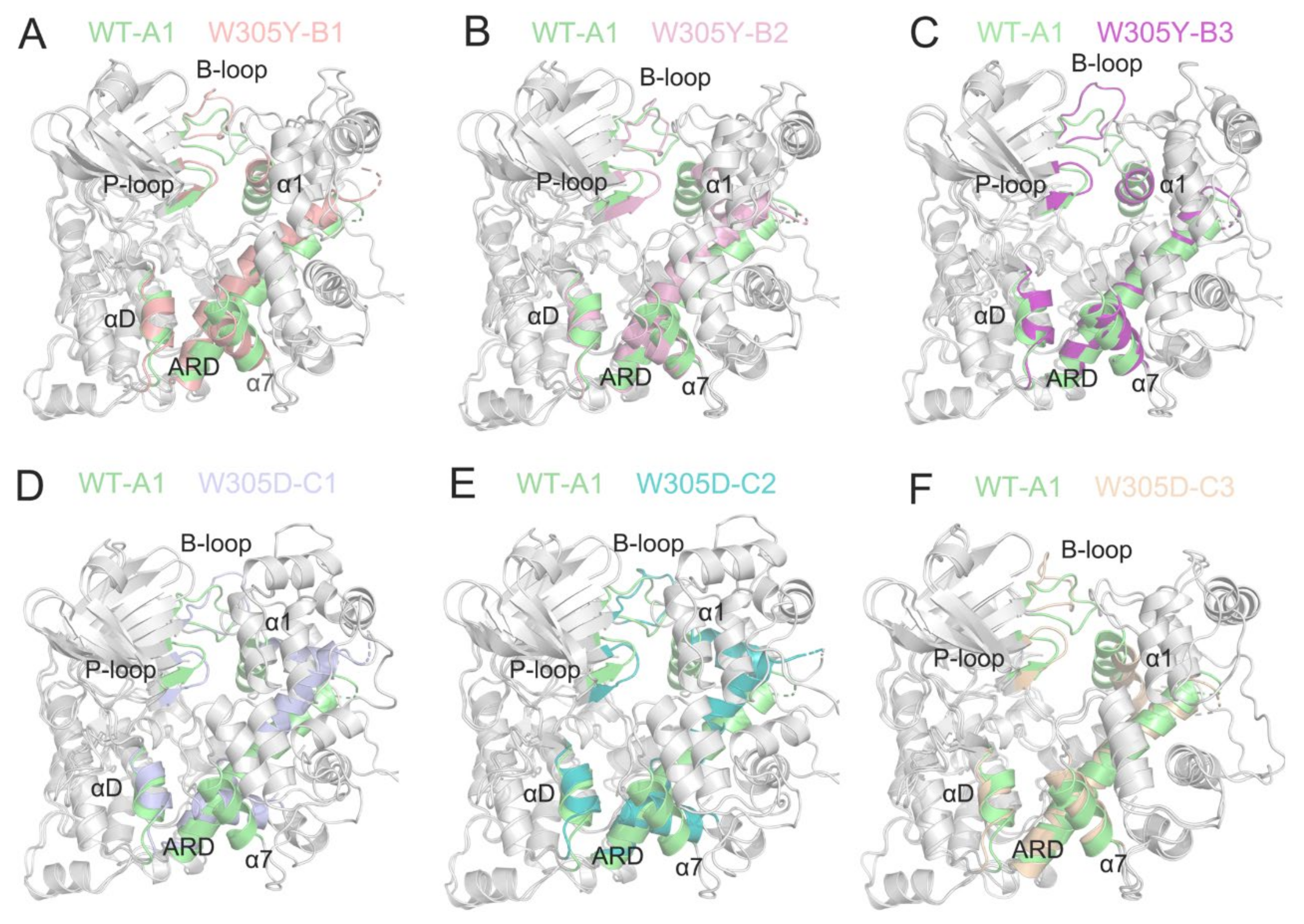
3.5. W305D Mutation Disturbs the DAPK1–CaM Interface
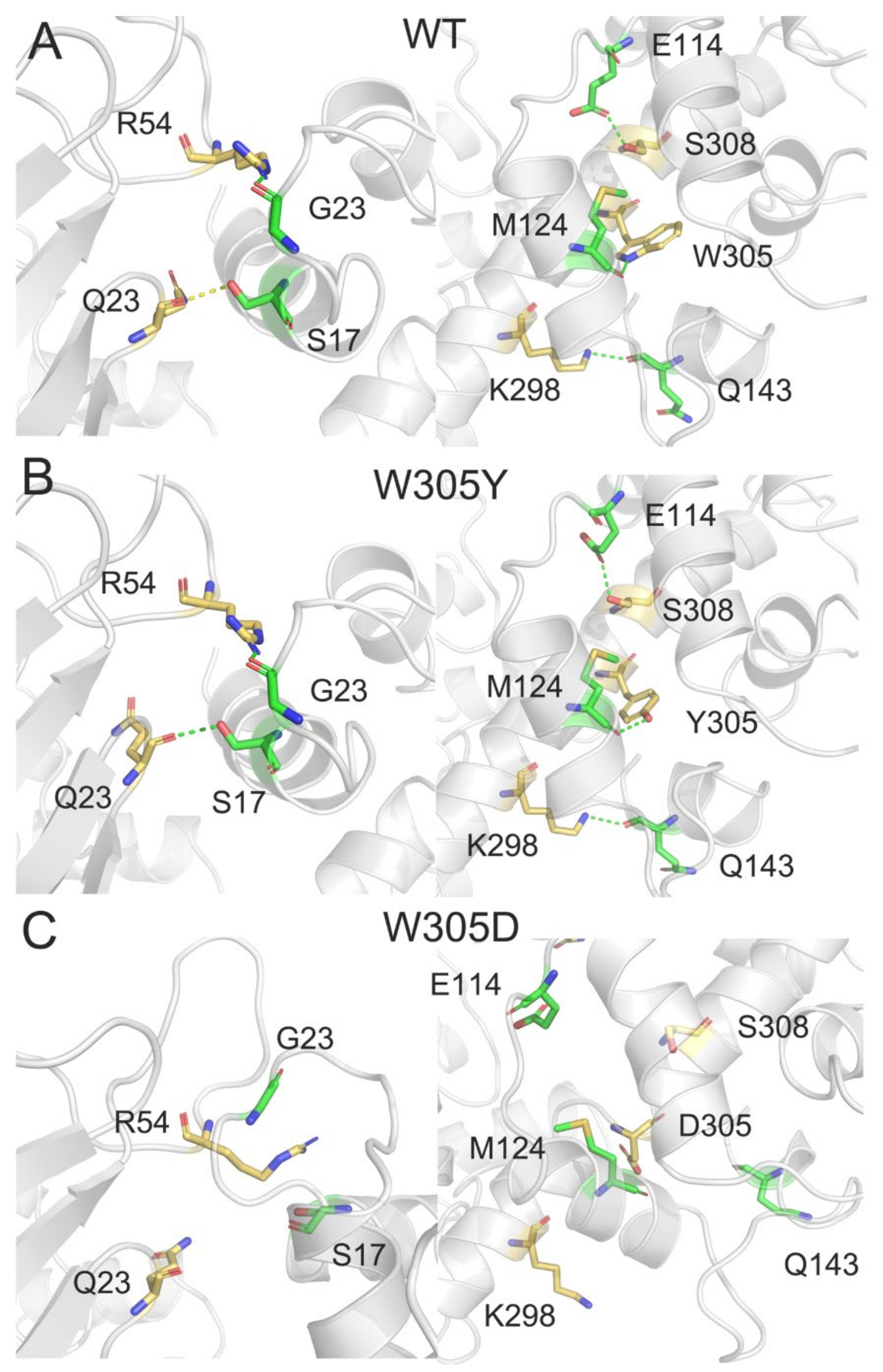
3.6. W305D Mutation Impairs the DAPK1–CaM Interactions
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bialik, S.; Kimchi, A. The death-associated protein kinases: Structure, function, and beyond. Annu. Rev. Biochem. 2006, 75, 189–210. [Google Scholar] [CrossRef] [PubMed]
- Hupp, T.R. Death-associated protein kinase (DAPK) and signal transduction. FEBS J. 2010, 277, 47. [Google Scholar] [CrossRef] [PubMed]
- Farag, A.K.; Roh, E.J. Death-associated protein kinase (DAPK) family modulators: Current and future therapeutic outcomes. Med. Res. Rev. 2019, 39, 349–385. [Google Scholar] [CrossRef] [PubMed]
- Simon, B.; Huart, A.S.; Wilmanns, M. Molecular mechanisms of protein kinase regulation by calcium/calmodulin. Bioorganic Med. Chem. 2015, 23, 2749–2760. [Google Scholar] [CrossRef]
- Elbadawy, M.; Usui, T.; Yamawaki, H.; Sasaki, K. Novel Functions of Death-Associated Protein Kinases through Mitogen-Activated Protein Kinase-Related Signals. Int. J. Mol. Sci. 2018, 19, 3031. [Google Scholar] [CrossRef]
- Xiang, H.; Zhang, J.; Lin, C.; Zhang, L.; Liu, B.; Ouyang, L. Targeting autophagy-related protein kinases for potential therapeutic purpose. Acta Pharm. Sin. B 2020, 10, 569–581. [Google Scholar] [CrossRef]
- Chen, D.; Zhou, X.Z.; Lee, T.H. Death-Associated Protein Kinase 1 as a Promising Drug Target in Cancer and Alzheimer’s Disease. Recent Pat. Anticancer Drug Discov. 2018, 14, 144–157. [Google Scholar] [CrossRef]
- Steinmann, S.; Scheibe, K.; Erlenbach-Wuensch, K.; Neufert, C.; Schneider-Stock, R. Death-associated protein kinase: A molecule with functional antagonistic duality and a potential role in inflammatory bowel disease (Review). Int. J. Oncol. 2015, 47, 5–15. [Google Scholar] [CrossRef]
- Chao, L.H.; Stratton, M.M.; Lee, I.H.; Rosenberg, O.S.; Levitz, J.; Mandell, D.J.; Kortemme, T.; Groves, J.T.; Schulman, H.; Kuriyan, J. A mechanism for tunable autoinhibition in the structure of a human Ca2+/calmodulin-dependent kinase II holoenzyme. Cell 2011, 146, 732–745. [Google Scholar] [CrossRef]
- Jin, Y.; Blue, E.K.; Gallagher, P.J. Control of death-associated protein kinase (DAPK) activity by phosphorylation and proteasomal degradation. J. Biol. Chem. 2006, 281, 39033–39040. [Google Scholar] [CrossRef]
- De Diego, I.; Kuper, J.; Bakalova, N.; Kursula, P.; Wilmanns, M. Molecular basis of the death-associated protein kinase-calcium/calmodulin regulator complex. Sci. Signal. 2010, 3, ra6. [Google Scholar] [CrossRef]
- Lu, S.; Ni, D.; Wang, C.; He, X.; Lin, H.; Wang, Z.; Zhang, J. Deactivation Pathway of Ras GTPase Underlies Conformational Substates as Targets for Drug Design. ACS Catal. 2019, 9, 7188–7196. [Google Scholar] [CrossRef]
- Lu, S.; He, X.; Yang, Z.; Chai, Z.; Zhou, S.; Wang, J.; Rehman, A.U.; Ni, D.; Pu, J.; Sun, J.; et al. Activation pathway of a G protein-coupled receptor uncovers conformational intermediates as targets for allosteric drug design. Nat. Commun. 2021, 12, 4721. [Google Scholar] [CrossRef]
- Lu, S.; Chen, Y.; Wei, J.; Zhao, M.; Ni, D.; He, X.; Zhang, J. Mechanism of allosteric activation of SIRT6 revealed by the action of rationally designed activators. Acta Pharm. Sin. B 2021, 11, 1355–1361. [Google Scholar] [CrossRef]
- Maloney, R.C.; Zhang, M.; Jang, H.; Nussinov, R. The mechanism of activation of monomeric B-Raf V600E. Comput. Struct. Biotechnol. J. 2021, 19, 3349–3363. [Google Scholar] [CrossRef]
- Jang, H.; Zhang, M.; Nussinov, R. The quaternary assembly of KRas4B with Raf-1 at the membrane. Comput. Struct. Biotechnol. J. 2020, 18, 737–748. [Google Scholar] [CrossRef]
- Ni, D.; Wei, J.; He, X.; Rehman, A.U.; Li, X.; Qiu, Y.; Pu, J.; Lu, S.; Zhang, J. Discovery of cryptic allosteric sites using reversed allosteric communication by a combined computational and experimental strategy. Chem. Sci. 2021, 12, 464–476. [Google Scholar] [CrossRef]
- An, X.; Bai, Q.; Bing, Z.; Liu, H.; Yao, X. Insights into the molecular mechanism of positive cooperativity between partial agonist MK-8666 and full allosteric agonist AP8 of hGPR40 by Gaussian accelerated molecular dynamics (GaMD) simulations. Comput. Struct. Biotechnol. J. 2021, 19, 3978–3989. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, Y.; Leung, E.L.H.; Yao, X. In silico study of intrinsic dynamics of full-length apo-ACE2 and RBD-ACE2 complex. Comput. Struct. Biotechnol. J. 2021, 19, 5455–5465. [Google Scholar] [CrossRef]
- Qiu, Y.; Yin, X.; Li, X.; Wang, Y.; Fu, Q.; Huang, R.; Lu, S. Untangling Dual-Targeting Therapeutic Mechanism of Epidermal Growth Factor Receptor (EGFR) Based on Reversed Allosteric Communication. Pharmaceutics 2021, 13, 747. [Google Scholar] [CrossRef]
- Wang, Y.; Ji, D.; Lei, C.; Chen, Y.; Qiu, Y.; Li, X.; Li, M.; Ni, D.; Pu, J.; Zhang, J.; et al. Mechanistic insights into the effect of phosphorylation on Ras conformational dynamics and its interactions with cell signaling proteins. Comput. Struct. Biotechnol. J. 2021, 19, 1184–1199. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N.long(N)method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Wu, X.; Brooks, B.R. Self-guided Langevin dynamics simulation method. Chem. Phys. Lett. 2003, 381, 512–518. [Google Scholar] [CrossRef]
- Saha, A.; Arantes, P.R.; Hsu, R.V.; Narkhede, Y.B.; Jinek, M.; Palermo, G. Molecular Dynamics Reveals a DNA-Induced Dynamic Switch Triggering Activation of CRISPR-Cas12a. J. Chem. Inf. Model. 2020, 60, 6427–6437. [Google Scholar] [CrossRef]
- Masterson, L.R.; Shi, L.; Metcalfe, E.; Gao, J.; Taylor, S.S.; Veglia, G. Dynamically committed, uncommitted, and quenched states encoded in protein kinase A revealed by NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2011, 108, 6969–69674. [Google Scholar] [CrossRef]
- Grant, B.J.; Rodrigues, A.P.C.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S.D. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Wang, Q.; Qi, X.; Liu, Y.; Li, G.; Lu, S.; Mou, L.; Chen, X. Deciphering the Mechanism of Gilteritinib Overcoming Lorlatinib Resistance to the Double Mutant I1171N/F1174I in Anaplastic Lymphoma Kinase. Front. Cell Dev. Biol. 2021, 9, 808864. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, C.; Peng, T.; Chai, Z.; Ni, D.; Liu, Y.; Zhang, J.; Chen, T.; Lu, S. Atomic-scale insights into allosteric inhibition and evolutional rescue mechanism of Streptococcus thermophilus Cas9 by the anti-CRISPR protein AcrIIA6. Comput. Struct. Biotechnol. J. 2021, 19, 6108–6124. [Google Scholar] [CrossRef] [PubMed]
- Palermo, G.; Miao, Y.; Walker, R.C.; Jinek, M.; McCammon, J.A. Striking plasticity of CRISPR-Cas9 and key role of non-target DNA, as revealed by molecular simulations. ACS Cent. Sci. 2016, 2, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- He, X.; Huang, N.; Qiu, Y.; Zhang, J.; Liu, Y.; Yin, X.-L.; Lu, S. Conformational Selection Mechanism Provides Structural Insights into the Optimization of APC-Asef Inhibitors. Molecules 2021, 26, 962. [Google Scholar] [CrossRef]
- Hassan, B.A.; Milicaj, J.; Ramirez-Mondragon, C.A.; Sham, Y.Y.; Taylor, E.A. Ligand-Induced Conformational and Dynamical Changes in a GT-B Glycosyltransferase: Molecular Dynamics Simulations of Heptosyltransferase i Complexes. J. Chem. Inf. Model. 2022, 62, 324–339. [Google Scholar] [CrossRef]
- Li, X.; Dai, J.; Ni, D.; He, X.; Zhang, H.; Zhang, J.; Fu, Q.; Liu, Y.; Lu, S. Insight into the mechanism of allosteric activation of PI3Kα by oncoprotein K-Ras4B. Int. J. Biol. Macromol. 2020, 144, 643–655. [Google Scholar] [CrossRef]
- Movilla, S.; Martí, S.; Roca, M.; Moliner, V. Unrevealing the Proteolytic Activity of RgpB Gingipain from Computational Simulations. J. Chem. Inf. Model. 2021, 61, 4582–4593. [Google Scholar] [CrossRef]
- Gosu, V.; Sasidharan, S.; Saudagar, P.; Lee, H.-K.; Shin, D. Computational Insights into the Structural Dynamics of MDA5 Variants Associated with Aicardi–Goutières Syndrome and Singleton–Merten Syndrome. Biomolecules 2021, 11, 1251. [Google Scholar] [CrossRef]
- Sultana, K.N.; Kuldeep, J.; Siddiqi, M.I.; Srivastava, S.K. Crystallographic and molecular dynamics simulation analysis of NAD synthetase from methicillin resistant Staphylococcus aureus (MRSA). Int. J. Biol. Macromol. 2020, 165, 2349–2362. [Google Scholar] [CrossRef]
- Baidya, A.T.; Kumar, A.; Kumar, R.; Darreh-Shori, T. Allosteric Binding Sites of Aβ Peptides on the Acetylcholine Synthesizing Enzyme ChAT as Deduced by In Silico Molecular Modeling. Int. J. Mol. Sci. 2022, 23, 6073. [Google Scholar] [CrossRef]
- Alaofi, A.L.; Shahid, M. Mutations of SARS-CoV-2 RBD may alter its molecular structure to improve its infection efficiency. Biomolecules 2021, 11, 1273. [Google Scholar] [CrossRef]
- Kumari, A.; Mittal, L.; Srivastava, M.; Pathak, D.P.; Asthana, S. Conformational Characterization of the Co-Activator Binding Site Revealed the Mechanism to Achieve the Bioactive State of FXR. Front. Mol. Biosci. 2021, 8, 658312. [Google Scholar] [CrossRef]
- Wu, D.; Xiao, J.; Salsbury, F.R. Light Chain Mutation Effects on the Dynamics of Thrombin. J. Chem. Inf. Model. 2021, 61, 950–965. [Google Scholar] [CrossRef]
- Wang, Y.; Li, M.; Liang, W.; Shi, X.; Fan, J.; Kong, R.; Liu, Y.; Zhang, J.; Chen, T.; Lu, S. Delineating the activation mechanism and conformational landscape of a class B G protein-coupled receptor glucagon receptor. Comput. Struct. Biotechnol. J. 2022, 20, 628–639. [Google Scholar] [CrossRef]
- Chen, J.; Wang, W.; Pang, L.; Zhu, W. Unveiling conformational dynamics changes of H-Ras induced by mutations based on accelerated molecular dynamics. Phys. Chem. Chem. Phys. 2020, 22, 21238–21250. [Google Scholar] [CrossRef]
- Neves Cruz, J.; De Oliveira, M.S.; Gomes Silva, S.; Da Silva Souza Filho, A.P.; Santiago Pereira, D.; Lima e Lima, A.H.; De Aguiar Andrade, E.H. Insight into the Interaction Mechanism of Nicotine, NNK, and NNN with Cytochrome P450 2A13 Based on Molecular Dynamics Simulation. J. Chem. Inf. Model. 2020, 60, 766–776. [Google Scholar] [CrossRef]
- Qu, Z.; Li, K.; Geng, X.; Huang, B.; Gao, J. Computational Insights Into the Effects of the R190K and N121Q Mutations on the SARS-CoV-2 Spike Complex With Biliverdin. Front. Mol. Biosci. 2021, 8, 791885. [Google Scholar] [CrossRef]
- Liu, C.; Li, Z.; Liu, Z.; Yang, S.; Wang, Q.; Chai, Z. Understanding the P-Loop Conformation in the Determination of Inhibitor Selectivity Toward the Hepatocellular Carcinoma-Associated Dark Kinase STK17B. Front. Mol. Biosci. 2022, 9, 901603. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, Y.; Zhang, Y.; Liu, Z.; Mao, F.; Chai, Z. Mechanistic Insights into the Mechanism of Inhibitor Selectivity toward the Dark Kinase STK17B against Its High Homology STK17A. Molecules 2022, 27, 4655. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ni, D.; Fan, J.; Li, M.; Zhang, J.; Hua, C.; Nussinov, R.; Lu, S. Markov State Models and Molecular Dynamics Simulations Reveal the Conformational Transition of the Intrinsically Disordered Hypervariable Region of K-Ras4B to the Ordered Conformation. J. Chem. Inf. Model. 2022, 63, 4222–4231. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, Y.; Fan, J.; Zhuang, H.; Liu, Y.; Ji, D.; Lu, S. Mechanistic Insights into the Long-Range Allosteric Regulation of KRAS Via Neurofibromatosis Type 1 (NF1) Scaffold Upon SPRED1 Loading. J. Mol. Biol. 2022, 434, 167730. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT | W305Y | W305D | |
|---|---|---|---|
| ΔEvdW | −189.72 ± 11.76 | −184.08 ± 11.64 | −189.00 ± 10.64 |
| ΔEele | −1743.98 ± 106.00 | −1732.59 ± 120.61 | −1357.90 ± 118.32 |
| ΔGSA | −29.49 ± 1.36 | −30.30 ± 1.60 | −29.34 ± 1.38 |
| ΔGGB | 1817.26 ± 98.24 | 1797.89 ± 112.31 | 1454.53 ± 108.30 |
| ΔGbinding | −116.44 ± 15.60 | −118.77 ± 16.86 | −92.38 ± 17.54 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Y.-P.; Gao, X.-Y.; Xu, G.-H.; Qin, Z.-F.; Ju, H.-X.; Li, D.-C.; Ma, D.-N. Computational Dissection of the Role of Trp305 in the Regulation of the Death-Associated Protein Kinase–Calmodulin Interaction. Biomolecules 2022, 12, 1395. https://doi.org/10.3390/biom12101395
Zhu Y-P, Gao X-Y, Xu G-H, Qin Z-F, Ju H-X, Li D-C, Ma D-N. Computational Dissection of the Role of Trp305 in the Regulation of the Death-Associated Protein Kinase–Calmodulin Interaction. Biomolecules. 2022; 12(10):1395. https://doi.org/10.3390/biom12101395
Chicago/Turabian StyleZhu, Yu-Ping, Xin-Yi Gao, Guo-Hui Xu, Zhao-Fu Qin, Hai-Xing Ju, De-Chuan Li, and De-Ning Ma. 2022. "Computational Dissection of the Role of Trp305 in the Regulation of the Death-Associated Protein Kinase–Calmodulin Interaction" Biomolecules 12, no. 10: 1395. https://doi.org/10.3390/biom12101395
APA StyleZhu, Y.-P., Gao, X.-Y., Xu, G.-H., Qin, Z.-F., Ju, H.-X., Li, D.-C., & Ma, D.-N. (2022). Computational Dissection of the Role of Trp305 in the Regulation of the Death-Associated Protein Kinase–Calmodulin Interaction. Biomolecules, 12(10), 1395. https://doi.org/10.3390/biom12101395