
Drug-Disease Association Prediction Using Heterogeneous Networks for Computational Drug Repositioning


Abstract
1. Introduction
2. Review of Network-Based Drug-Repositioning Approaches
2.1. Graph Mining Algorithms
2.1.1. Heterogeneous Network Clustering
2.1.2. TL_HGBI
2.1.3. DrugNet
2.1.4. MBiRW
2.1.5. TP-NRWRH
2.1.6. DR-IBRW
2.1.7. EMP-SVD
- Drug → (treats) → Disease
- Drug → (binds to) → Protein → (causes) → Disease
- Drug → (binds to) → Protein → (binds to) → Drug → (treats) → Disease
- Drug → (treats) → Disease → (treated by) → Drug → (treats) → Disease
- Drug → (treats) → Disease → (caused by) → Protein → (causes) → Disease
2.1.8. BGMSDDA
2.2. Matrix Factorization or Matrix Completion
2.2.1. DRRS
2.2.2. OMC
2.2.3. DRIMC
2.2.4. MSBMF
2.2.5. NTD-DR
2.3. Deep Learning
2.3.1. deepDR
2.3.2. ANMF
2.3.3. NEDD
2.3.4. SNF-NN
2.3.5. SAEROF
2.3.6. LAGCN
3. Experiments
3.1. Experimental Data
3.1.1. Drug Network
3.1.2. Disease Network
3.1.3. Gene Network
3.1.4. Associations
3.2. Experimental Settings
4. Results
4.1. Accuracy Comparison with Network-CS
4.2. Accuracy Comparison with Network-ATC
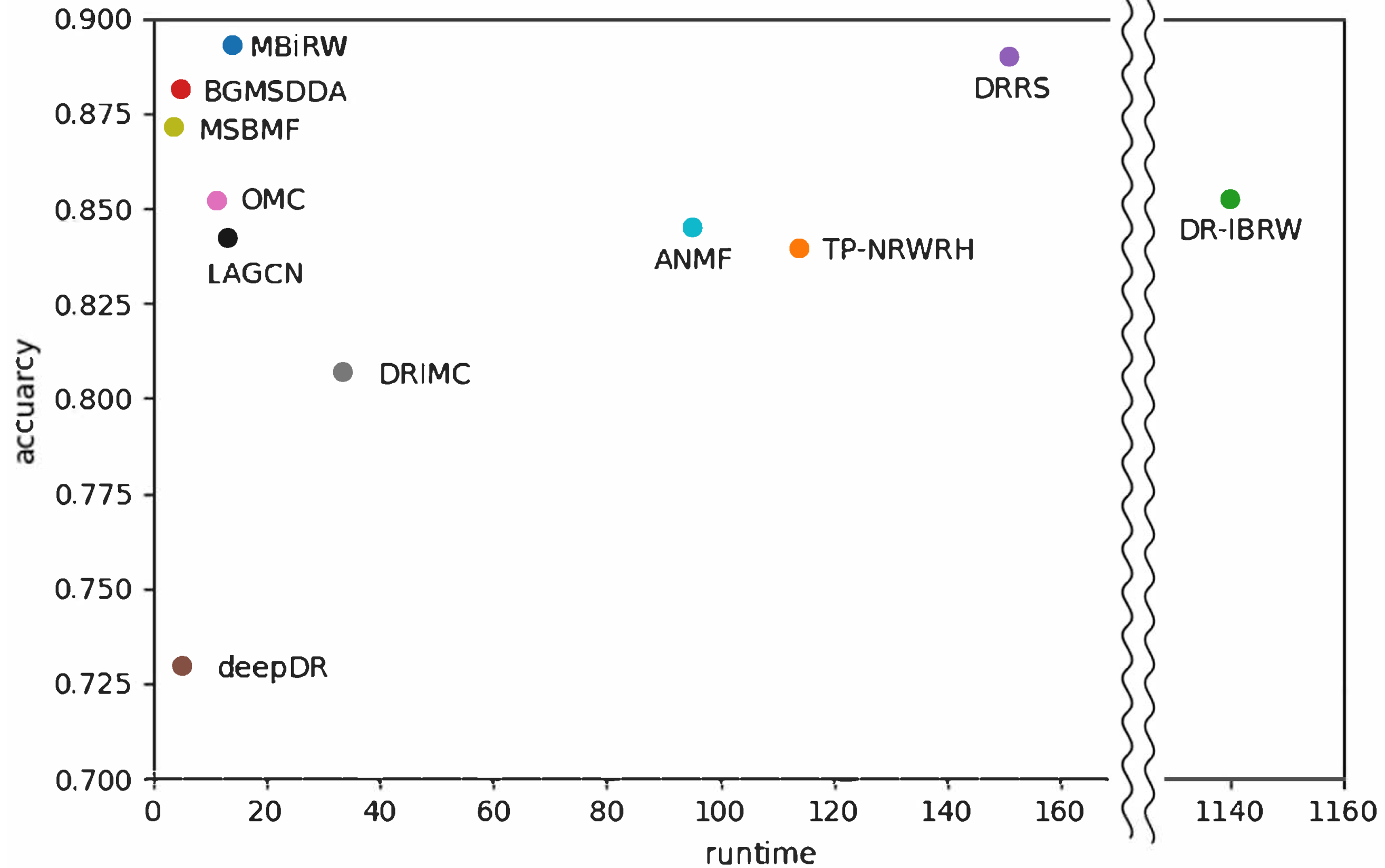
4.3. Robustness Comparison
4.4. Efficiency Comparison
5. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CS | Chemical structures |
| ATC | Anatomic Therapeutic Chemical |
| PPI | Protein-protein interaction |
| DTI | Drug-target interaction |
| ROC | Receiver operating characteristic |
| AUC | The area under the ROC curve |
| AUPR | The area under the precision-recall curve |
| TP | True positives |
| TPR | True positive Rate |
| FP | False positives |
| FPR | False positive Rate |
References
- Chan, H.C.S.; Shan, H.; Dahoun, T.; Vogel, H.; Yuan, S. Advancing drug discovery via artificial intelligence. Trends Pharmacol. Sci. 2019, 40, 801. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, I.; Lue, T.F.; Padma-Nathan, H.; Rosen, R.C.; Steers, W.D.; Wicker, P.A. Oral sildenafil in the treatment of erectile dysfunction. N. Engl. J. Med. 1998, 338, 1397–1404. [Google Scholar] [CrossRef]
- Singh, T.U.; Parida, S.; Lingaraju, M.C.; Kesavan, M.; Kumar, D.; Singh, R.K. Drug repurposing approach to fight COVID-19. Pharmacol. Rep. 2020, 72, 1479–1508. [Google Scholar] [CrossRef]
- Shim, J.S.; Liu, J.O. Recent advances in drug repositioning for the discovery of new anticancer drugs. Int. J. Biol. Sci. 2014, 10, 654–663. [Google Scholar] [CrossRef]
- Kim, T.W. Drug repositioning approaches for the discovery of new therapeutics for Alzheimer’s disease. Neurotherapeutics 2015, 12, 132–142. [Google Scholar] [CrossRef]
- Shahreza, M.L.; Ghadiri, N.; Mousavi, S.R.; Varshosaz, J.; Green, J.R. A review of network-based approaches to drug repositioning. Briefings Bioinform. 2018, 19, 878–892. [Google Scholar] [CrossRef]
- Xue, H.; Li, J.; Xie, H.; Wang, Y. Review of drug repositioning approaches and resources. Int. J. Biol. Sci. 2018, 14, 1232–1244. [Google Scholar] [CrossRef]
- Gysi, D.M.; do Valle, I.; Zitnik, M.; Ameli, A.; Gan, X.; Varol, O.; Ghiassian, S.D.; Patten, J.J.; Davey, R.A.; Loscalzo, J.; et al. Network medicine framework for identifying drug-repurposing opportunities for COVID-19. Proc. Natl. Acad. Sci. USA 2021, 118, e2025581118. [Google Scholar] [CrossRef]
- Hopkins, A.L. Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef]
- Yildirim, M.A.; Goh, K.-I.; Cusick, M.E.; Barabasi, A.-L.; Vidal, M. Drug-target network. Nat. Biotechnol. 2007, 25, 1119–1126. [Google Scholar] [CrossRef]
- Yamanishi, Y.; Araki, M.; Gutteridge, A.; Honda, W.; Kanehisa, M. Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 2008, 24, i232–i240. [Google Scholar] [CrossRef]
- Luo, Y.; Zhao, X.; Zhou, J.; Yang, J.; Zhang, Y.; Kuang, W.; Peng, J.; Chen, L.; Zeng, J. A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nat. Commun. 2017, 8, 573. [Google Scholar] [CrossRef]
- Wan, F.; Hong, L.; Xiao, A.; Jiang, T.; Zeng, J. NeoDTI: Neural integration of neighbor information from a heterogeneous network for discovering new drug–target interactions. Bioinformatics 2019, 35, 104–111. [Google Scholar] [CrossRef]
- Zeng, X.; Zhu, S.; Lu, W.; Liu, Z.; Huang, J.; Zhou, Y.; Fang, J.; Huang, Y.; Guo, H.; Li, L.; et al. Target identification among known drugs by deep learning from heterogeneous networks. Chem. Sci. 2020, 11, 1775. [Google Scholar] [CrossRef]
- Ding, H.; Takigawa, I.; Mamitsuka, H.; Zhu, S. Similarity-based machine learning methods for predicting drug–target interactions: A brief review. Briefings Bioinform. 2014, 15, 734–747. [Google Scholar] [CrossRef]
- Chen, R.; Liu, X.; Jin, S.; Lin, J.; Liu, J. Machine learning for drug-target interaction prediction. Molecules 2018, 23, 2208. [Google Scholar] [CrossRef]
- Bagherian, M.; Sabeti, E.; Wang, K.; Sartor, M.A.; Nikolovska-Coleska, Z.; Najarian, K. Machine learning approaches and databases for prediction of drug-target interaction: A survey paper. Briefings Bioinform. 2021, 22, 247–269. [Google Scholar] [CrossRef]
- Ru, X.; Ye, X.; Sakurai, T.; Zou, Q.; Xu, L.; Lin, C. Current status and future prospects of drug-target interaction prediction. Briefings Funct. Genom. 2021, 20, 312–322. [Google Scholar] [CrossRef]
- Jung, Y.-S.; Kim, Y.; Cho, Y.-R. Comparative analysis of network-based approaches and machine learning algorithms for predicting drug-target interactions. Methods 2022, 198, 19–31. [Google Scholar] [CrossRef]
- Barabasi, A.-L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef]
- Ata, S.K.; Wu, M.; Fang, Y.; Ou-Yang, L.; Kwoh, C.K.; Li, X.-L. Recent advances in network-based methods for disease gene prediction. Briefings Bioinform. 2021, 22, bbaa303. [Google Scholar] [CrossRef]
- Kim, Y.; Park, J.-H.; Cho, Y.-R. Network-based approaches for disease-gene association prediction using protein–protein interaction networks. Int. J. Mol. Sci. 2022, 23, 7411. [Google Scholar] [CrossRef]
- Wu, C.; Gudivada, R.C.; Aronow, B.J.; Jegga, A.G. Computational drug repositioning through heterogeneous network clustering. Bmc Syst. Biol. 2013, 7, S6. [Google Scholar] [CrossRef]
- Wang, W.; Yang, S.; Zhang, X.; Li, J. Drug repositioning by integrating target information through a heterogeneous network model. Bioinformatics 2014, 30, 2923–2930. [Google Scholar] [CrossRef]
- Martinez, V.; Navarro, C.; Cano, C.; Fajardo, W.; Blanco, A. DrugNet: Network-based drug-disease prioritization by integrating heterogeneous data. Artif. Intell. Med. 2015, 63, 41–49. [Google Scholar] [CrossRef]
- Luo, H.; Wang, J.; Li, M.; Luo, J.; Peng, X.; Wu, F.-X.; Pan, Y. Drug repositioning based on comprehensive similarity measures and Bi-Random walk algorithm. Bioinformatics 2016, 32, 2664–2671. [Google Scholar] [CrossRef]
- Liu, H.; Song, Y.; Guan, J.; Luo, L.; Zhuang, Z. Inferring new indications for approved drugs via random walk on drug-disease heterogenous networks. Bmc Bioinform. 2016, 17, 539. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, M.; Ren, Y.; Jia, L.; Yu, G. Drug repositioning based on individual bi-random walks on a heterogeneous network. BMC Bioinform. 2019, 20, 547. [Google Scholar] [CrossRef]
- Wu, G.; Liu, J.; Yue, X. Prediction of drug-disease associations based on ensemble meta paths and singular value decomposition. BMC Bioinform. 2019, 20, 134. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Li, J.; Gu, G.; Sun, Y.; Lin, Z.; Zhu, Y.; Wang, W. BGMSDDA: A bipartite graph diffusion algorithm with multiple similarity integration for drug–disease association prediction. Mol. Omics 2021, 17, 997–1011. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Li, M.; Wang, S.; Liu, Q.; Li, Y.; Wang, J. Computational drug repositioning using low-rank matrix approximation and randomized algorithms. Bioinformatics 2018, 34, 1904–1912. [Google Scholar] [CrossRef]
- Yang, M.; Luo, H.; Li, Y.; Wu, F.-X.; Wang, J. Overlap matrix completion for predicting drug-associated indications. PLoS Comput. Biol. 2019, 15, e1007541. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xu, H.; Li, X.; Gao, Q.; Wang, L. DRIMC: An improved drug repositioning approach using Bayesian inductive matrix completion. Bioinformatics 2020, 36, 2839–2847. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wu, G.; Zhao, Q.; Li, Y.; Wang, J. Computational drug repositioning based on multi-similarities bilinear matrix factorization. Briefings Bioinform. 2021, 22, bbaa267. [Google Scholar] [CrossRef] [PubMed]
- Jamali, A.A.; Tan, Y.; Kusalik, A.; Wu, F.-X. NTD-DR: Nonnegative tensor decomposition for drug repositioning. PLoS ONE 2022, 17, e0270852. [Google Scholar]
- Zeng, X.; Zhu, S.; Liu, X.; Zhou, Y.; Nussinov, R.; Cheng, F. deepDR: A network-based deep learning approach to in silico drug repositioning. Bioinformatics 2019, 35, 5191–5198. [Google Scholar] [CrossRef]
- Yang, X.; Zamit, L.; Liu, Y.; He, J. Additional neural matrix factorization model for computational drug repositioning. BMC Bioinform. 2019, 20, 423. [Google Scholar] [CrossRef]
- Zhou, R.; Lu, Z.; Luo, H.; Xiang, J.; Zeng, M.; Li, M. NEDD: A network embedding based method for predicting drug-disease associations. BMC Bioinform. 2020, 21, 387. [Google Scholar] [CrossRef]
- Jarada, T.N.; Rokne, J.G.; Alhajj, R. SNF-NN: Computational method to predict drug-disease interactions using similarity network fusion and neural networks. BMC Bioinform. 2021, 22, 28. [Google Scholar] [CrossRef]
- Jiang, H.-J.; Huang, Y.-A.; You, Z.-H. SAEROF: An ensemble approach for large-scale drug-disease association prediction by incorporating rotation forest and sparse autoencoder deep neural network. Sci. Rep. 2020, 10, 4972. [Google Scholar] [CrossRef]
- Yu, Z.; Huang, F.; Zhao, X.; Xiao, W.; Zhang, W. Predicting drug–disease associations through layer attention graph convolutional network. Briefings Bioinform. 2021, 22, bbaa243. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Nepusz, T.; Yu, H.; Paccanaro, A. Detecting overlapping protein complexes in protein–protein interaction networks. Nat. Methods 2012, 9, 471–472. [Google Scholar] [CrossRef]
- Martinez, V.; Cano, C.; Blanco, A. ProphNet: A generic prioritization method through propagation of information. BMC Bioinform. 2014, 15, S5. [Google Scholar] [CrossRef]
- Chen, X.; Liu, M.-X.; Yan, G.-Y. Drug-target interaction prediction by random walk on the heterogeneous network. Mol. Biosyst. 2012, 8, 1970–1978. [Google Scholar] [CrossRef]
- Yang, M.; Luo, H.; Li, Y.; Wang, J. Drug repositioning based on bounded nuclear norm regularization. Bioinformatics 2019, 35, i455–i463. [Google Scholar] [CrossRef]
- Wang, B.; Mezlini, A.M.; Demir, F.; Fiume, M.; Tu, Z.; Brudno, M.; Haibe-Kains, B.; Goldenberg, A. Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods 2014, 11, 333–337. [Google Scholar] [CrossRef]
- Boyd, S.; Parikh, N.; Chu, E.; Peleato, B.; Eckstein, J. Distributed optimization and statistical learning via the alternating direction method of multipliers. Found. Trends® Mach. Learn. 2011, 3, 1–122. [Google Scholar]
- Bullinaria, J.A.; Levy, J.P. Extracting semantic representations from word co-occurrence statistics: A computational study. Behav. Res. Methods 2007, 39, 510–526. [Google Scholar] [CrossRef]
- Chen, Y.; de Rijke, M. A collective variational autoencoder for top-N recommendation with side information. In Proceedings of the 3rd Workshop on Deep Learning for Recommender Systems, Vancouver, BC, Canada, 6 October 2018; pp. 3–9. [Google Scholar]
- Fu, T.; Lee, W.-C.; Lei, Z. HIN2Vec: Explore meta-paths in heterogeneous information networks for representation learning. In Proceedings of the 2017 ACM Conference on Information and Knowledge Management (CIKM), Singapore, 6–10 November 2017; pp. 1797–1806. [Google Scholar]
- Grover, A.; Leskovec, J. node2vec: Scalable feature learning for networks. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 855–864. [Google Scholar]
- Rodriguez, J.J.; Kuncheva, L.I.; Alonso, C.J. Rotation Forest: A new classifier ensemble method. IEEE Trans. Pattern Anal. Mach. Intell. 2006, 28, 1619–1630. [Google Scholar] [CrossRef]
- Köhler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 2021, 49, D1207–D1217. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1998, 28, 31–36. [Google Scholar] [CrossRef]
- Steinbeck, C.; Han, Y.; Kuhn, S.; Horlacher, O.; Luttmann, E.; Willighagen, E. The Chemistry Development Kit (CDK): An open-source Java library for chemo- and bioinformatics. J. Chem. Inf. Comput. Sci. 2003, 43, 493–500. [Google Scholar] [CrossRef]
- Olson, T.; Singh, R. Predicting anatomic therapeutic chemical classification codes using tiered learning. BMC Bioinform. 2017, 18, 266. [Google Scholar] [CrossRef] [PubMed]
- Amberger, J.S.; Bocchini, C.A.; Scott, A.F.; Hamosh, A. OMIM.org: Leveraging knowledge across phenotype–gene relationships. Nucleic Acids Res. 2019, 47, D1038–D1043. [Google Scholar] [CrossRef] [PubMed]
- Wakap, S.N.; Lambert, D.M.; Olry, A.; Rodwell, C.; Gueydan, C.; Lanneau, V.; Murphy, D.; Le Cam, Y.; Rath, A. Estimating cumulative point prevalence of rare diseases: Analysis of the Orphanet database. Eur. J. Hum. Genet. 2009, 28, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Bragin, E.; Chatzimichali, E.A.; Wright, C.F.; Hurles, M.E.; Firth, H.V.; Bevan, A.P.; Swaminathan, G.J. DECIPHER: Database for the interpretation of phenotype-linked plausibly pathogenic sequence and copy-number variation. Nucleic Acids Res. 2014, 42, D993–D1000. [Google Scholar] [CrossRef]
- Pesquita, C.; Faria, D.; Falcao, A.O.; Lord, P.; Couto, F.M. Semantic similarity in biomedical ontologies. PLoS Comput. Biol. 2009, 5, e1000443. [Google Scholar] [CrossRef]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.-J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef]
- Gottlieb, A.; Stein, G.Y.; Ruppin, E.; Sharan, R. PREDICT: A method for inferring novel drug indications with application to personalized medicine. Mol. Syst. Biol. 2011, 7, 496. [Google Scholar] [CrossRef]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; Wiegers, J.; Wiegers, T.C.; Mattingly, C.J. Comparative Toxicogenomics Database (CTD): Update 2021. Nucleic Acids Res. 2021, 49, D1138–D1143. [Google Scholar] [CrossRef]
- Zhang, L.; Tan, J.; Han, D.; Zhu, H. From machine learning to deep learning: Progress in machine intelligence for rational drug discovery. Drug Discov. Today 2017, 22, 1680–1685. [Google Scholar] [CrossRef]
- Min, S.; Lee, B.; Yoon, S. Deep learning in bioinformatics. Briefings Bioinform. 2017, 18, 851–869. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Method | Algorithms | Features (or Tools) Used for Network Construction | ||
|---|---|---|---|---|---|
| Drug Network | Disease Network | Gene Network | |||
| Graph Mining | Wu et al. [25] | graph clustering | biological processes, pathways, phenotypes | biological processes, pathways, phenotypes | - |
| TL_HGBI [26] | propagation | chemical structures, DTIs | MimMiner | protein sequences | |
| DrugNet [27] | propagation | ATC codes | semantic sim.(DO) | PPIs | |
| MBiRW [28] | bi-random walk | chemical structures, drug-disease assoc. | MimMiner, drug-disease assoc. | - | |
| TP-NRWRH [29] | random walk | chemical structures, drug-disease assoc. | MimMiner, drug-disease assoc. | - | |
| DR-IBRW [30] | bi-random walk | chemical structures, drug-disease assoc. | symptoms, drug-disease assoc. | - | |
| EMP-SVD [31] | meta-path search | chemical structures | MimMiner | protein sequences | |
| BGMSDDA [32] | graph diffusion | chemical structures | MimMiner | - | |
| Matrix Factorization / Matrix Completion | DRRS [33] | nuclear norm minimization | chemical structures | MimMiner | - |
| OMC [34] | nuclear norm minimization | chemical structures, DTIs | MimMiner, disease-gene assoc. | - | |
| DRIMC [35] | logistic matrix factorization | chemical structures, target domain, target annotation | MimMiner | - | |
| MSBMF [36] | bilinear matrix factorization | chemical structures, ATC codes, side effects, DDIs, target profiles | MimMiner, semantic sim.(DO) | - | |
| NTD-DR [37] | tensor decomposition | chemical structures, ATC codes, target sequences, semantic sim.(GO), pathways | drug-disease assoc., disease-gene assoc., semantic sim.(GO), PPIs | protein sequences, semantic sim.(GO), PPIs | |
| Deep Learning | deepDR [38] | MDA, cVAE | DDIs, DTIs, chemical structures, target sequences, semantic sim.(GO), side effects, etc. | - | - |
| ANMF [39] | autoencoder | chemical structures | MimMiner | - | |
| NEDD [40] | HIN2vec | chemical structures | MimMiner | - | |
| SNF-NN [41] | SNF, neural networks | chemical structures, DTIs, side effects | disease-gene assoc., disease-miRNA assoc., phenotypes | - | |
| SAEROF [42] | autoencoder, rotation forest | chemical structures | semantic sim.(MeSH) | - | |
| LAGCN [43] | GCN | target features, chemical structures, DDIs, pathways, etc. | semantic sim.(MeSH) | - | |
| Associations | Number of Drugs | Number of Diseases | Number of Genes | Number of Edges | |
|---|---|---|---|---|---|
| Network-CS | Drug–Disease | 615 | 285 | - | 1728 |
| Drug–Gene | 450 | - | 14,430 | 170,652 | |
| Disease–Gene | - | 180 | 323 | 390 | |
| Network-ATC | Drug–Disease | 593 | 282 | - | 1681 |
| Drug–Gene | 437 | - | 14,430 | 167,535 | |
| Disease–Gene | - | 180 | 323 | 390 |
| Method | Prediction on the Drug Side | Prediction of the Disease Side | |||||
|---|---|---|---|---|---|---|---|
| AUC | AUPR | AUPR* | AUC | AUPR | AUPR* | ||
| Graph Mining | MBiRW | 0.753 | 0.046 | 0.762 | 0.692 | 0.195 | 0.769 |
| TP-NRWRH | 0.746 | 0.043 | 0.753 | 0.751 | 0.084 | 0.795 | |
| DR-IBRW | 0.747 | 0.046 | 0.758 | 0.704 | 0.162 | 0.776 | |
| BGMSDDA | 0.790 | 0.078 | 0.804 | 0.694 | 0.089 | 0.760 | |
| Matrix Factorization/ Matrix Completion | DRRS | 0.761 | 0.048 | 0.768 | 0.731 | 0.081 | 0.763 |
| OMC | 0.813 | 0.076 | 0.820 | 0.751 | 0.029 | 0.715 | |
| DRIMC | 0.749 | 0.051 | 0.764 | 0.700 | 0.051 | 0.732 | |
| MSBMF | 0.805 | 0.176 | 0.842 | 0.669 | 0.048 | 0.708 | |
| Deep Learning | deepDR | 0.685 | 0.024 | 0.686 | 0.606 | 0.016 | 0.613 |
| ANMF | 0.646 | 0.030 | 0.678 | 0.673 | 0.037 | 0.692 | |
| LAGCN | 0.751 | 0.042 | 0.756 | 0.643 | 0.031 | 0.677 | |
| Method | Prediction on the Drug Side | Prediction of the Disease Side | |||||
|---|---|---|---|---|---|---|---|
| AUC | AUPR | AUPR* | AUC | AUPR | AUPR* | ||
| Graph Mining | MBiRW | 0.893 | 0.390 | 0.917 | 0.768 | 0.207 | 0.819 |
| TP-NRWRH | 0.840 | 0.140 | 0.855 | 0.775 | 0.090 | 0.809 | |
| DR-IBRW | 0.853 | 0.309 | 0.887 | 0.720 | 0.174 | 0.786 | |
| BGMSDDA | 0.881 | 0.340 | 0.701 | 0.705 | 0.139 | 0.765 | |
| Matrix Factorization/ Matrix Completion | DRRS | 0.890 | 0.291 | 0.909 | 0.755 | 0.117 | 0.796 |
| OMC | 0.852 | 0.343 | 0.883 | 0.813 | 0.214 | 0.845 | |
| DRIMC | 0.807 | 0.080 | 0.820 | 0.699 | 0.043 | 0.726 | |
| MSBMF | 0.872 | 0.300 | 0.902 | 0.702 | 0.053 | 0.735 | |
| Deep Learning | deepDR | 0.730 | 0.028 | 0.714 | 0.614 | 0.017 | 0.620 |
| ANMF | 0.845 | 0.199 | 0.868 | 0.739 | 0.050 | 0.743 | |
| LAGCN | 0.842 | 0.079 | 0.843 | 0.742 | 0.044 | 0.753 | |
| Method | Edge Removal | AUC | Edge Addition | |||
|---|---|---|---|---|---|---|
| −20% | −10% | 10% | 20% | |||
| Graph Mining | MBiRW | −1.47% | 0.16% | 0.884 | −0.41% | −2.48% |
| TP−NRWRH | −3.39% | −1.90% | 0.923 | −2.24% | −4.34% | |
| DR−IBRW | −4.93% | −0.26% | 0.871 | −1.52% | −5.06% | |
| BGMSDDA | −4.45% | −3.28% | 0.832 | 0.10% | −1.55% | |
| Matrix Factorization/ Matrix Completion | DRRS | −2.93% | −0.37% | 0.913 | −1.19% | −2.27% |
| OMC | −3.61% | 4.86% | 0.941 | −1.18% | −2.67% | |
| DRIMC | −2.78% | −0.91% | 0.878 | −0.44% | −2.80% | |
| MSBMF | −3.31% | −1.20% | 0.901 | −0.59% | −2.56% | |
| Deep Learning | deepDR | −4.80% | −3.44% | 0.850 | −2.53% | −4.21% |
| ANMF | −4.81% | −0.75% | 0.935 | −1.64% | −1.82% | |
| LAGCN | −1.00% | −1.00% | 0.718 | −0.21% | −1.00% | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.; Jung, Y.-S.; Park, J.-H.; Kim, S.-J.; Cho, Y.-R. Drug-Disease Association Prediction Using Heterogeneous Networks for Computational Drug Repositioning. Biomolecules 2022, 12, 1497. https://doi.org/10.3390/biom12101497
Kim Y, Jung Y-S, Park J-H, Kim S-J, Cho Y-R. Drug-Disease Association Prediction Using Heterogeneous Networks for Computational Drug Repositioning. Biomolecules. 2022; 12(10):1497. https://doi.org/10.3390/biom12101497
Chicago/Turabian StyleKim, Yoonbee, Yi-Sue Jung, Jong-Hoon Park, Seon-Jun Kim, and Young-Rae Cho. 2022. "Drug-Disease Association Prediction Using Heterogeneous Networks for Computational Drug Repositioning" Biomolecules 12, no. 10: 1497. https://doi.org/10.3390/biom12101497
APA StyleKim, Y., Jung, Y.-S., Park, J.-H., Kim, S.-J., & Cho, Y.-R. (2022). Drug-Disease Association Prediction Using Heterogeneous Networks for Computational Drug Repositioning. Biomolecules, 12(10), 1497. https://doi.org/10.3390/biom12101497