

Cisplatin Resistance: Genetic and Epigenetic Factors Involved




Abstract
1. Introduction
2. Cisplatin: Mechanism of Action
3. Resistance to Cisplatin Treatment
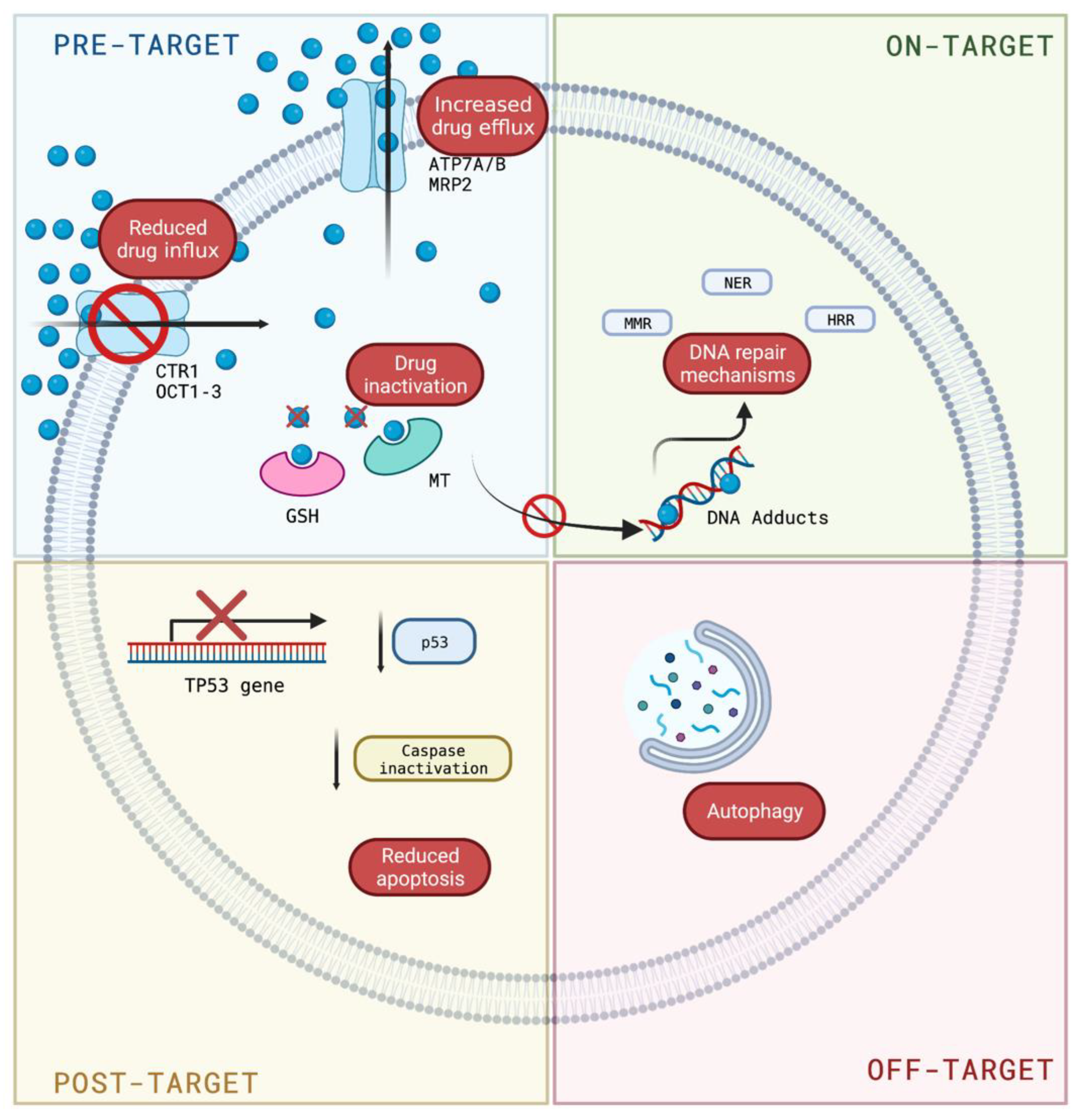
3.1. Mechanisms of Cisplatin Resistance
3.1.1. Pre-Target Resistance
3.1.2. On-Target Resistance
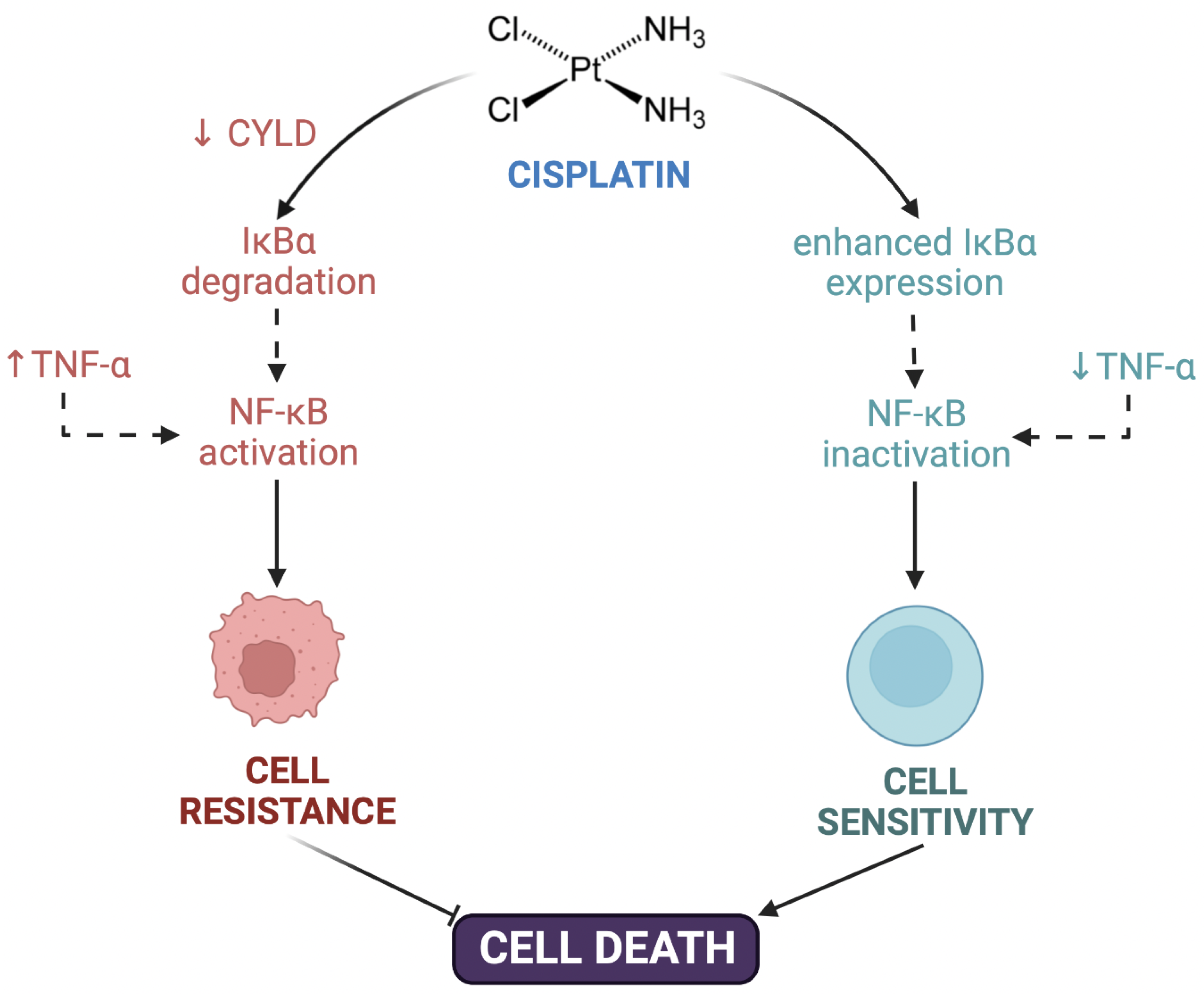
3.1.3. Post-Target Resistance
3.1.4. Off-Target Resistance
4. Epigenetics and Resistance to Treatment
4.1. Epigenetic Mechanisms Associated with Pre-Target, on-Target, and Post-Target Resistance
4.2. Epigenetic Mechanisms Associated with Off-Target Resistance

{kind=link}
{kind=link}
| Type | Cisplatin Resistance | Molecule Involved | Reference |
|---|---|---|---|
| Genetic factors | Pre-target | Decreased CTR1 expression | [32,33,34,35,36] |
| Enhanced ATP7A, ATP7B, and MRP2 expression | [35,37,38,39] | ||
| Intracellular inactivation of cisplatin by GSH or metallothioneins | [40,41] | ||
| On-target | Enhanced ERCC1 endonuclease | [42,43,44,45,46] | |
| Downregulation of MSH2 and MLH1 expression | [16] | ||
| Enhanced homologous recombination mechanism | [48] | ||
| Augmented polymerases β, η, & ζ activity | [16,49,50] | ||
| Post-target | Inactivation of TP53 gene | [51] | |
| Inactivation of caspases | [52,53,54,55,56] | ||
| CYLD Lysine 63 deubiquitinase downregulation | [57] | ||
| NF-κB hyperactivation | [57] | ||
| Off-target | Overexpression of human epidermal growth factor receptor and dual specificity tyrosine phosphorylation regulated kinase 1B | [60] | |
| Enhanced antioxidant enzymes such as ferroxidase and superoxide dismutase | [61] | ||
| Autophagy | [62,63] | ||
| Epigenetic factors | Pre-target | miR-38 (regulating ABCB1/MDR1) | [70] |
| miR-148a (regulating ATP7A) | [12] | ||
| lncRNA ROR (targeting miR-153-3p/ABCB1) | [71] | ||
| On target | miR-200c (targeting ERCC3/ERCC4) | [72] | |
| Post-target | Methylation of miR-200b enhances IGFBP3, ICAM1, and TNFSF10 gene expression | [73] | |
| Downregulation of miR-100 and miR-214 (targeting PTEN) | [74] | ||
| LncRNA ROR (targeting TP53) | [75,76] | ||
| miR-335-3p (regulates apoptosis) | [77] | ||
| Off-target | Demethylation of the regulatory region of the FOXF1 gene | [89] | |
| Epigenetic silencing of spermidine/spermine N1-acetyltransferase and argininosuccinate synthase 1 | [90] | ||
| Enhanced HDAC6 expression | [92] | ||
| Dysregulation of miR-7, miR-132, and miR-148a | [94,95,96] | ||
| Overexpression of lncRNA HOTTIP activates Wnt/β-catenin pathway | [98,99] | ||
| LncRNA UCA1 promotes autophagy | [100,101] | ||
| LncRNA TUG1 suppress PDCD4 expression | [102] |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- WHO. World Health Statistics 2021: Monitoring Health for the SDGs, Sustainable Development Goals; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef]
- Vermorken, J.B.; Remenar, E.; Van Herpen, C.; Gorlia, T.; Mesia, R.; Degardin, M.; Stewart, J.S.; Jelic, S.; Betka, J.; Preiss, J.H. Cisplatin, fluorouracil, and docetaxel in unresectable head and neck cancer. N. Engl. J. Med. 2007, 357, 1695–1704. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Sheinfeld, J.; Mazumdar, M.; Bajorin, D.F.; Bosl, G.J.; Herr, H.; Lyn, P.; Vlamis, V. Etoposide and cisplatin adjuvant therapy for patients with pathologic stage II germ cell tumors. J. Clin. Oncol. 1995, 13, 2700–2704. [Google Scholar] [CrossRef]
- Li, D.; Zhang, Y.; Xie, Y.; Xiang, J.; Zhu, Y.; Yang, J. Enhanced tumor suppression by adenoviral PTEN gene therapy combined with cisplatin chemotherapy in small-cell lung cancer. Cancer Gene Ther. 2013, 20, 251–259. [Google Scholar] [CrossRef]
- Magali, L.; Pascal, F.; Serge, A.; Mathieu, B.; Ayoube, Z.; Claire, T.; Christiane, M. Better survival in impaired renal function patients with metastatic non-small cell lung cancer treated by cisplatin-pemetrexed. Eur. J. Clin. Pharmacol. 2020, 76, 1573–1580. [Google Scholar] [CrossRef]
- Armstrong, D.K.; Bundy, B.; Wenzel, L.; Huang, H.Q.; Baergen, R.; Lele, S.; Copeland, L.J.; Walker, J.L.; Burger, R.A. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N. Engl. J. Med. 2006, 354, 34–43. [Google Scholar] [CrossRef]
- Moore, K.N.; Herzog, T.J.; Lewin, S.; Giuntoli, R.L.; Armstrong, D.K.; Rocconi, R.P.; Spannuth, W.A.; Gold, M.A. A comparison of cisplatin/paclitaxel and carboplatin/paclitaxel in stage IVB, recurrent or persistent cervical cancer. Gynecol. Oncol. 2007, 105, 299–303. [Google Scholar] [CrossRef]
- Coppin, C.; Gospodarowicz, M.K.; James, K.; Tannock, I.F.; Zee, B.; Carson, J.; Pater, J.; Sullivan, L.D. Improved local control of invasive bladder cancer by concurrent cisplatin and preoperative or definitive radiation. The National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 1996, 14, 2901–2907. [Google Scholar] [CrossRef]
- Yu, Z.; Cao, W.; Ren, Y.; Zhang, Q.; Liu, J. ATPase copper transporter A, negatively regulated by miR-148a-3p, contributes to cisplatin resistance in breast cancer cells. Clin. Transl. Med. 2020, 10, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Basu, A.; Krishnamurthy, S. Cellular responses to Cisplatin-induced DNA damage. J. Nucleic Acids 2010, 2010, 201367. [Google Scholar] [CrossRef]
- Florea, A.-M.; Büsselberg, D. Cisplatin as an anti-tumor drug: Cellular mechanisms of activity, drug resistance and induced side effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.-W.; Pouliot, L.M.; Hall, M.D.; Gottesman, M.M. Cisplatin resistance: A cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol. Rev. 2012, 64, 706–721. [Google Scholar] [CrossRef]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef]
- Fuertes, M.; Castilla, J.; Alonso, C.; Prez, J. Cisplatin biochemical mechanism of action: From cytotoxicity to induction of cell death through interconnections between apoptotic and necrotic pathways. Curr. Med. Chem. 2003, 10, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Kartal-Yandim, M.; Adan-Gokbulut, A.; Baran, Y. Molecular mechanisms of drug resistance and its reversal in cancer. Crit. Rev. Biotechnol. 2016, 36, 716–726. [Google Scholar] [CrossRef]
- Steding, C.E. Creating chemotherapeutic-resistant breast cancer cell lines: Advances and future perspectives. Future Oncol. 2016, 12, 1517–1527. [Google Scholar] [CrossRef]
- Pogribny, I.P.; Filkowski, J.N.; Tryndyak, V.P.; Golubov, A.; Shpyleva, S.I.; Kovalchuk, O. Alterations of microRNAs and their targets are associated with acquired resistance of MCF-7 breast cancer cells to cisplatin. Int. J. Cancer 2010, 127, 1785–1794. [Google Scholar] [CrossRef] [PubMed]
- Velasco, G.; Sánchez, C.; Guzmán, M. Anticancer mechanisms of cannabinoids. Curr. Oncol. 2016, 23, 23–32. [Google Scholar] [CrossRef]
- Aye, Y.; Li, M.; Long, M.; Weiss, R. Ribonucleotide reductase and cancer: Biological mechanisms and targeted therapies. Oncogene 2015, 34, 2011–2021. [Google Scholar] [CrossRef]
- McDermott, M.; Eustace, A.; Busschots, S.; Breen, L.; Clynes, M.; O’Donovan, N.; Stordal, B. In vitro development of chemotherapy and targeted therapy drug-resistant cancer cell lines: A practical guide with case studies. Front. Oncol. 2014, 4, 40. [Google Scholar] [CrossRef]
- Lukyanova, N.Y.; Rusetskya, N.; Tregubova, N.; Chekhun, V. Molecular profile and cell cycle in MCF-7 cells resistant to cisplatin and doxorubicin. Exp. Oncol. 2009, 31, 87–91. [Google Scholar]
- Puspita, N.A.; Bedford, A. Morphological Changes of Cisplatin-resistant Human Breast Cancer MCF-7 Cell Line. Int. J. Integr. Health Sci. 2017, 5, 8–14. [Google Scholar] [CrossRef]
- Hinojosa-García, L.M.; Dueñas-González, A. Papel de la quimioterapia en el tratamiento del carcinoma cervicouterino. Rev. Inst. Nal. Cancerol. (Mex) 2000, 46, 47–57. [Google Scholar]
- Aleksakhina, S.N.; Kashyap, A.; Imyanitov, E.N. Mechanisms of acquired tumor drug resistance. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2019, 1872, 188310. [Google Scholar] [CrossRef]
- Lund, R.J.; Huhtinen, K.; Salmi, J.; Rantala, J.; Nguyen, E.V.; Moulder, R.; Goodlett, D.R.; Lahesmaa, R.; Carpén, O. DNA methylation and transcriptome changes associated with cisplatin resistance in ovarian cancer. Sci. Rep. 2017, 7, 1469. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Lips, K.S.; Metzner, L.; Neubert, R.H.; Koepsell, H.; Brandsch, M. Drug specificity and intestinal membrane localization of human organic cation transporters (OCT). Biochem. Pharmacol. 2005, 70, 1851–1860. [Google Scholar] [CrossRef] [PubMed]
- Ciarimboli, G. Organic Cation Transporters 2 as Mediators of Cisplatin Nephrotoxicity. In Platinum and Other Heavy Metal Compounds in Cancer Chemotherapy; Bonetti, A., Leone, R., Muggia, F.M., Howell, S.B., Eds.; Cancer Drug Discovery and Development. Humana Press: Totowa, NJ, USA, 2009; pp. 353–358. [Google Scholar]
- Safaei, R. Role of copper transporters in the uptake and efflux of platinum containing drugs. Cancer Lett. 2006, 234, 34–39. [Google Scholar] [CrossRef]
- Kuo, M.T.; Fu, S.; Savaraj, N.; Chen, H.H. Role of the human high-affinity copper transporter in copper homeostasis regulation and cisplatin sensitivity in cancer chemotherapy. Cancer Res. 2012, 72, 4616–4621. [Google Scholar] [CrossRef] [PubMed]
- Köberle, B.; Brenner, W.; Albers, A.; Usanova, S.; Thüroff, J.W.; Kaina, B. ERCC1 and XPF expression in human testicular germ cell tumors. Oncol. Rep. 2010, 23, 223–227. [Google Scholar] [CrossRef][Green Version]
- Liang, Z.D.; Long, Y.; Tsai, W.-B.; Fu, S.; Kurzrock, R.; Gagea-Iurascu, M.; Zhang, F.; Chen, H.H.; Hennessy, B.T.; Mills, G.B. Mechanistic basis for overcoming platinum resistance using copper chelating agents. Mol. Cancer Ther. 2012, 11, 2483–2494. [Google Scholar] [CrossRef]
- Korita, P.V.; Wakai, T.; Shirai, Y.; Matsuda, Y.; Sakata, J.; Takamura, M.; Yano, M.; Sanpei, A.; Aoyagi, Y.; Hatakeyama, K. Multidrug resistance-associated protein 2 determines the efficacy of cisplatin in patients with hepatocellular carcinoma. Oncol. Rep. 2010, 23, 965–972. [Google Scholar]
- Yamasaki, M.; Makino, T.; Masuzawa, T.; Kurokawa, Y.; Miyata, H.; Takiguchi, S.; Nakajima, K.; Fujiwara, Y.; Matsuura, N.; Mori, M. Role of multidrug resistance protein 2 (MRP2) in chemoresistance and clinical outcome in oesophageal squamous cell carcinoma. Br. J. Cancer 2011, 104, 707–713. [Google Scholar] [CrossRef]
- Chen, H.H.; Kuo, M.T. Role of glutathione in the regulation of Cisplatin resistance in cancer chemotherapy. Met.-Based Drugs 2010, 2010, 430939. [Google Scholar] [CrossRef]
- Knipp, M.; Karotki, A.V.; Chesnov, S.; Natile, G.; Sadler, P.J.; Brabec, V.; Vašák, M. Reaction of Zn7Metallothionein with cis-and trans-[Pt (N-donor) 2Cl2] anticancer complexes: Trans-PtII complexes retain their N-donor ligands. J. Med. Chem. 2007, 50, 4075–4086. [Google Scholar] [CrossRef]
- Martin, L.P.; Hamilton, T.C.; Schilder, R.J. Platinum resistance: The role of DNA repair pathways. Clin. Cancer Res. 2008, 14, 1291–1295. [Google Scholar] [CrossRef]
- Saldivar, J.S.; Wu, X.; Follen, M.; Gershenson, D. Nucleotide excision repair pathway review I: Implications in ovarian cancer and platinum sensitivity. Gynecol. Oncol. 2007, 107, S56–S71. [Google Scholar] [CrossRef] [PubMed]
- Chang, I.-Y.; Kim, M.-H.; Kim, H.B.; Kim, S.-H.; Kim, H.-Y.; You, H.J. Small interfering RNA-induced suppression of ERCC1 enhances sensitivity of human cancer cells to cisplatin. Biochem. Biophys. Res. Commun. 2005, 327, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Usanova, S.; Piée-Staffa, A.; Sied, U.; Thomale, J.; Schneider, A.; Kaina, B.; Köberle, B. Cisplatin sensitivity of testis tumour cells is due to deficiency in interstrand-crosslink repair and low ERCC1-XPF expression. Mol. Cancer 2010, 9, 248. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, M.; Sato, Y.; Ohnuma, H.; Takayama, T.; Sagawa, T.; Nobuoka, T.; Harada, K.; Miyamoto, H.; Sato, Y.; Takahashi, Y. A phase II study of neoadjuvant combination chemotherapy with docetaxel, cisplatin, and S-1 for locally advanced resectable gastric cancer: Nucleotide excision repair (NER) as potential chemoresistance marker. Cancer Chemother. Pharmacol. 2013, 71, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Narod, S.A.; Foulkes, W.D. BRCA1 and BRCA2: 1994 and beyond. Nat. Rev. Cancer 2004, 4, 665–676. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Bassett, E.; Vaisman, A.; Tropea, K.A.; McCall, C.M.; Masutani, C.; Hanaoka, F.; Chaney, S.G. Frameshifts and deletions during in vitro translesion synthesis past Pt–DNA adducts by DNA polymerases β and η. DNA Repair 2002, 1, 1003–1016. [Google Scholar] [CrossRef]
- Albertella, M.R.; Green, C.M.; Lehmann, A.R.; O’Connor, M.J. A role for polymerase η in the cellular tolerance to cisplatin-induced damage. Cancer Res. 2005, 65, 9799–9806. [Google Scholar] [CrossRef]
- Martinez-Rivera, M.; Siddik, Z.H. Resistance and gain-of-resistance phenotypes in cancers harboring wild-type p53. Biochem. Pharmacol. 2012, 83, 1049–1062. [Google Scholar] [CrossRef]
- Ding, Z.; Yang, X.; Pater, A.; Tang, S.-C. Resistance to apoptosis is correlated with the reduced caspase-3 activation and enhanced expression of antiapoptotic proteins in human cervical multidrug-resistant cells. Biochem. Biophys. Res. Commun. 2000, 270, 415–420. [Google Scholar] [CrossRef]
- Duiker, E.W.; Meijer, A.; van der Bilt, A.R.; Meersma, G.J.; Kooi, N.; van der Zee, A.G.; De Vries, E.; de Jong, S. Drug-induced caspase 8 upregulation sensitises cisplatin-resistant ovarian carcinoma cells to rhTRAIL-induced apoptosis. Br. J. Cancer 2011, 104, 1278–1287. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.K.; Mahidhara, R.; Seol, D.-W. The role of caspase-8 in resistance to cancer chemotherapy. Drug Resist. Updates 2001, 4, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, D.; Tsutsumi, K.; Oyake, D.; Ohta, T.; Nishikawa, H.; Koizuka, I. Inhibition of caspase-9 activity and Apaf-1 expression in cisplatin-resistant head and neck squamous cell carcinoma cells. Auris Nasus Larynx 2003, 30, 85–88. [Google Scholar] [CrossRef]
- Nikounezhad, N.; Nakhjavani, M.; Shirazi, F.H. Generation of cisplatin-resistant ovarian cancer cell lines. Iran. J. Pharm. Sci. 2016, 12, 11–20. [Google Scholar]
- Suenaga, N.; Kuramitsu, M.; Komure, K.; Kanemaru, A.; Takano, K.; Ozeki, K.; Nishimura, Y.; Yoshida, R.; Nakayama, H.; Shinriki, S.; et al. Loss of Tumor Suppressor CYLD Expression Triggers Cisplatin Resistance in Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 5194. [Google Scholar] [CrossRef]
- Kim, S.B.; Kim, J.S.; Lee, J.H.; Yoon, W.J.; Lee, D.S.; Ko, M.S.; Kwon, B.S.; Choi, D.H.; Cho, H.R.; Lee, B.J.; et al. NF-kappaB activation is required for cisplatin-induced apoptosis in head and neck squamous carcinoma cells. FEBS Lett. 2006, 580, 311–318. [Google Scholar] [CrossRef]
- Wang, L.H.; Li, Y.; Yang, S.N.; Wang, F.Y.; Hou, Y.; Cui, W.; Chen, K.; Cao, Q.; Wang, S.; Zhang, T.Y.; et al. Gambogic acid synergistically potentiates cisplatin-induced apoptosis in non-small-cell lung cancer through suppressing NF-kappaB and MAPK/HO-1 signalling. Br. J. Cancer 2014, 110, 341–352. [Google Scholar] [CrossRef]
- Deng, X.; Ewton, D.Z.; Friedman, E. Mirk/Dyrk1B maintains the viability of quiescent pancreatic cancer cells by reducing levels of reactive oxygen species. Cancer Res. 2009, 69, 3317–3324. [Google Scholar] [CrossRef]
- Fijołek, J.; Wiatr, E.; Rowińska-Zakrzewska, E.; Giedronowicz, D.; Langfort, R.; Chabowski, M.; Orłowski, T.; Roszkowski, K. p53 and HER2/neu expression in relation to chemotherapy response in patients with non-small cell lung cancer. Int. J. Biol. Markers 2006, 21, 81–87. [Google Scholar] [CrossRef]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef]
- Ren, J.-H.; He, W.-S.; Nong, L.; Zhu, Q.-Y.; Hu, K.; Zhang, R.-G.; Huang, L.-L.; Zhu, F.; Wu, G. Acquired cisplatin resistance in human lung adenocarcinoma cells is associated with enhanced autophagy. Cancer Biother. Radiopharm. 2010, 25, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.-W.; Huang, K.; Yang, C.; Kang, C.-S. Non-coding RNAs as regulators in epigenetics. Oncol. Rep. 2017, 37, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Neganova, M.E.; Klochkov, S.G.; Aleksandrova, Y.R.; Aliev, G. Histone modifications in epigenetic regulation of cancer: Perspectives and achieved progress. Semin. Cancer Biol. 2022, 83, 452–471. [Google Scholar] [CrossRef] [PubMed]
- Vafadar, A.; Shabaninejad, Z.; Movahedpour, A.; Mohammadi, S.; Fathullahzadeh, S.; Mirzaei, H.R.; Namdar, A.; Savardashtaki, A.; Mirzaei, H. Long non-coding RNAs as epigenetic regulators in cancer. Curr. Pharm. Des. 2019, 25, 3563–3577. [Google Scholar] [CrossRef]
- Arif, K.; Elliott, E.K.; Haupt, L.M.; Griffiths, L.R. Regulatory mechanisms of epigenetic miRNA relationships in human cancer and potential as therapeutic targets. Cancers 2020, 12, 2922. [Google Scholar] [CrossRef]
- Gulyaeva, L.F.; Kushlinskiy, N.E. Regulatory mechanisms of microRNA expression. J. Transl. Med. 2016, 14, 143. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Yi, D.; Xu, L.; Wang, R.; Lu, X.; Sang, J. miR-381 overcomes cisplatin resistance in breast cancer by targeting MDR1. Cell Biol. Int. 2019, 43, 12–21. [Google Scholar] [CrossRef]
- Cheng, F.; Zhao, Z.; Liu, W. Long non-coding RNA ROR regulated ABCB1 to induce cisplatin resistance in osteosarcoma by sponging miR-153-3p. Eur. Rev. Med. Pharm. Sci. 2019, 23, 7256–7265. [Google Scholar]
- Li, M.; Gao, M.; Xie, X.; Zhang, Y.; Ning, J.; Liu, P.; Gu, K. MicroRNA-200c reverses drug resistance of human gastric cancer cells by targeting regulation of the NER-ERCC3/4 pathway. Oncol. Lett. 2019, 18, 145–152. [Google Scholar] [CrossRef]
- Shindo, T.; Niinuma, T.; Nishiyama, N.; Shinkai, N.; Kitajima, H.; Kai, M.; Maruyama, R.; Tokino, T.; Masumori, N.; Suzuki, H. Epigenetic silencing of miR-200b is associated with cisplatin resistance in bladder cancer. Oncotarget 2018, 9, 24457. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Kong, W.; He, L.; Zhao, J.-J.; O’Donnell, J.D.; Wang, J.; Wenham, R.M.; Coppola, D.; Kruk, P.A.; Nicosia, S.V. MicroRNA expression profiling in human ovarian cancer: miR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res. 2008, 68, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Gu, M.; You, B.; Shi, S.; Shan, Y.; Bao, L.; You, Y. Long non-coding RNA ROR promotes proliferation, migration and chemoresistance of nasopharyngeal carcinoma. Cancer Sci. 2016, 107, 1215–1222. [Google Scholar] [CrossRef]
- Shi, H.; Pu, J.; Zhou, X.-L.; Ning, Y.-Y.; Bai, C. Silencing long non-coding RNA ROR improves sensitivity of non-small-cell lung cancer to cisplatin resistance by inhibiting PI3K/Akt/mTOR signaling pathway. Tumor Biol. 2017, 39, 1010428317697568. [Google Scholar] [CrossRef]
- Sun, Y.-F.; Wang, Y.; Li, X.-D.; Wang, H. SNHG15, a p53-regulated lncRNA, suppresses cisplatin-induced apoptosis and ROS accumulation through the miR-335-3p/ZNF32 axis. Am. J. Cancer Res. 2022, 12, 816. [Google Scholar] [PubMed]
- Liu, R.; Guo, H.; Lu, S. MiR-335-5p restores cisplatin sensitivity in ovarian cancer cells through targeting BCL2L2. Cancer Med. 2018, 7, 4598–4609. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, M.; Ciccarone, F.; Calabrese, R.; Franceschi, C.; Bürkle, A.; Caiafa, P. Reconfiguration of DNA methylation in aging. Mech. Ageing Dev. 2015, 151, 60–70. [Google Scholar] [CrossRef]
- Flanagan, J.M.; Wilson, A.; Koo, C.; Masrour, N.; Gallon, J.; Loomis, E.; Flower, K.; Wilhelm-Benartzi, C.; Hergovich, A.; Cunnea, P. Platinum-based chemotherapy induces methylation changes in blood DNA associated with overall survival in patients with ovarian cancer. Clin. Cancer Res. 2017, 23, 2213–2222. [Google Scholar] [CrossRef]
- De Caceres, I.I.; Cortes-Sempere, M.; Moratilla, C.; Machado-Pinilla, R.; Rodriguez-Fanjul, V.; Manguan-Garcia, C.; Cejas, P.; López-Ríos, F.; Paz-Ares, L.; De Castrocarpeño, J. IGFBP-3 hypermethylation-derived deficiency mediates cisplatin resistance in non-small-cell lung cancer. Oncogene 2010, 29, 1681–1690. [Google Scholar] [CrossRef]
- Toh, T.B.; Lim, J.J.; Chow, E.K.-H. Epigenetics in cancer stem cells. Mol. Cancer 2017, 16, 29. [Google Scholar] [CrossRef]
- Easwaran, H.; Tsai, H.-C.; Baylin, S.B. Cancer epigenetics: Tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell 2014, 54, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Suresh, R.; Ali, S.; Ahmad, A.; Philip, P.A.; Sarkar, F.H. The role of cancer stem cells in recurrent and drug-resistant lung cancer. Lung Cancer Pers. Med. Nov. Ther. Clin. Manag. 2016, 890, 57–74. [Google Scholar]
- Lopez-Ayllon, B.D.; Moncho-Amor, V.; Abarrategi, A.; de Cáceres, I.I.; Castro-Carpeño, J.; Belda-Iniesta, C.; Perona, R.; Sastre, L. Cancer stem cells and cisplatin-resistant cells isolated from non-small-lung cancer cell lines constitute related cell populations. Cancer Med. 2014, 3, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Bora-Singhal, N.; Perumal, D.; Nguyen, J.; Chellappan, S. Gli1-mediated regulation of Sox2 facilitates self-renewal of stem-like cells and confers resistance to EGFR inhibitors in non–small cell lung cancer. Neoplasia 2015, 17, 538–551. [Google Scholar] [CrossRef] [PubMed]
- Milewski, D.; Pradhan, A.; Wang, X.; Cai, Y.; Le, T.; Turpin, B.; Kalinichenko, V.V.; Kalin, T.V. FoxF1 and FoxF2 transcription factors synergistically promote rhabdomyosarcoma carcinogenesis by repressing transcription of p21 Cip1 CDK inhibitor. Oncogene 2017, 36, 850–862. [Google Scholar] [CrossRef]
- Ran, L.; Chen, Y.; Sher, J.; Wong, E.W.; Murphy, D.; Zhang, J.Q.; Li, D.; Deniz, K.; Sirota, I.; Cao, Z. FOXF1 defines the core-regulatory circuitry in gastrointestinal stromal tumor. Cancer Discov. 2018, 8, 234–251. [Google Scholar] [CrossRef]
- Zhao, J.; Xue, X.; Fu, W.; Dai, L.; Jiang, Z.; Zhong, S.; Deng, B.; Yin, J. Epigenetic activation of FOXF1 confers cancer stem cell properties to cisplatin-resistant non-small cell lung cancer. Int. J. Oncol. 2020, 56, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Yeon, A.; You, S.; Kim, M.; Gupta, A.; Park, M.H.; Weisenberger, D.J.; Liang, G.; Kim, J. Rewiring of cisplatin-resistant bladder cancer cells through epigenetic regulation of genes involved in amino acid metabolism. Theranostics 2018, 8, 4520. [Google Scholar] [CrossRef]
- Ha, Y.-N.; Sung, H.Y.; Yang, S.-D.; Chae, Y.J.; Ju, W.; Ahn, J.-H. Epigenetic modification of α-N-acetylgalactosaminidase enhances cisplatin resistance in ovarian cancer. Korean J. Physiol. Pharmacol. 2018, 22, 43–51. [Google Scholar] [CrossRef]
- Wang, L.; Xiang, S.; Williams, K.A.; Dong, H.; Bai, W.; Nicosia, S.V.; Khochbin, S.; Bepler, G.; Zhang, X. Depletion of HDAC6 enhances cisplatin-induced DNA damage and apoptosis in non-small cell lung cancer cells. PLoS ONE 2012, 7, e44265. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Sempere, M.; De Miguel, M.; Pernia, O.; Rodriguez, C.; de Castro Carpeno, J.; Nistal, M.; Conde, E.; López-Ríos, F.; Belda-Iniesta, C.; Perona, R. IGFBP-3 methylation-derived deficiency mediates the resistance to cisplatin through the activation of the IGFIR/Akt pathway in non-small cell lung cancer. Oncogene 2013, 32, 1274–1283. [Google Scholar] [CrossRef] [PubMed]
- Vera Puente, O.; Jiménez Hernández, J.; Pernía, O.; Rodriguez-Antolín, C.; Rodriguez, C.; Sanchez Cabo, F.; Soto Romero, J.; Rosas, R.; Lopez-Magallon, S.; Esteban Rodriguez, I. DNA methylation of miR-7 is a mechanism involved in platinum response through MAFG overexpression in cancer cells. Theranostics 2017, 7, 4118–4134. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhu, Q.; Lu, L.; Liu, Y. MiR-132 inhibits migration and invasion and increases chemosensitivity of cisplatin-resistant oral squamous cell carcinoma cells via targeting TGF-β1. Bioengineered 2020, 11, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Xi, J.; Xu, X.; Peng, B.; Zhang, B. MiR-148a suppressed cell invasion and migration via targeting WNT10b and modulating β-catenin signaling in cisplatin-resistant colorectal cancer cells. Biomed. Pharmacother. 2019, 109, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Vera, O.; Rodriguez-Antolin, C.; de Castro, J.; Karreth, F.A.; Sellers, T.A.; de Caceres, I.I. An epigenomic approach to identifying differential overlapping and cis-acting lncRNAs in cisplatin-resistant cancer cells. Epigenetics 2018, 13, 251–263. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, L.; Wang, Q. Overexpression of long non-coding RNA HOTTIP increases chemoresistance of osteosarcoma cell by activating the Wnt/β-catenin pathway. Am. J. Transl. Res. 2016, 8, 2385. [Google Scholar]
- Li, F.; Cao, L.; Hang, D.; Wang, F.; Wang, Q. Long non-coding RNA HOTTIP is up-regulated and associated with poor prognosis in patients with osteosarcoma. Int. J. Clin. Exp. Pathol. 2015, 8, 11414. [Google Scholar]
- Fan, Y.; Shen, B.; Tan, M.; Mu, X.; Qin, Y.; Zhang, F.; Liu, Y. Long non-coding RNA UCA 1 increases chemoresistance of bladder cancer cells by regulating Wnt signaling. FEBS J. 2014, 281, 1750–1758. [Google Scholar] [CrossRef]
- Zhou, B.; Zhuang, X.; Wang, Y.; Lin, Z.; Zhang, D.; Fan, S.; Huang, Z.; Li, J.; Chen, W. Long non-coding RNA UCA1 increases chemoresistance of tongue cancer cells by regulating autophagy signaling. Int. J. Oral Maxillofac. Surg. 2017, 46, 139. [Google Scholar] [CrossRef]
- Xu, C.; Guo, Y.; Liu, H.; Chen, G.; Yan, Y.; Liu, T. TUG1 confers cisplatin resistance in esophageal squamous cell carcinoma by epigenetically suppressing PDCD4 expression via EZH2. Cell Biosci. 2018, 8, 61. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lugones, Y.; Loren, P.; Salazar, L.A. Cisplatin Resistance: Genetic and Epigenetic Factors Involved. Biomolecules 2022, 12, 1365. https://doi.org/10.3390/biom12101365
Lugones Y, Loren P, Salazar LA. Cisplatin Resistance: Genetic and Epigenetic Factors Involved. Biomolecules. 2022; 12(10):1365. https://doi.org/10.3390/biom12101365
Chicago/Turabian StyleLugones, Yuliannis, Pía Loren, and Luis A. Salazar. 2022. "Cisplatin Resistance: Genetic and Epigenetic Factors Involved" Biomolecules 12, no. 10: 1365. https://doi.org/10.3390/biom12101365
APA StyleLugones, Y., Loren, P., & Salazar, L. A. (2022). Cisplatin Resistance: Genetic and Epigenetic Factors Involved. Biomolecules, 12(10), 1365. https://doi.org/10.3390/biom12101365