
Myricetin as a Potential Adjuvant in Chemotherapy: Studies on the Inhibition of Human Glutathione Transferase A1–1

 , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Heterologous Expression and Purification of Recombinant hGSTA1–1
2.3. Assay of hGSTA1–1 Activity and Inhibition Analysis
2.4. Kinetic Inhibition Studies
2.5. Irreversible Inactivation of hGSTA1–1 by Phenethyl Isothiocyanate (PEITC)
2.6. Molecular Modeling
2.7. Statistical Analysis
3. Results and Discussion
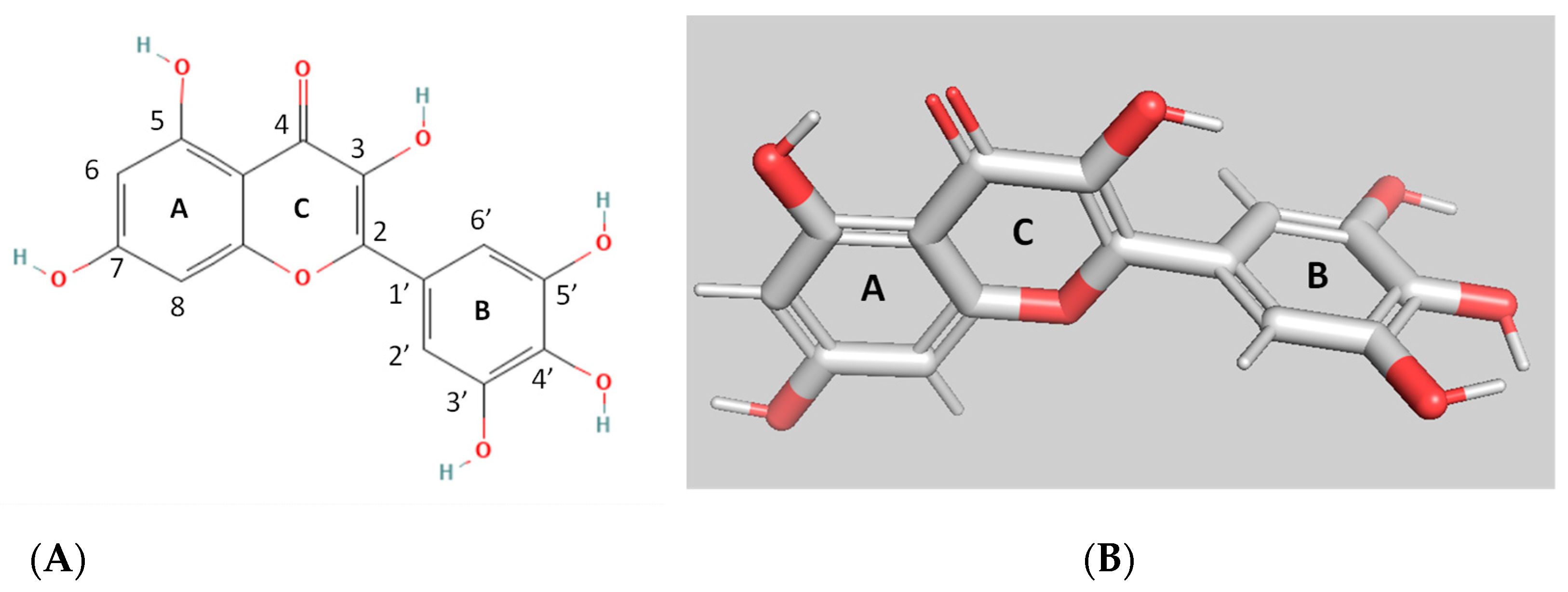
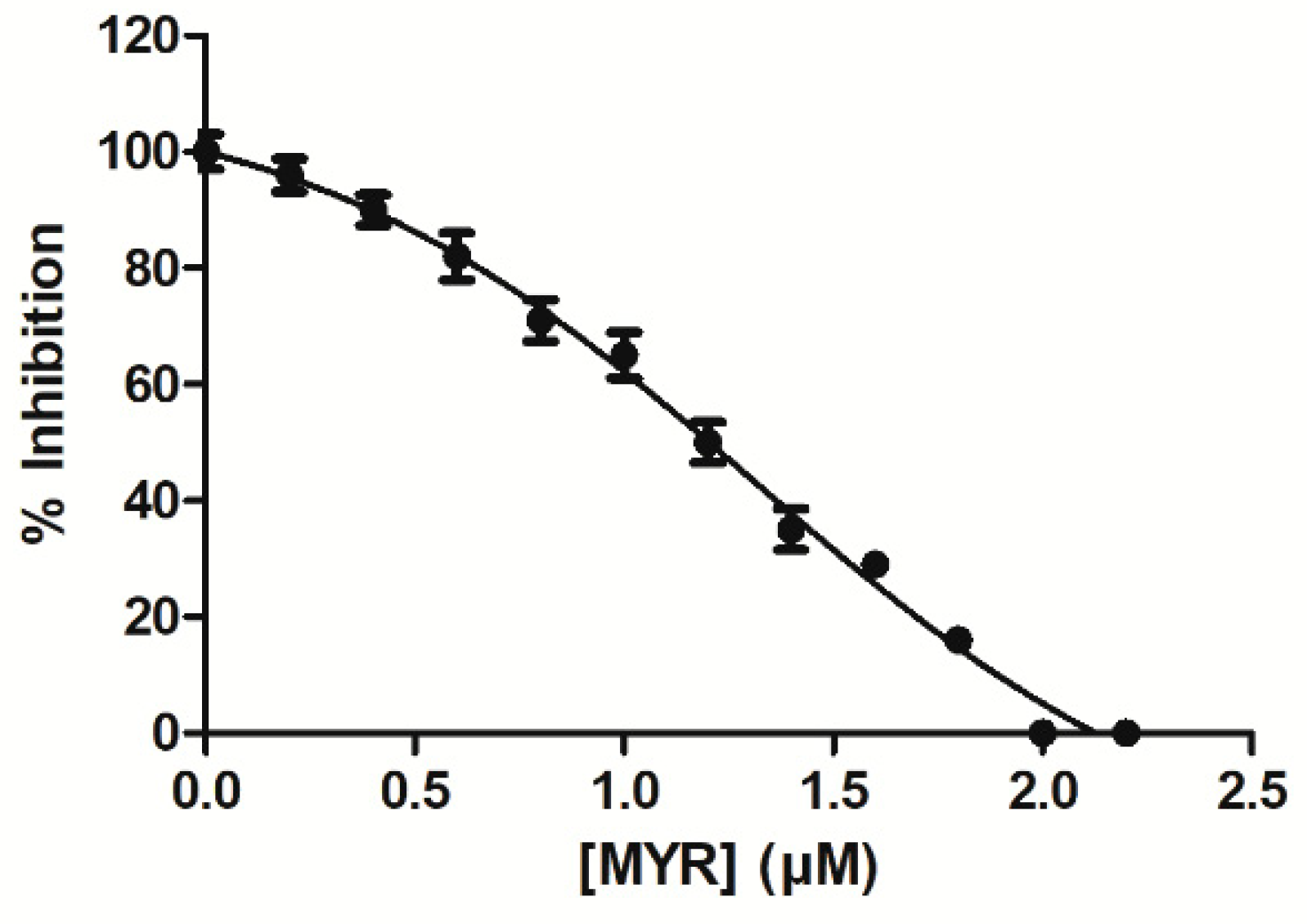
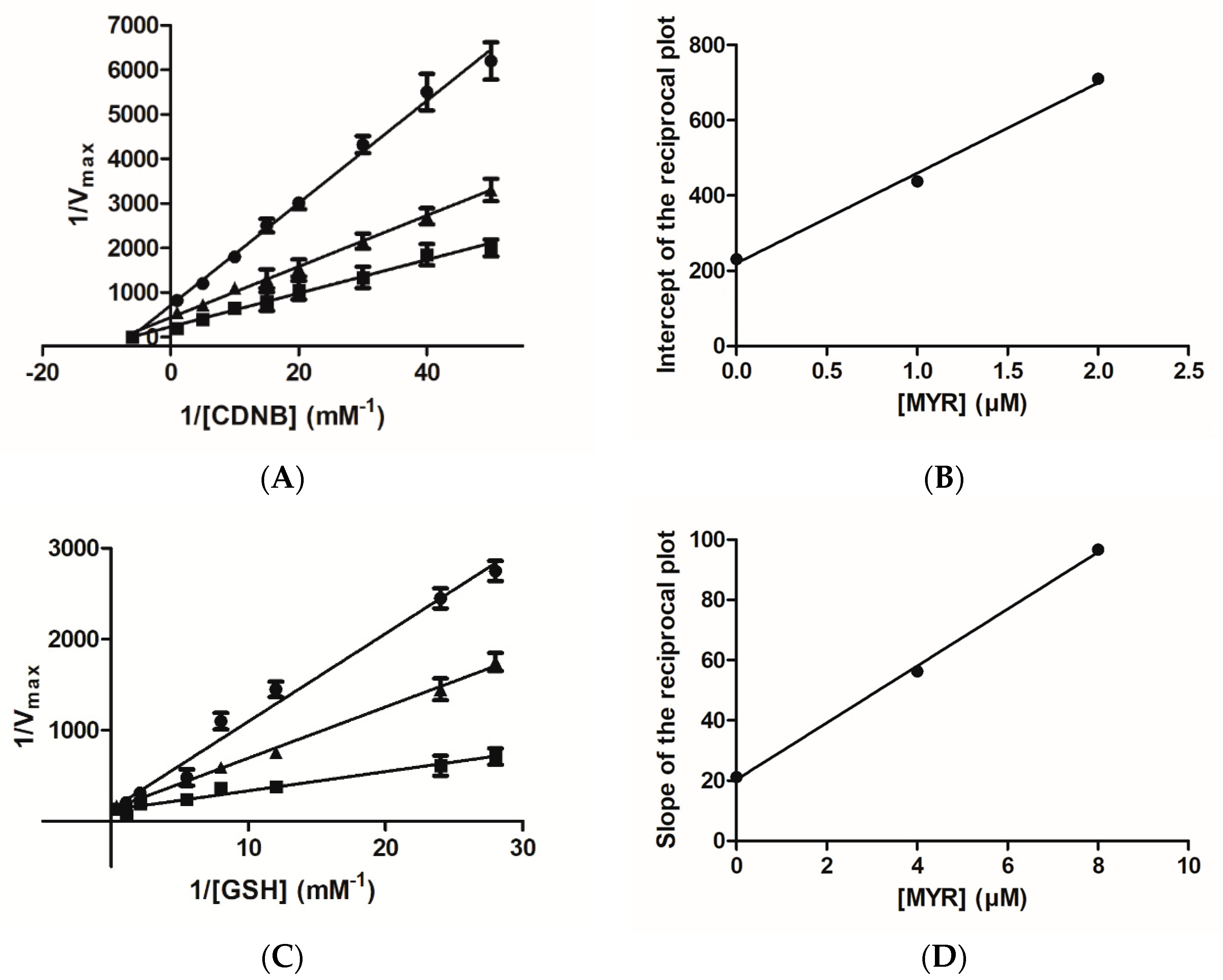
3.1. Kinetics Inhibition Analysis of the Interaction of MYR with hGSTA1–1
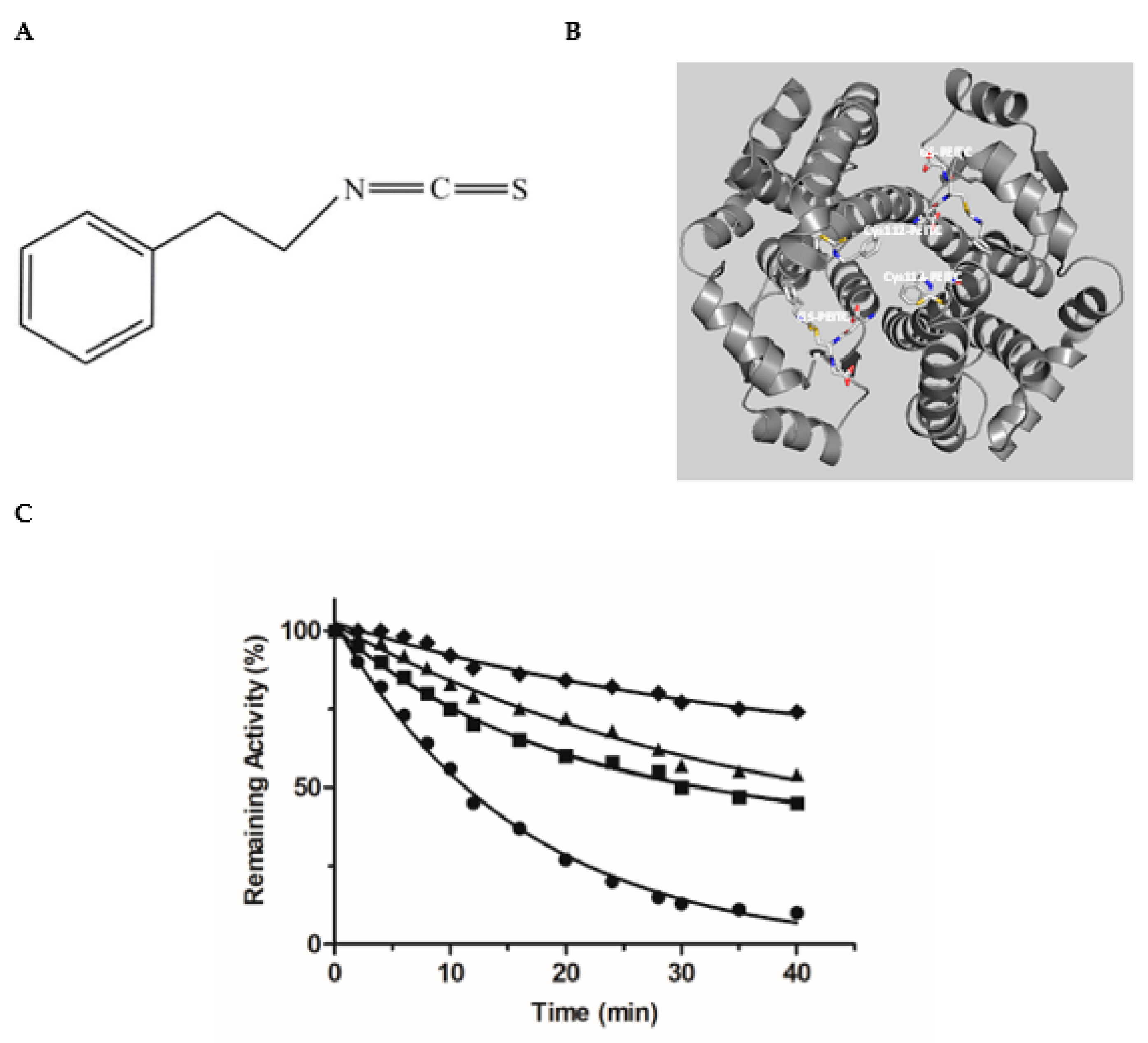
3.2. Irreversible Inactivation of hGSTA1–1 by Phenethyl Isothiocyanate (PEITC)
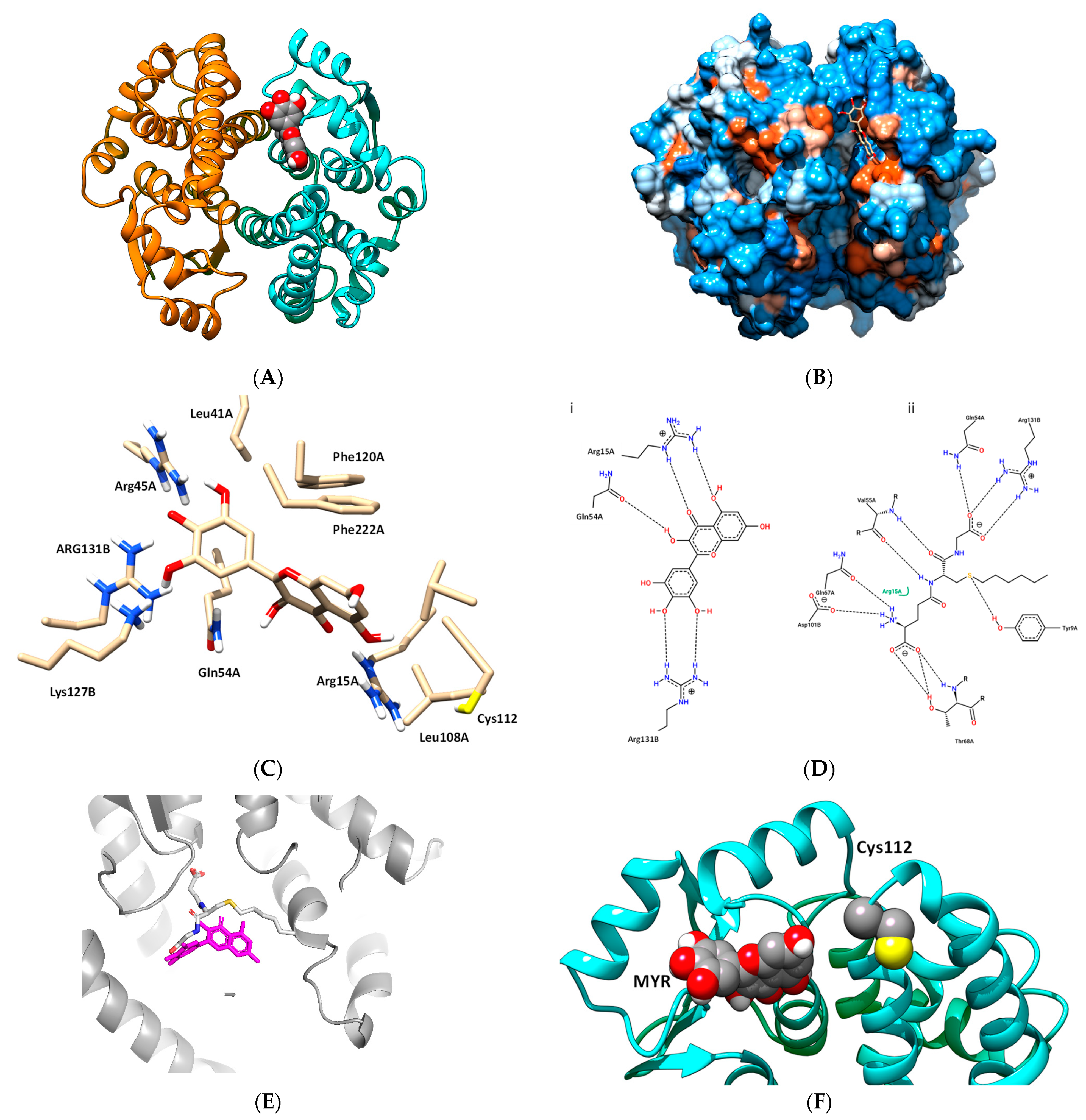
3.3. Study of the Interaction of hGSTA1–1 and MYR by In Silico Molecular Docking
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Perperopoulou, F.; Pouliou, F.; Labrou, N.E. Recent advances in protein engineering and biotechnological applications of glutathione transferases. Crit. Rev. Biotechnol. 2018, 38, 511–528. [Google Scholar] [CrossRef]
- Mannervik, B.; Ismail, A.; Lindström, H.; Sjödin, B.; Ing, N.H. Glutathione Transferases as Efficient Ketosteroid Isomerases. Front. Mol. Biosci. 2021, 8, 765970. [Google Scholar] [CrossRef] [PubMed]
- Pljesa-Ercegovac, M.; Savic-Radojevic, A.; Matic, M.; Coric, V.; Djukic, T.; Radic, T.; Simic, T. Glutathione Transferases: Potential Targets to Overcome Chemoresistance in Solid Tumors. Int. J. Mol. Sci. 2018, 19, 3785. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Gupta, S.K.; Singh, P.; Ali, V.; Kumar, V.; Verma, M. Drug-metabolizing enzymes: Role in drug resistance in cancer. Clin. Transl. Oncol. 2020, 22, 1667–1680. [Google Scholar] [CrossRef]
- Singh, S. Cytoprotective and regulatory functions of glutathione S-transferases in cancer cell proliferation and cell death. Cancer Chemother. Pharmacol. 2015, 75, 1–15. [Google Scholar] [CrossRef]
- Ściskalska, M.; Milnerowicz, H. The role of GSTπ isoform in the cells signalling and anticancer therapy. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 8537–8550. [Google Scholar] [CrossRef]
- Vaish, S.; Gupta, D.; Mehrotra, R.; Mehrotra, S.; Basantani, M.K. Glutathione S-transferase: A versatile protein family. 3 Biotech 2020, 10, 321. [Google Scholar] [CrossRef]
- Cui, J.; Li, G.; Yin, J.; Li, L.; Tan, Y.; Wei, H.; Liu, B.; Deng, L.; Tang, J.; Chen, Y.; et al. GSTP1 and cancer: Expression, methylation, polymorphisms and signaling (Review). Int. J. Oncol. 2020, 56, 867–878. [Google Scholar] [CrossRef]
- Nebert, D.W.; Vasiliou, V. Analysis of the glutathione S-transferase (GST) gene family. Hum. Genom. 2004, 1, 460–464. [Google Scholar] [CrossRef]
- Coles, B.F.; Kadlubar, F.F. Human Alpha Class Glutathione S-Transferases: Genetic Polymorphism, Expression, and Susceptibility to Disease. Methods Enzymol. 2005, 401, 9–42. [Google Scholar] [CrossRef] [PubMed]
- Tew, K.D. Glutathione-associated Enzymes in Anticancer Drug Resistance. Cancer Res. 1994, 54, 4313–4320. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-Y.; Kong, R.-J.; Li, Y.-M.; Huang, J.-Q.; Zhou, X.; Li, S.; Cheng, H. Carrier-free nanomedicine for enhanced photodynamic tumor therapy through glutathione S-transferase inhibition. Chem. Commun. 2022, 58, 3917–3920. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, F.; Wang, C.; Wang, C.; Tang, Y.; Jiang, Z. Glutathione S-transferase A1 mediates nicotine-induced lung cancer cell metastasis by promoting epithelial-mesenchymal transition. Exp. Ther. Med. 2017, 14, 1783–1788. [Google Scholar] [CrossRef]
- Zou, M.; Hu, X.; Xu, B.; Tong, T.; Jing, Y.; Xi, L.; Zhou, W.; Lu, J.; Wang, X.; Yang, X.; et al. Glutathione S-transferase isozyme alpha 1 is predominantly involved in the cisplatin resistance of common types of solid cancer. Oncol. Rep. 2019, 41, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yang, Z.; Zang, L.; Wang, G.; Zhou, S.; Jin, G.; Yang, Z.; Pan, X. Downregulation of Glutathione S-transferase A1 suppressed tumor growth and induced cell apoptosis in A549 cell line. Oncol. Lett. 2018, 16, 467–474. [Google Scholar] [CrossRef]
- Teslenko, I.; Trudeau, J.; Luo, S.; Watson, C.J.; Chen, G.; Truica, C.I.; Lazarus, P. Influence of glutathione-S-transferase A1*B allele on the metabolism of the aromatase inhibitor, exemestane, in human liver cytosols and in patients treated with exemestane. J. Pharmacol. Exp. Ther. 2022, 382, 327–334. [Google Scholar] [CrossRef]
- Kashyap, D.; Garg, V.K.; Tuli, H.S.; Yerer, M.B.; Sak, K.; Sharma, A.K.; Kumar, M.; Aggarwal, V.; Sandhu, S.S. Fisetin and Quercetin: Promising Flavonoids with Chemopreventive Potential. Biomolecules 2019, 9, 174. [Google Scholar] [CrossRef]
- Sachdeva, V.; Roy, A.; Bharadvaja, N. Current prospects of nutraceuticals: A review. Curr. Pharm. Biotechnol. 2020, 21, 884–896. [Google Scholar] [CrossRef]
- Chen, W.; Wang, S.; Wu, Y.; Shen, X.; Xu, S.; Guo, Z.; Zhang, R.; Xing, D. The physiologic activity and mechanism of querce-tin-like natural plant flavonoids. Curr. Pharm. Biotechnol. 2020, 21, 654–658. [Google Scholar] [CrossRef]
- Hosseinzadeh, E.; Hassanzadeh, A.; Marofi, F.; Alivand, M.R.; Solali, S. Flavonoid-Based Cancer Therapy: An Updated Review. Anti-Cancer Agents Med. Chem. 2020, 20, 1398–1414. [Google Scholar] [CrossRef]
- Xie, Y.; Wang, Y.; Xiang, W.; Wang, Q.; Cao, Y. Molecular Mechanisms of the Action of Myricetin in Cancer. Mini Rev. Med. Chem. 2020, 20, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Agraharam, G.; Girigoswami, A.; Girigoswami, K. Myricetin: A Multifunctional Flavonol in Biomedicine. Curr. Pharmacol. Rep. 2022, 8, 48–61. [Google Scholar] [CrossRef]
- Semwal, D.K.; Semwal, R.B.; Combrinck, S.; Viljoen, A. Myricetin: A Dietary Molecule with Diverse Biological Activities. Nutrients 2016, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Zhu, M.; Wang, L.; Yu, S. Anti-tumor effects and associated molecular mechanisms of myricetin. Biomed. Pharmacother. 2019, 120, 109506. [Google Scholar] [CrossRef] [PubMed]
- Taheri, Y.; Suleria, H.A.R.; Martins, N.; Sytar, O.; Beyatli, A.; Yeskaliyeva, B.; Seitimova, G.; Salehi, B.; Semwal, P.; Painuli, S.; et al. Myricetin bioactive effects: Moving from preclinical evidence to potential clinical applications. BMC Complement. Med. Ther. 2020, 20, 241. [Google Scholar] [CrossRef] [PubMed]
- Javed, Z.; Khan, K.; Herrera-Bravo, J.; Naeem, S.; Iqbal, M.J.; Raza, Q.; Sadia, H.; Raza, S.; Bhinder, M.; Calina, D.; et al. Myricetin: Targeting signaling networks in cancer and its implication in chemotherapy. Cancer Cell Int. 2022, 22, 239. [Google Scholar] [CrossRef]
- Devi, K.P.; Rajavel, T.; Habtemariam, S.; Nabavi, S.F. Molecular mechanisms underlying anticancer effects of myricetin. Life Sci. 2015, 142, 19–25. [Google Scholar] [CrossRef]
- Song, X.; Rao, H.; Guo, C.; Yang, B.; Ren, Y.; Wang, M.; Li, Y.; Cao, Z.; Pei, J. Myricetin exhibit selective anti-lymphoma activity by targeting BTK and is effective via oral administration in vivo. Phytomedicine 2021, 93, 153802. [Google Scholar] [CrossRef]
- Zheng, A.-W.; Chen, Y.-Q.; Zhao, L.-Q.; Feng, J.-G. Myricetin induces apoptosis and enhances chemosensitivity in ovarian cancer cells. Oncol. Lett. 2017, 13, 4974–4978. [Google Scholar] [CrossRef]
- Huang, H.; Chen, A.Y.; Ye, X.; Li, B.; Rojanasakul, Y.; Rankin, G.O.; Chen, Y.C. Myricetin inhibits proliferation of cisplatin-resistant cancer cells through a p53-dependent apoptotic pathway. Int. J. Oncol. 2015, 47, 1494–1502. [Google Scholar] [CrossRef]
- Axarli, I.; Labrou, N.E.; Petrou, C.; Rassias, N.; Cordopatis, P.; Clonis, Y.D. Sulphonamide-based bombesin prodrug analogues for glutathione transferase, useful in targeted cancer chemotherapy. Eur. J. Med. Chem. 2009, 44, 2009–2016. [Google Scholar] [CrossRef] [PubMed]
- Alqarni, M.; Foudah, A.; Muharram, M.; Labrou, N. The Interaction of the Flavonoid Fisetin with Human Glutathione Transferase A1-1. Metabolites 2021, 11, 190. [Google Scholar] [CrossRef] [PubMed]
- Alqarni, M.; Foudah, A.; Muharram, M.; Labrou, N. The Interaction of Human Glutathione Transferase GSTA1-1 with Reactive Dyes. Molecules 2021, 26, 2399. [Google Scholar] [CrossRef] [PubMed]
- Kumari, V.; Dyba, M.A.; Holland, R.J.; Liang, Y.-H.; Singh, S.V.; Ji, X. Irreversible Inhibition of Glutathione S-Transferase by Phenethyl Isothiocyanate (PEITC), a Dietary Cancer Chemopreventive Phytochemical. PLoS ONE 2016, 11, e0163821. [Google Scholar] [CrossRef]
- Platis, M.; Vlachakis, D.; Foudah, A.I.; Muharram, M.M.; Alqarni, M.H.; Papageorgiou, A.; Labrou, N.E. The Interaction of Schistosoma Japonicum Glutathione Transferase with Cibacron Blue 3GA and its Fragments. Med. Chem. 2021, 17, 332–343. [Google Scholar] [CrossRef]
- Bodourian, C.S.; Poudel, N.; Papageorgiou, A.C.; Antoniadi, M.; Georgakis, N.D.; Abe, H.; Labrou, N.E. Ligandability Assessment of Human Glutathione Transferase M1-1 Using Pesticides as Chemical Probes. Int. J. Mol. Sci. 2022, 23, 3606. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Le Trong, I.; Stenkamp, R.E.; Ibarra, C.; Atkins, W.M.; Adman, E.T. 1.3-Å resolution structure of human glutathione S-transferase with S-hexyl glutathione bound reveals possible extended ligandin binding site. Proteins Struct. Funct. Bioinform. 2002, 48, 618–627. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 2.0; Schrödinger, LLC.: New York, NY, USA, 2018.
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Honaker, M.T.; Acchione, M.; Zhang, W.; Mannervik, B.; Atkins, W.M. Enzymatic Detoxication, Conformational Selection, and the Role of Molten Globule Active Sites. J. Biol. Chem. 2013, 288, 18599–18611. [Google Scholar] [CrossRef]
- Georgakis, N.D.; Karagiannopoulos, D.A.; Thireou, T.N.; Eliopoulos, E.E.; Labrou, N.E.; Tsoungas, P.G.; Koutsilieris, M.N.; Clonis, Y.D. Concluding the trilogy: The interaction of 2,2′-dihydroxy-benzophenones and their carbonyl N-analogues with human glu-tathione transferase M1-1 face to face with the P1-1 and A1-1 isoenzymes involved in MDR. Chem. Biol. Drug Des. 2017, 90, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Premetis, G.; Marugas, P.; Fanos, G.; Vlachakis, D.; Chronopoulou, E.G.; Perperopoulou, F.; Dubey, K.K.; Shukla, P.; Foudah, A.I.; Muharram, M.M.; et al. The Interaction of the Microtubule Targeting Anticancer Drug Colchicine with Human Glutathione Transferases. Curr. Pharm. Des. 2020, 26, 5205–5212. [Google Scholar] [CrossRef] [PubMed]
- Pouliou, F.M.; Thireou, T.N.; Eliopoulos, E.E.; Tsoungas, P.G.; Labrou, N.E.; Clonis, Y.D. Isoenzyme- and allozyme-specific inhibitors: 2,2′-dihydroxybenzophenones and their carbonyl N-analogues that discriminate between human glutathione transferase A1-1 and P1-1 allozymes. Chem. Biol. Drug Des. 2015, 86, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Koutsoumpli, G.E.; Dimaki, V.D.; Thireou, T.N.; Eliopoulos, E.E.; Labrou, N.E.; Varvounis, G.I.; Clonis, Y.D. Synthesis and study of 2-(pyrrolesulfonylmethyl)-N-arylimines: A new class of inhibitors for human glutathione transferase A1-1. J. Med. Chem. 2012, 55, 6802–6813. [Google Scholar] [CrossRef][Green Version]
- Zoi, O.G.; Thireou, T.N.; Rinotas, V.E.; Tsoungas, P.G.; Eliopoulos, E.E.; Douni, E.K.; Labrou, N.E.; Clonis, Y.D. Designer xanthone: An inhibitor scaffold for MDR-involved human glutathione transferase isoenzyme A1-1. J. Biomol. Screen. 2013, 18, 1092–1102. [Google Scholar] [CrossRef][Green Version]
- Ploemen, J.; Van Ommen, B.; Bogaards, J.J.P.; Van Bladeren, P.J. Ethacrynic acid and its glutathione conjugate as inhibitors of glutathione S-transferases. Xenobiotica 1993, 23, 913–923. [Google Scholar] [CrossRef]
- Tew, K.D.; Bomber, A.M.; Hoffman, S.J. Ethacrynic acid and piriprost as enhancers of cytotoxicity in drug resistant and sensitive cell lines. Cancer Res. 1988, 48, 3622–3625. [Google Scholar]
- Petrini, M.; Conte, A.; Caracciolo, F.; Sabbatini, A.R.M.; Grassi, B.; Ronca, G. Reversing of chlorambucil resistance by ethacrynic acid in a B-CLL patient. Br. J. Haematol. 1993, 85, 409–410. [Google Scholar] [CrossRef]
- Zhang, J.; Ye, Z.-W.; Janssen-Heininger, Y.; Townsend, D.M.; Tew, K.D. Development of Telintra as an Inhibitor of Glutathione S-Transferase P. Handb. Exp. Pharmacol. 2021, 264, 71–91. [Google Scholar] [CrossRef]
- Karpusas, M.; Axarli, I.; Chiniadis, L.; Papakyriakou, A.; Bethanis, K.; Scopelitou, K.; Clonis, Y.D.; Labrou, N.E. The Interaction of the Chemotherapeutic Drug Chlorambucil with Human Glutathione Transferase A1-1: Kinetic and Structural Analysis. PLoS ONE 2013, 8, e56337. [Google Scholar] [CrossRef]
- Axarli, I.; Muleta, A.W.; Chronopoulou, E.G.; Papageorgiou, A.C.; Labrou, N.E. Directed evolution of glutathione transferases towards a selective glutathione-binding site and improved oxidative stability. Biochim. et Biophys. Acta (BBA)—Gen. Subj. 2017, 1861, 3416–3428. [Google Scholar] [CrossRef] [PubMed]
- Geib, T.; Lento, C.; Wilson, D.J.; Sleno, L. Liquid Chromatography-Tandem Mass Spectrometry Analysis of Acetaminophen Covalent Binding to Glutathione S-Transferases. Front. Chem. 2019, 7, 558. [Google Scholar] [CrossRef] [PubMed]
- Stierand, K.; Maass, P.C.; Rarey, M. Molecular complexes at a glance: Automated generation of two-dimensional complex diagrams. Bioinformatics 2006, 22, 1710–1716. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Competing Ligand | kobs (min −1) | Half-Life (min) |
|---|---|---|
| - | 0.062 ± 0.004 | 11.23 |
| S-nitrobenzyl-GSH (1 mM) | 0.049 ± 0.004 | 14.05 |
| MYR (10 μΜ) | 0.027 ± 0.006 | 25.84 |
| ΜΥΡ (20 μΜ) | 0.02589 ± 0.009 | 27.85 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alqarni, M.H.; Foudah, A.I.; Muharram, M.M.; Alam, A.; Labrou, N.E. Myricetin as a Potential Adjuvant in Chemotherapy: Studies on the Inhibition of Human Glutathione Transferase A1–1. Biomolecules 2022, 12, 1364. https://doi.org/10.3390/biom12101364
Alqarni MH, Foudah AI, Muharram MM, Alam A, Labrou NE. Myricetin as a Potential Adjuvant in Chemotherapy: Studies on the Inhibition of Human Glutathione Transferase A1–1. Biomolecules. 2022; 12(10):1364. https://doi.org/10.3390/biom12101364
Chicago/Turabian StyleAlqarni, Mohammed Hamed, Ahmed Ibrahim Foudah, Magdy Mohamed Muharram, Aftab Alam, and Nikolaos E. Labrou. 2022. "Myricetin as a Potential Adjuvant in Chemotherapy: Studies on the Inhibition of Human Glutathione Transferase A1–1" Biomolecules 12, no. 10: 1364. https://doi.org/10.3390/biom12101364
APA StyleAlqarni, M. H., Foudah, A. I., Muharram, M. M., Alam, A., & Labrou, N. E. (2022). Myricetin as a Potential Adjuvant in Chemotherapy: Studies on the Inhibition of Human Glutathione Transferase A1–1. Biomolecules, 12(10), 1364. https://doi.org/10.3390/biom12101364