
Dysregulation of DNA Methylation and Epigenetic Clocks in Prostate Cancer among Puerto Rican Men

 , , , ,
, , , ,  , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. IRB Approval, and Tissue Sample Selection
2.2. DNA Methylation Analysis
2.2.1. Illumina EPIC Methylation and DNA Samples
2.2.2. Quality Control and Normalization for Data Obtained from Epigenome-Wide Methylation Assays
2.2.3. Detection of Differentially Methylated Regions (DMRs)
2.3. Analysis for Ancestry Structure
2.4. Estimation of Epigenetic Mitotic Clocks
3. Results
3.1. Demographic and Clinicopathological Characteristics of Study Group
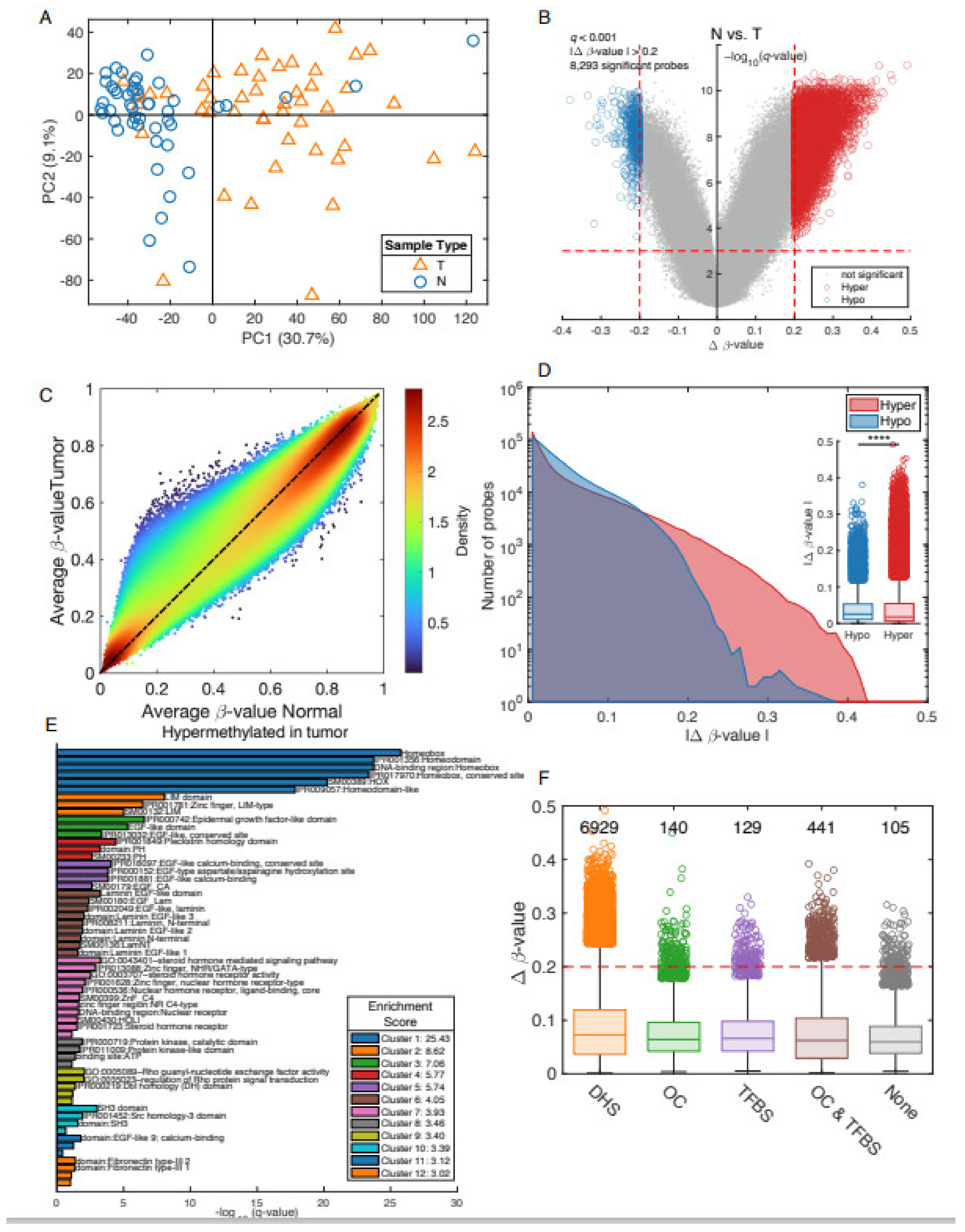
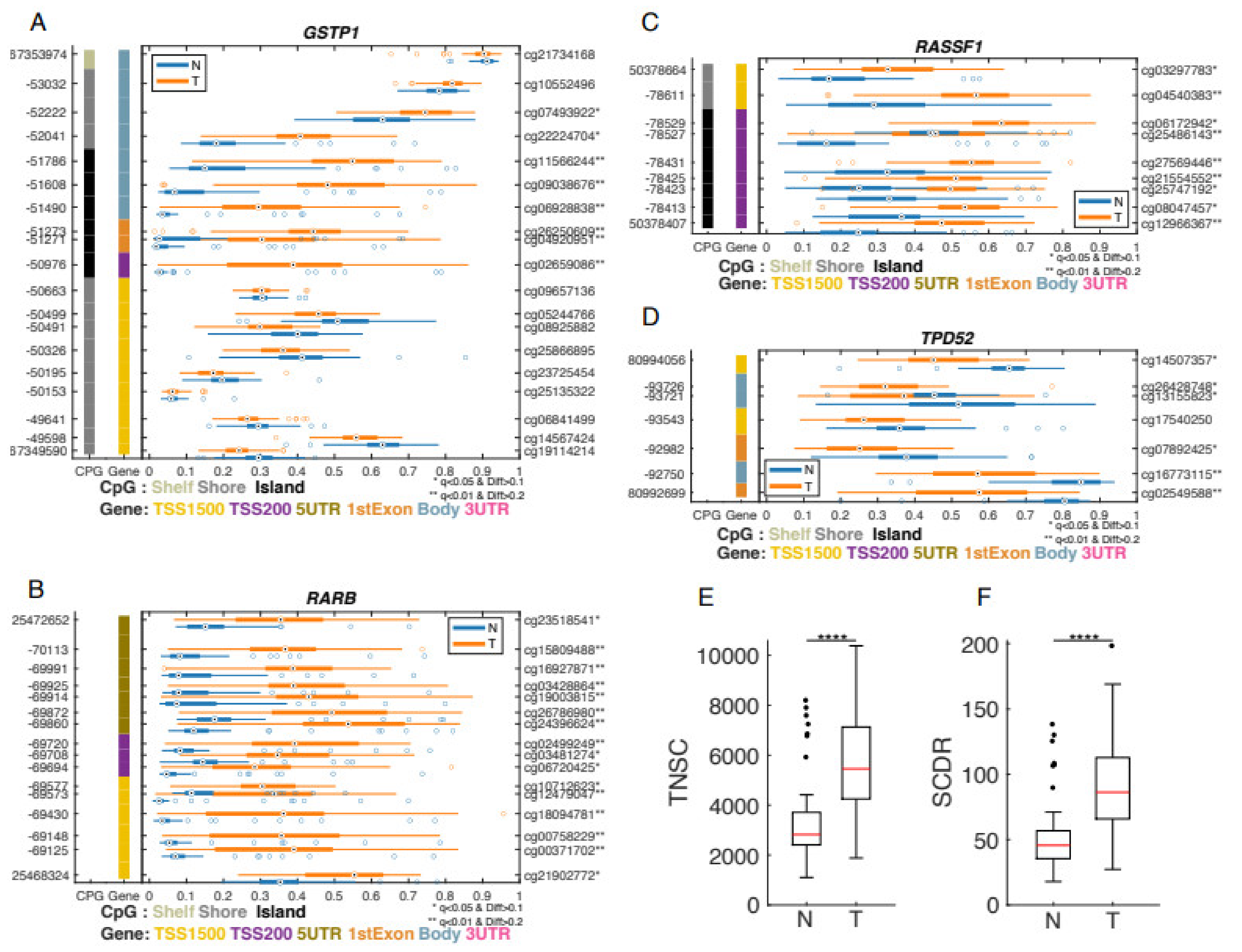
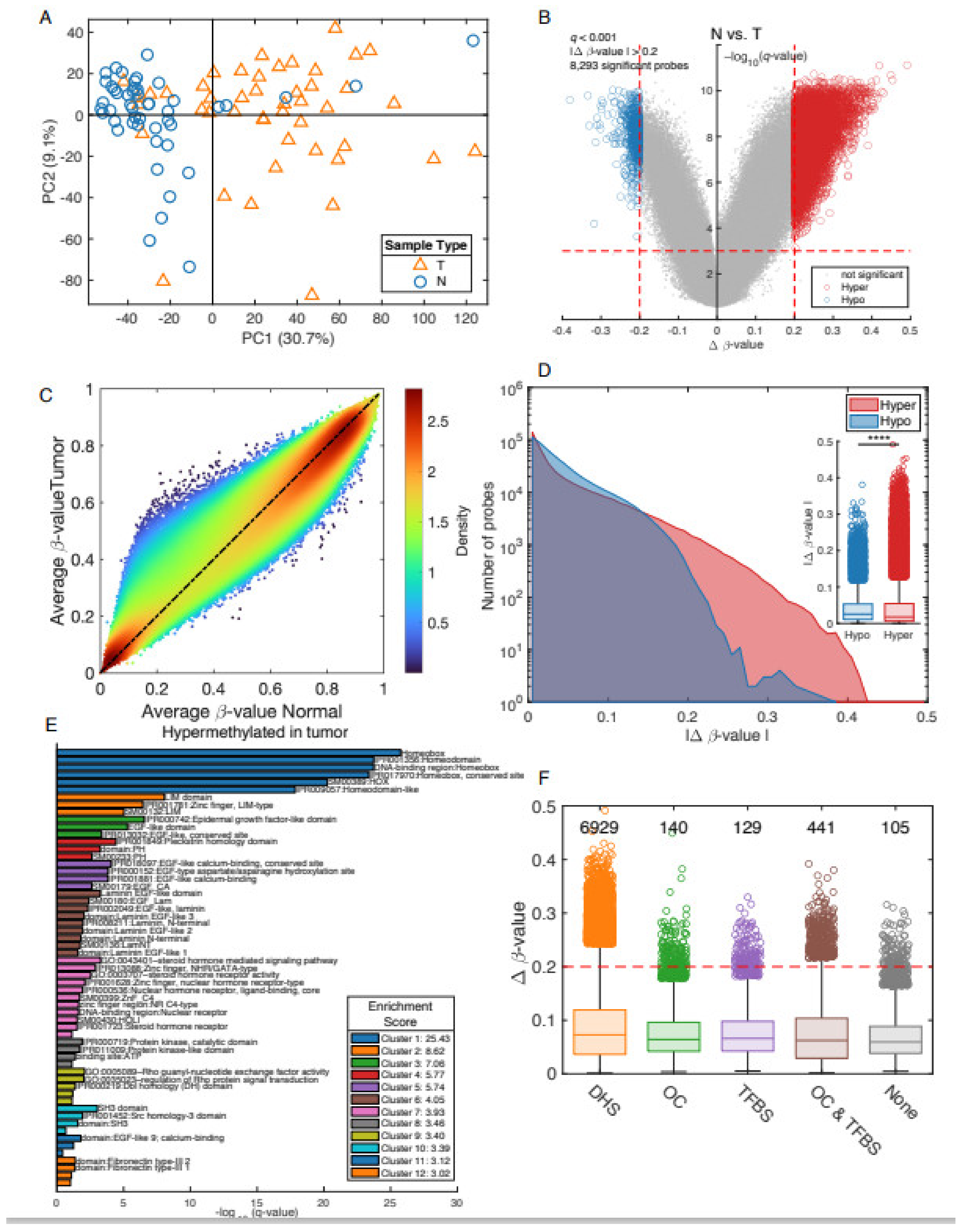
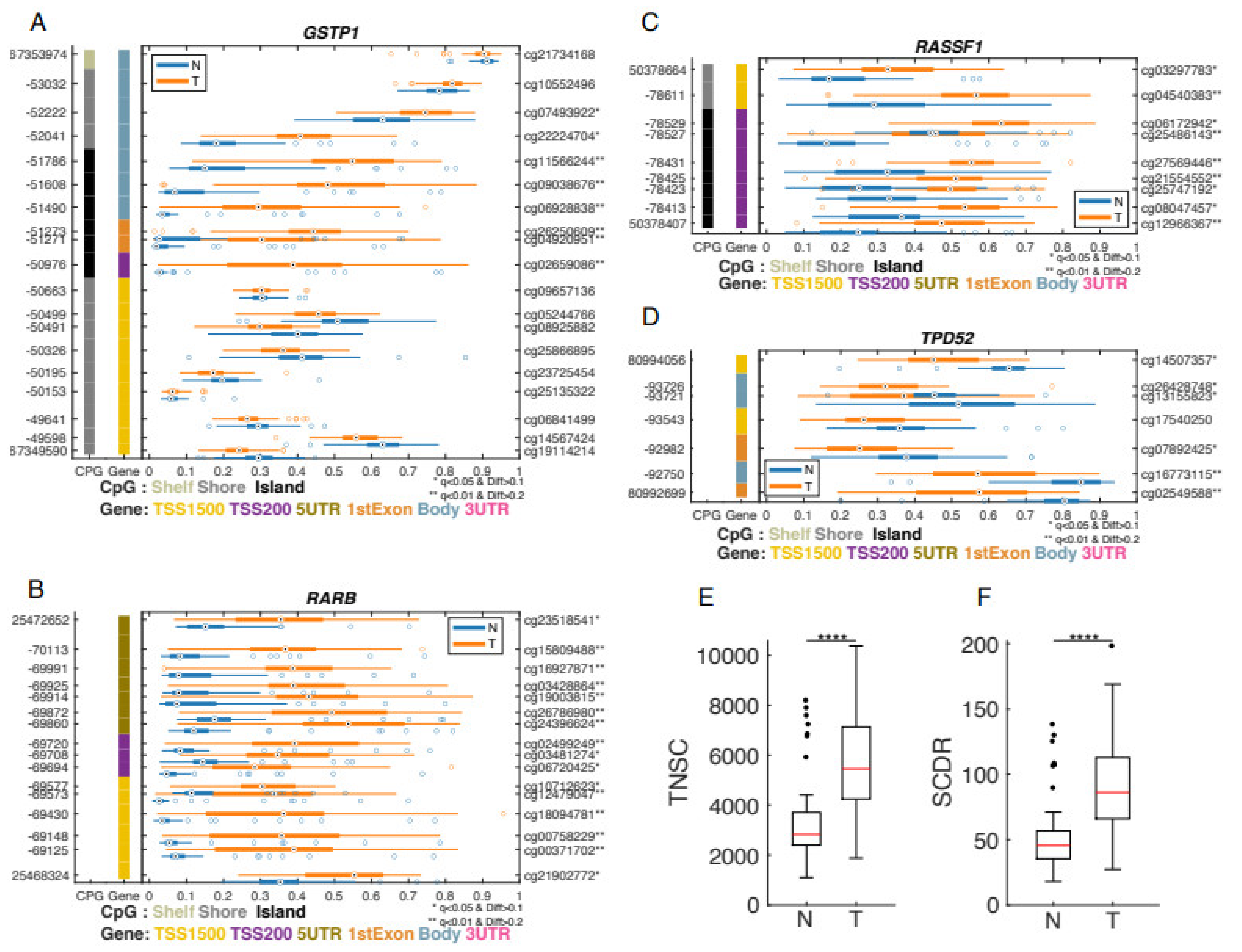
3.2. Differential Methylation between Prostate Cancer Tumors and Adjacent Non-Involved Tissue
3.3. Comparison of Epigenetic Clocks between Tumor vs. Adjacent Normal Tissues
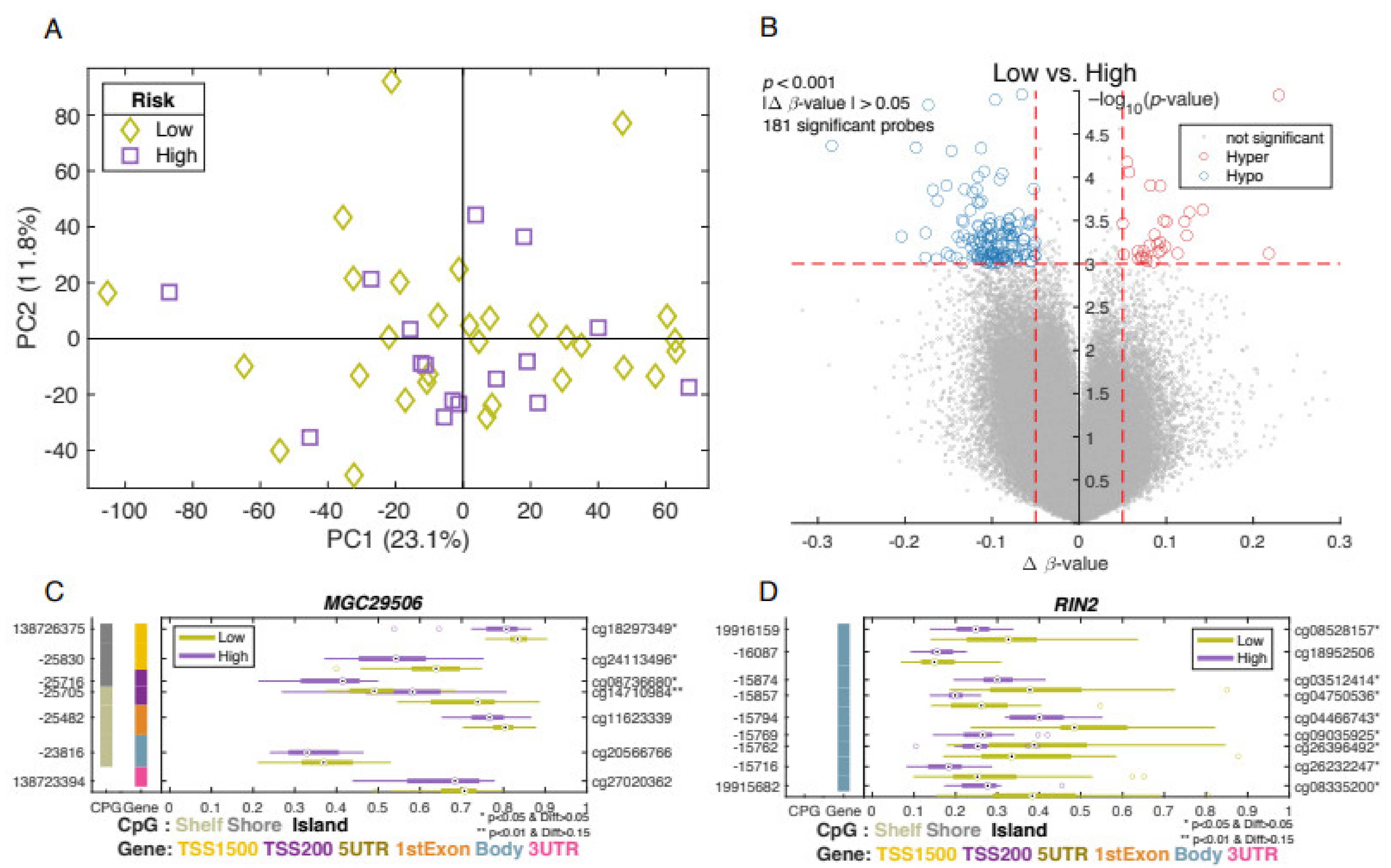
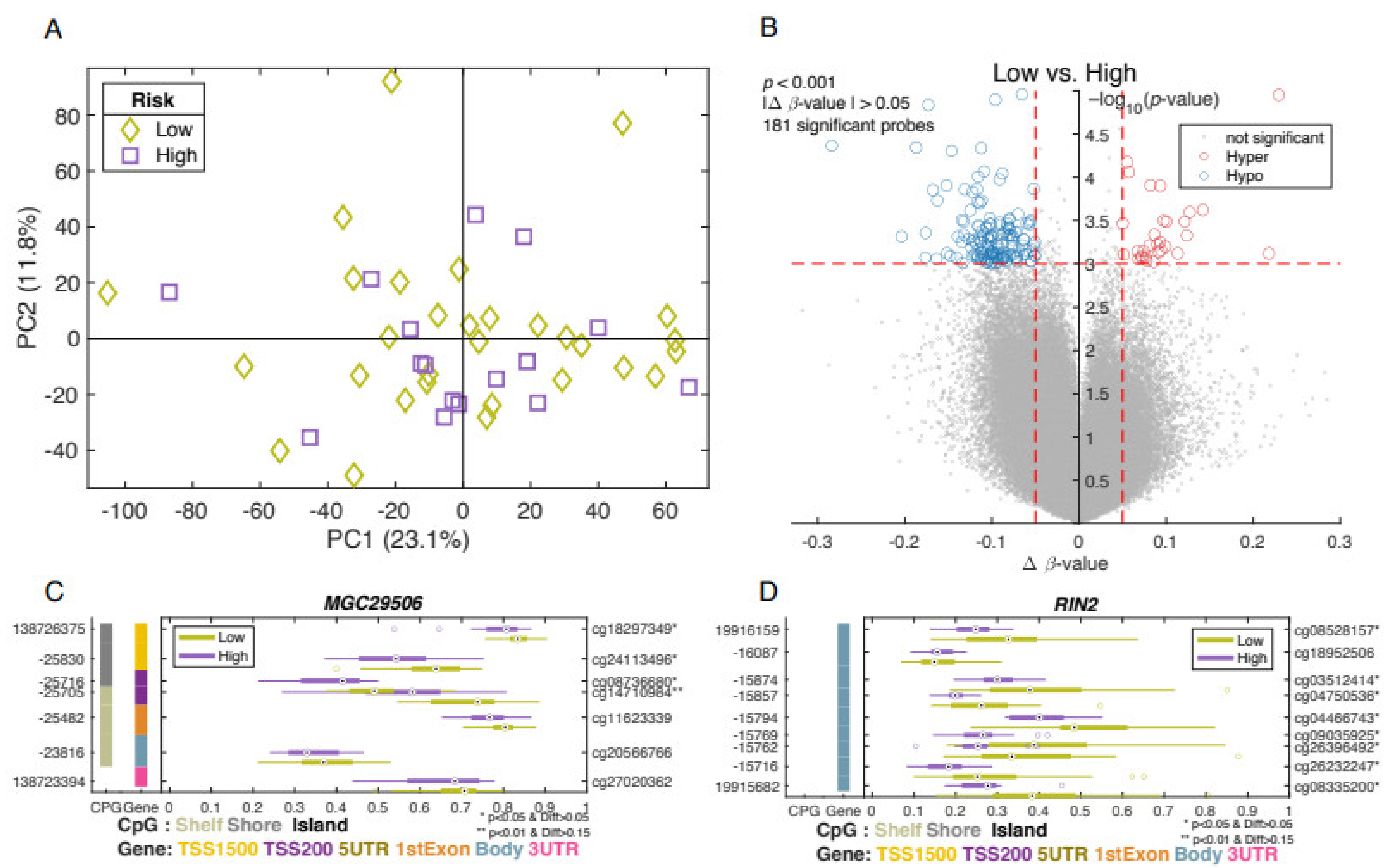
3.4. Differentially Methylated Genes Associated with Aggressive Type of Prostate Cancer
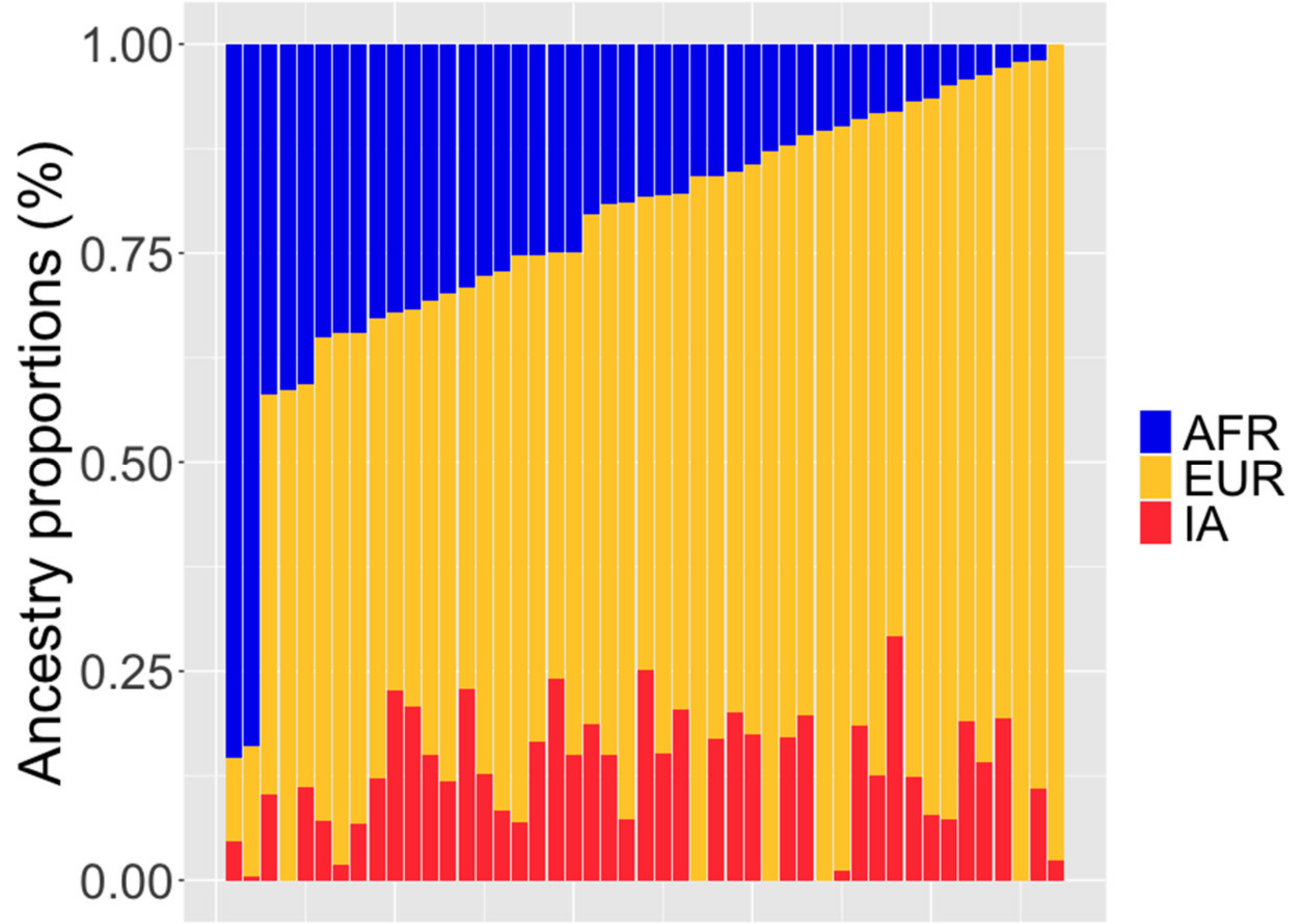
3.5. Ancestry Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- ACS. Cancer Facts & Figures 2021; American Cancer Society: Atlanta, GA, USA, 2021. [Google Scholar]
- NCI. Cancer Stat Facts: Prostate Cancer; Surveillance, Epidemiology, and End Results Program; National Cancer Institute: Bethesda, MD, USA, 2018. [Google Scholar]
- Chinea, F.M.; Patel, V.N.; Kwon, D.; Lamichhane, N.; Lopez, C.; Punnen, S.; Kobetz, E.N.; Abramowitz, M.C.; Pollack, A. Ethnic heterogeneity and prostate cancer mortality in Hispanic/Latino men: A population-based study. Oncotarget 2017, 8, 69709–69721. [Google Scholar] [CrossRef] [Green Version]
- Tortolero-Luna, G.; Zavala-Zegarra, D.; Pérez-Ríos, N.; Torres-Cintrón, C.R.; Ortiz-Ortiz, K.J.; Traverso-Ortiz, M.; Román-Ruiz, Y.; Veguilla-Rosario, I.; Vázquez-Cubano, N.; Merced-Vélez, M.F.; et al. Cancer in Puerto Rico, 2006–2010; Puerto Rico Central Cancer Registry: San Juan, PR, USA, 2013. [Google Scholar]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Rodrigues, D.N.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- Xu, N.; Wu, Y.P.; Ke, Z.B.; Liang, Y.C.; Cai, H.; Su, W.T.; Tao, X.; Chen, S.H.; Zheng, Q.S.; Wei, Y.; et al. Identification of key DNA methylation-driven genes in prostate adenocarcinoma: An integrative analysis of TCGA methylation data. J. Transl. Med. 2019, 17, 311. [Google Scholar] [CrossRef] [PubMed]
- Massie, C.E.; Mills, I.G.; Lynch, A.G. The importance of DNA methylation in prostate cancer development. J. Steroid Biochem. Mol. Biol. 2017, 166, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research. The molecular taxonomy of primary prostate cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carleton, N.M.; Zhu, G.; Gorbounov, M.; Miller, M.C.; Pienta, K.J.; Resar, L.M.S.; Veltri, R.W. PBOV1 as a potential biomarker for more advanced prostate cancer based on protein and digital histomorphometric analysis. Prostate 2018, 78, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Komura, K.; Sweeney, C.J.; Inamoto, T.; Ibuki, N.; Azuma, H.; Kantoff, P.W. Current treatment strategies for advanced prostate cancer. Int. J. Urol. 2018, 25, 220–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giudice, A.; Montella, M.; Boccellino, M.; Crispo, A.; D’Arena, G.; Bimonte, S.; Facchini, G.; Ciliberto, G.; Botti, G.; Quagliuolo, L.; et al. Epigenetic changes induced by green tea catechins are associated with prostate cancer. Curr. Mol. Med. 2017, 17, 405–420. [Google Scholar] [CrossRef]
- Pan, H.; Renaud, L.; Chaligne, R.; Bloehdorn, J.; Tausch, E.; Mertens, D.; Fink, A.M.; Fischer, K.; Zhang, C.; Betel, D.; et al. Discovery of Candidate DNA Methylation Cancer Driver Genes. Cancer Discov. 2021, 11, 2266–2281. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y. Promoter hypermethylation in prostate cancer. Cancer Control 2010, 17, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Park, J.Y. DNA methylation in promoter region as biomarkers in prostate cancer. Methods Mol. Biol. 2012, 863, 67–109. [Google Scholar] [PubMed] [Green Version]
- Devaney, J.M.; Wang, S.; Funda, S.; Long, J.; Taghipour, D.J.; Tbaishat, R.; Furbert-Harris, P.; Ittmann, M.; Kwabi-Addo, B. Identification of novel DNA-methylated genes that correlate with human prostate cancer and high-grade prostatic intraepithelial neoplasia. Prostate Cancer Prostatic Dis. 2013, 16, 292–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.W.; Kim, S.-T.; Turner, A.R.; Young, T.; Smith, S.; Liu, W.; Lindberg, J.; Egevad, L.; Gronberg, H.; Isaacs, W.B.; et al. Identification of new differentially methylated genes that have potential functional consequences in prostate cancer. PLoS ONE 2012, 7, e48455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geybels, M.S.; Zhao, S.; Wong, C.-J.; Bibikova, M.; Klotzle, B.; Wu, M.; Ostrander, E.A.; Fan, J.-B.; Feng, Z.; Stanford, J.L. Epigenomic profiling of DNA methylation in paired prostate cancer versus adjacent benign tissue. Prostate 2015, 75, 1941–1950. [Google Scholar] [CrossRef] [Green Version]
- Apprey, V.; Wang, S.; Tang, W.; Kittles, R.A.; Southerland, W.M.; Ittmann, M.; Kwabi-Addo, B. Association of genetic ancestry with DNA methylation changes in prostate cancer disparity. Anticancer Res. 2019, 39, 5861–5866. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Vogelstein, B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issa, J.P. Aging and epigenetic drift: A vicious cycle. J. Clin. Investig. 2014, 124, 24–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baylin, S.B.; Ohm, J.E. Epigenetic gene silencing in cancer—A mechanism for early oncogenic pathway addiction? Nat. Rev. Cancer 2006, 6, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Slieker, R.C.; BIOS Consortium; Van Iterson, M.; Luijk, R.; Beekman, M.; Zhernakova, D.V.; Moed, M.H.; Mei, H.; Van Galen, M.; Deelen, P.; et al. Age-related accrual of methylomic variability is linked to fundamental ageing mechanisms. Genome Biol. 2016, 17, 109. [Google Scholar] [CrossRef] [Green Version]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.-B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Dugué, P.-A.; Bassett, J.K.; Joo, J.E.; Jung, C.-H.; Wong, E.M.; Moreno-Betancur, M.; Schmidt, D.; Makalic, E.; Li, S.; Severi, G.; et al. DNA methylation-based biological aging and cancer risk and survival: Pooled analysis of seven prospective studies. Int. J. Cancer 2018, 142, 1611–1619. [Google Scholar] [CrossRef]
- Teschendorff, A.E. A comparison of epigenetic mitotic-like clocks for cancer risk prediction. Genome Med. 2020, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Deya, G.; Matta, J.; Encarnación-Medina, J.; Ortiz-Sanchéz, C.; Dutil, J.; Putney, R.; Berglund, A.; Dhillon, J.; Kim, Y.; Park, J.Y. Differential DNA methylation in prostate tumors from Puerto Rican men. Int. J. Mol. Sci. 2021, 22, 733. [Google Scholar] [CrossRef] [PubMed]
- Via, M.; Gignoux, C.R.; Roth, L.A.; Fejerman, L.; Galanter, J.; Choudhry, S.; Toro-Labrador, G.; Viera-Vera, J.; Oleksyk, T.K.; Beckman, K.; et al. History shaped the geographic distribution of genomic admixture on the island of Puerto Rico. PLoS ONE 2011, 6, e16513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Fortin, J.-P.; Triche, T.J., Jr.; Hansen, K.D. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics 2016, 33, 558–560. [Google Scholar] [CrossRef] [PubMed]
- Triche, T.J., Jr.; Weisenberger, D.J.; van den Berg, D.; Laird, P.W.; Siegmund, K.D. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res. 2013, 41, e90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortin, J.-P.; Labbe, A.; Lemire, M.; Zanke, B.W.; Hudson, T.J.; Fertig, E.J.; Greenwood, C.M.T.; Hansen, K.D. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014, 15, 503. [Google Scholar] [CrossRef] [Green Version]
- Du, P.; Zhang, X.; Huang, C.-C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2006, 8, 118–127. [Google Scholar] [CrossRef]
- Wilhelm-Benartzi, C.S.; Koestler, D.C.; Karagas, M.R.; Flanagan, J.M.; Christensen, B.C.; Kelsey, K.T.; Marsit, C.J.; Houseman, E.A.; Brown, R. Review of processing and analysis methods for DNA methylation array data. Br. J. Cancer 2013, 109, 1394–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaffe, A.E.; Murakami, P.; Lee, H.; Leek, J.T.; Fallin, M.D.; Feinberg, A.P.; Irizarry, R.A. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int. J. Epidemiol. 2012, 41, 200–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, E.M.; Berglund, A.E.; Riggs, B.M.; Eschrich, S.A.; Putney, R.M.; Ajidahun, A.O.; Coppola, D.; Shibata, D. Expanding epigenomics to archived FFPE tissues: An evaluation of DNA repair methodologies. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2622–2631. [Google Scholar] [CrossRef] [Green Version]
- Siegel, E.M.; Eschrich, S.; Winter, K.; Riggs, B.; Berglund, A.; Ajidahun, A.; Simko, J.; Moughan, J.; Ajani, J.; Magliocco, A.; et al. Epigenomic characterization of locally advanced anal. cancer: A radiation therapy oncology group 98–11 specimen study. Dis. Colon Rectum 2014, 57, 941–957. [Google Scholar] [CrossRef] [PubMed]
- Konno, H.; Yamauchi, S.; Berglund, A.; Putney, R.M.; Mulé, J.J.; Barber, G.N. Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene 2018, 37, 2037–2051. [Google Scholar] [CrossRef]
- Tsai, H.-J.; Choudhry, S.; Naqvi, M.; Rodriguez-Cintron, W.; Burchard, E.G.; Ziv, E. Comparison of three methods to estimate genetic ancestry and control for stratification in genetic association studies among admixed populations. Qual. Life Res. 2005, 118, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kryvenko, O.N.; Mitrache, N.; Do, K.C.; Jankowski, M.; Chitale, D.A.; Trudeau, S.; Rundle, A.; Belinsky, S.A.; Rybicki, B.A. Methylation of the RARB gene increases prostate cancer risk in black Americans. J. Urol. 2013, 190, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Chen, J.; Zhang, B.; Chen, X.; Huang, B.; Zhuang, J.; Mo, C.; Qiu, S. Association between RASSF1A promoter methylation and prostate cancer: A systematic review and meta-analysis. PLoS ONE 2013, 8, e75283. [Google Scholar]
- Cheong, A.; Zhang, X.; Cheung, Y.-Y.; Tang, W.-Y.; Chen, J.; Ye, S.-H.; Medvedovic, M.; Leung, Y.K.; Prins, G.S.; Ho, S.-M. DNA methylome changes by estradiol benzoate and bisphenol A links early-life environmental exposures to prostate cancer risk. Epigenetics 2016, 11, 674–689. [Google Scholar] [CrossRef]
- Kachroo, N.; Valencia, T.; Warren, A.Y.; Gnanapragasam, V.J. Evidence for downregulation of the negative regulator SPRED2 in clinical prostate cancer. Br. J. Cancer 2012, 108, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Portela, P.; Jobim, L.F.; Salim, P.H.; Koff, W.J.; Wilson, T.J.; Jobim, M.R.; Schwartsmann, G.; Roesler, R.; Jobim, M. Analysis of KIR gene frequencies and HLA class I genotypes in prostate cancer and control group. Int. J. Immunogenet. 2012, 39, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Sayaman, R.W.; Saad, M.; Thorsson, V.; Hu, D.; Hendrickx, W.; Roelands, J.; Porta-Pardo, E.; Mokrab, Y.; Farshidfar, F.; Kirchoff, T.; et al. Germline genetic contribution to the immune landscape of cancer. Immunity 2021, 54, 367.e8–386.e8. [Google Scholar] [CrossRef] [PubMed]
- Sakabe, T.; Tsuchiya, H.; Kanki, K.; Azumi, J.; Gonda, K.; Mizuta, Y.; Yamada, D.; Wada, H.; Shomori, K.; Nagano, H.; et al. Identification of the genes chemosensitizing hepatocellular carcinoma cells to interferon-alpha/5-fluorouracil and their clinical significance. PLoS ONE 2013, 8, e56197. [Google Scholar] [CrossRef]
- Arai, T.; Miyoshi, Y.; Kim, S.J.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Association of GSTP1 CpG islands hypermethylation with poor prognosis in human breast cancers. Breast Cancer Res. Treat. 2006, 100, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.; Shukla, S.; Gupta, S. Promoter demethylation and chromatin remodeling by green tea polyphenols leads to re-expression of GSTP1 in human prostate cancer cells. Int. J. Cancer 2010, 126, 2520–2533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mian, O.Y.; Khattab, M.H.; Hedayati, M.; Coulter, J.; Abubaker-Sharif, B.; Schwaninger, J.M.; Veeraswamy, R.K.; Brooks, J.D.; Hopkins, L.; Shinohara, D.B.; et al. GSTP1 Loss results in accumulation of oxidative DNA base damage and promotes prostate cancer cell survival following exposure to protracted oxidative stress. Prostate 2015, 76, 199–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enokida, H.; Shiina, H.; Urakami, S.; Igawa, M.; Ogishima, T.; Pookot, D.; Li, L.-C.; Tabatabai, Z.L.; Kawahara, M.; Nakagawa, M.; et al. Ethnic group-related differences in CpG hypermethylation of the GSTP1 gene promoter among African-American, Caucasian and Asian patients with prostate cancer. Int. J. Cancer 2005, 116, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.-Y.; Kim, B.-H.; Choi, M.; Yoo, E.J.; Moon, K.C.; Cho, Y.-M.; Kim, D.; Kang, G.H. Hypermethylation of CpG island loci and hypomethylation of LINE-1 and Alu repeats in prostate adenocarcinoma and their relationship to clinicopathological features. J. Pathol. 2006, 211, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y. Promoter hypermethylation as a biomarker in prostate adenocarcinoma. Method. Mol. Biol. 2015, 1238, 607–625. [Google Scholar]
- Kirby, M.K.; Ramaker, R.C.; Roberts, B.S.; Lasseigne, B.N.; Gunther, D.S.; Burwell, T.C.; Davis, N.S.; Gulzar, Z.G.; Absher, D.M.; Cooper, S.J.; et al. Genome-wide DNA methylation measurements in prostate tissues uncovers novel prostate cancer diagnostic biomarkers and transcription factor binding patterns. BMC Cancer 2017, 17, 273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, M.; Katoh, M. MGC29506 gene, frequently down-regulated in intestinal-type gastric cancer, encodes secreted-type protein with conserved cysteine residues. Int. J. Oncol. 2003, 23, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Shen, C.; Fu, Y.; Tian, L.; Chen, M. MGC29506 induces cell cycle arrest and is downregulated in gastric cancer. Cell. Immunol. 2013, 281, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-Q.; Xu, S.-W.; Wang, W.; Piao, S.-Z.; Mao, X.L.; Zhou, X.-B.; Wang, Y.; Wu, W.-D.; Ye, L.-P.; Li, S.-W. Identification and validation of a novel DNA damage and DNA repair related genes-based signature for colon cancer prognosis. Front. Genet. 2021, 12, 635863. [Google Scholar] [CrossRef] [PubMed]
- Alvi, M.A.; Liu, X.; O’Donovan, M.; Newton, R.; Wernisch, L.; Shannon, N.B.; Shariff, K.; di Pietro, M.; Bergman, J.J.G.H.M.; Ragunath , K.; et al. DNA methylation as an adjunct to histopathology to detect prevalent, inconspicuous dysplasia and early-stage neoplasia in Barrett’s esophagus. Clin. Cancer Res. 2013, 19, 878–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Carastro, L.M.; Lin, H.-Y.; Park, H.Y.; Kim, D.; Radlein, S.; Hampton, K.K.; Hakam, A.; Zachariah, B.; Pow-Sang, J.; Park, J.Y. Role of p73 dinucleotide polymorphism in prostate cancer and p73 protein isoform balance. Prostate Cancer 2014, 2014, 129582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M.P.; Borecki, I.B.; Zhang, Z.; Nguyen, L.; Ma, D.; Gao, X.; Qi, Y.; Carrington, M.; Rader, J.S. HLA-Cw group 1 ligands for KIR increase susceptibility to invasive cervical cancer. Immunogenetics 2010, 62, 761–765. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-W.; Wong, L.L.-Y.; Tse, E.Y.-T.; Liu, H.F.; Leong, V.Y.-L.; Lee, J.M.-F.; Hardie, D.G.; Ng, I.O.-L.; Ching, Y.-P. AMPK promotes p53 acetylation via phosphorylation and inactivation of SIRT1 in liver cancer cells. Cancer Res. 2012, 72, 4394–4404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- William, W.N.; Kim, J.-S.; Liu, D.D.; Solis, L.; Behrens, C.; Lee, J.J.; Lippman, S.M.; Kim, E.S.; Hong, W.K.; Wistuba, I.I.; et al. The impact of phosphorylated AMP-activated protein kinase expression on lung cancer survival. Ann. Oncol. 2012, 23, 78–85. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Risk | High | Low | p-Value |
|---|---|---|---|
| n = 17 | n = 32 | ||
| Age at Diagnosis | 64.6 ± 5.7 | 60.3 ± 9.1 | 0.085 |
| PSA | 7.72 ± 5.07 | 7.01 ± 6.46 | 0.724 |
| Gleason score | <0.0001 | ||
| 6 | 0 | 20 | |
| 7 (3 + 4) | 0 | 12 | |
| 7 (4 + 3) | 11 | 0 | |
| 8–9 | 6 | 0 | |
| Stage | <0.0001 | ||
| T1c | 1 | 1 | |
| T2a | 2 | 9 | |
| T2c | 8 | 20 | |
| T3a | 1 | 1 | |
| T3b | 5 | 1 | |
| Surgical margins | 0.71 | ||
| Yes | 1 | 3 | |
| No | 14 | 27 | |
| Missing | 2 | 2 |
| Ancestral Population | Average | SD | Maximum | Minimum |
|---|---|---|---|---|
| African | 0.219 | 0.178 | 0.853 | 0.00001 |
| European | 0.658 | 0.179 | 0.978 | 0.0995 |
| Indigenous American | 0.123 | 0.0784 | 0.292 | 0.00001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berglund, A.; Matta, J.; Encarnación-Medina, J.; Ortiz-Sanchéz, C.; Dutil, J.; Linares, R.; Marcial, J.; Abreu-Takemura, C.; Moreno, N.; Putney, R.; et al. Dysregulation of DNA Methylation and Epigenetic Clocks in Prostate Cancer among Puerto Rican Men. Biomolecules 2022, 12, 2. https://doi.org/10.3390/biom12010002
Berglund A, Matta J, Encarnación-Medina J, Ortiz-Sanchéz C, Dutil J, Linares R, Marcial J, Abreu-Takemura C, Moreno N, Putney R, et al. Dysregulation of DNA Methylation and Epigenetic Clocks in Prostate Cancer among Puerto Rican Men. Biomolecules. 2022; 12(1):2. https://doi.org/10.3390/biom12010002
Chicago/Turabian StyleBerglund, Anders, Jaime Matta, Jarline Encarnación-Medina, Carmen Ortiz-Sanchéz, Julie Dutil, Raymond Linares, Joshua Marcial, Caren Abreu-Takemura, Natasha Moreno, Ryan Putney, and et al. 2022. "Dysregulation of DNA Methylation and Epigenetic Clocks in Prostate Cancer among Puerto Rican Men" Biomolecules 12, no. 1: 2. https://doi.org/10.3390/biom12010002
APA StyleBerglund, A., Matta, J., Encarnación-Medina, J., Ortiz-Sanchéz, C., Dutil, J., Linares, R., Marcial, J., Abreu-Takemura, C., Moreno, N., Putney, R., Chakrabarti, R., Lin, H.-Y., Yamoah, K., Osterman, C. D., Wang, L., Dhillon, J., Kim, Y., Kim, S. J., Ruiz-Deya, G., & Park, J. Y. (2022). Dysregulation of DNA Methylation and Epigenetic Clocks in Prostate Cancer among Puerto Rican Men. Biomolecules, 12(1), 2. https://doi.org/10.3390/biom12010002