
Exploring the Value of BRD9 as a Biomarker, Therapeutic Target and Co-Target in Prostate Cancer


Abstract
:1. Introduction
1.1. Prostate Cancer and Its Biomarkers
1.2. Current Therapies for PCa
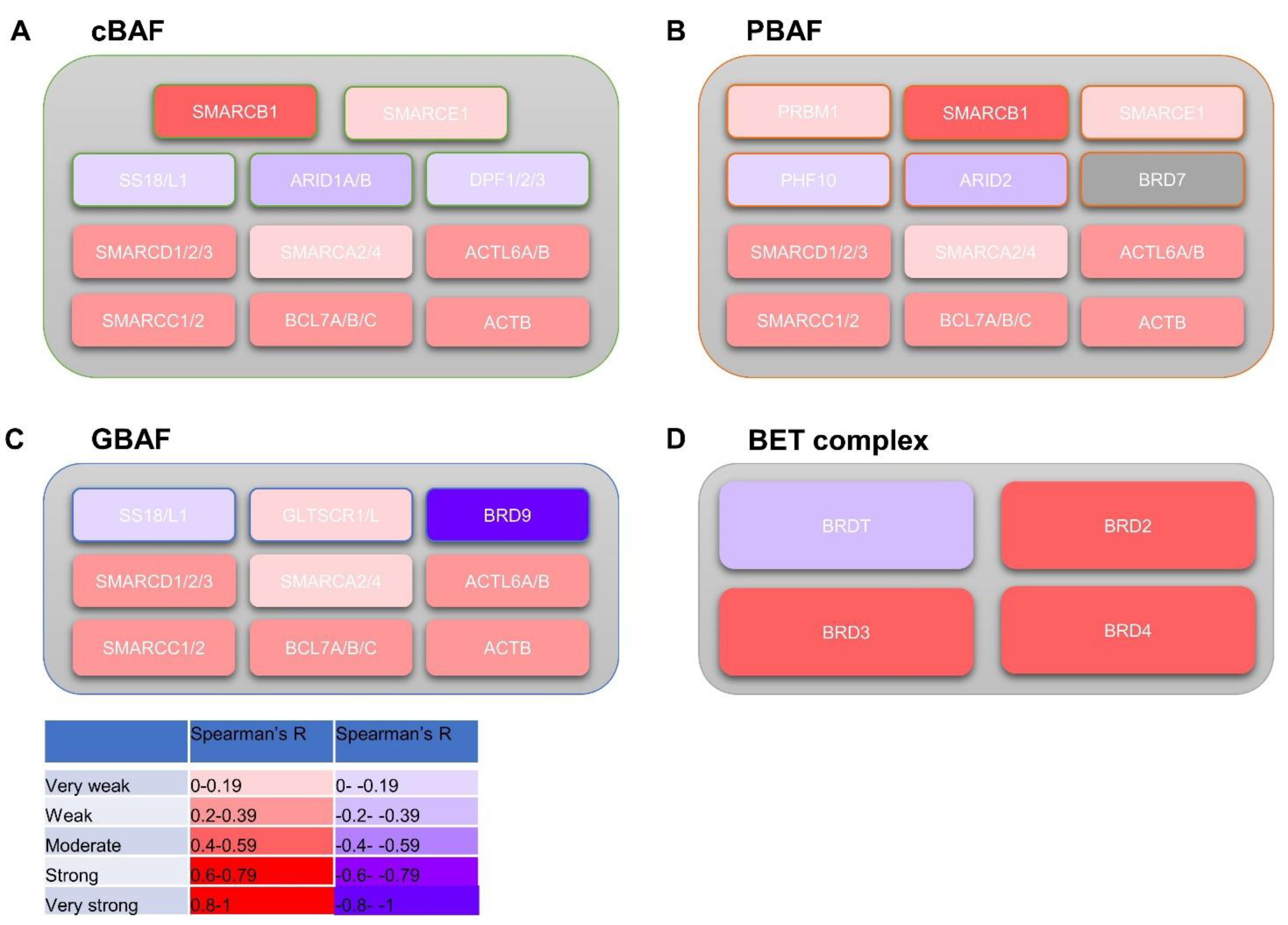
1.3. The Relationship between BRD9 and SWI/SNF and BET Complexes
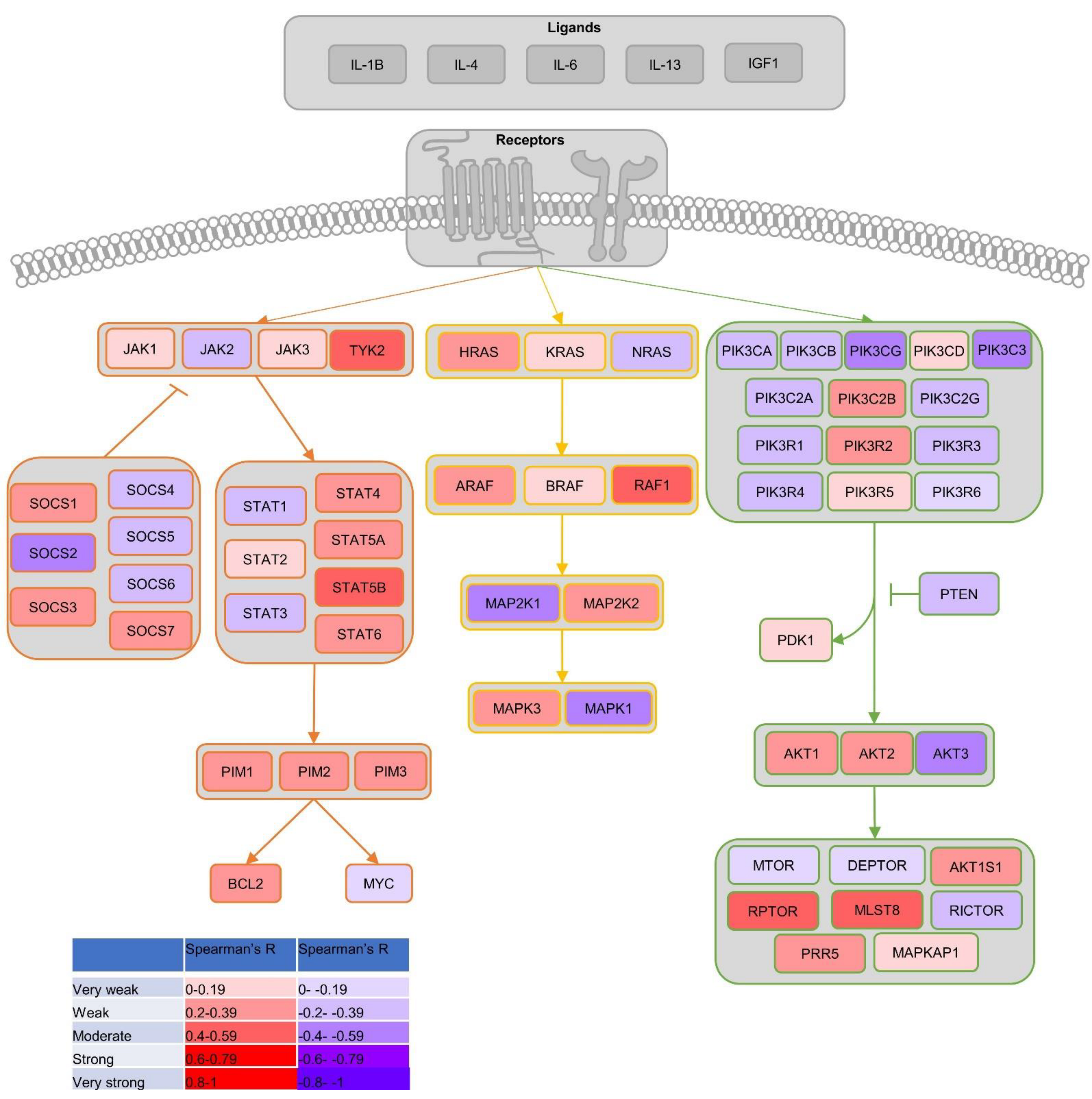
1.4. Common Signalling Pathways in PCa
1.5. BRD9 and PCa
2. Materials and Methods
2.1. Cancertool, cBioPortal and Genemania
2.2. Gene Correlations and Co-Expression Analysis
2.3. Comparing Continuous Data
2.4. Correlations
2.5. Survival Analysis
2.6. Comparing Mutation Distribution
3. Results
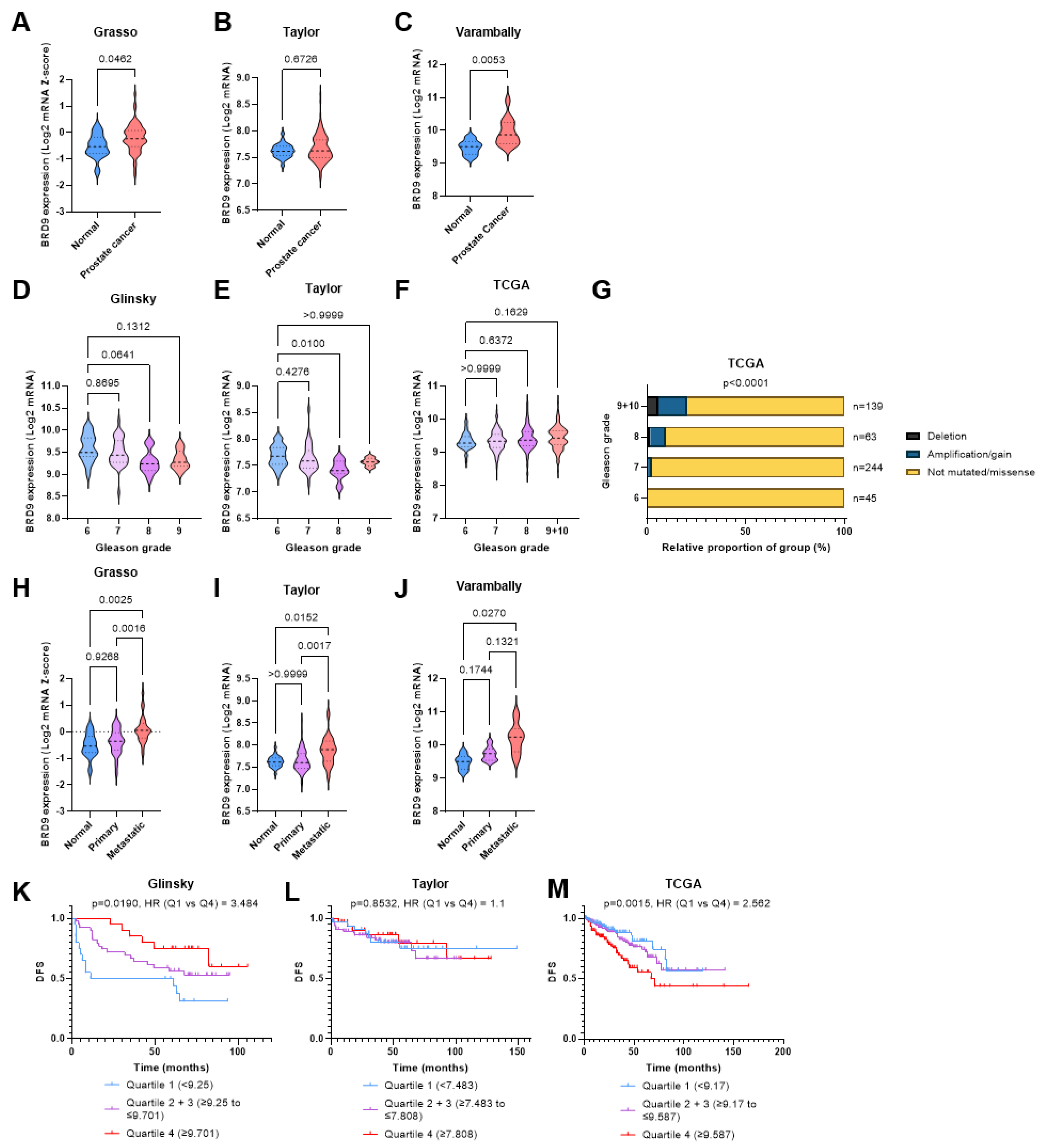
3.1. BRD9 Has Potential as a Diagnostic and Prognostic Biomarker in PCa
3.2. BRD9 Is Not Overexpressed in Patients with Advanced Stage PCa
3.3. BRD9 Does Not Appear to Play a Role as a Predictive Biomarker in PCa
3.4. BRD9 May Play a Role as a Therapeutic Target in CRPC
3.5. BRD9 Correlates with Genes in the SWI/SNF and BET Complexes
3.6. BRD9 Correlates with Genes Involved in Common PCa Proliferation-Driving Pathways
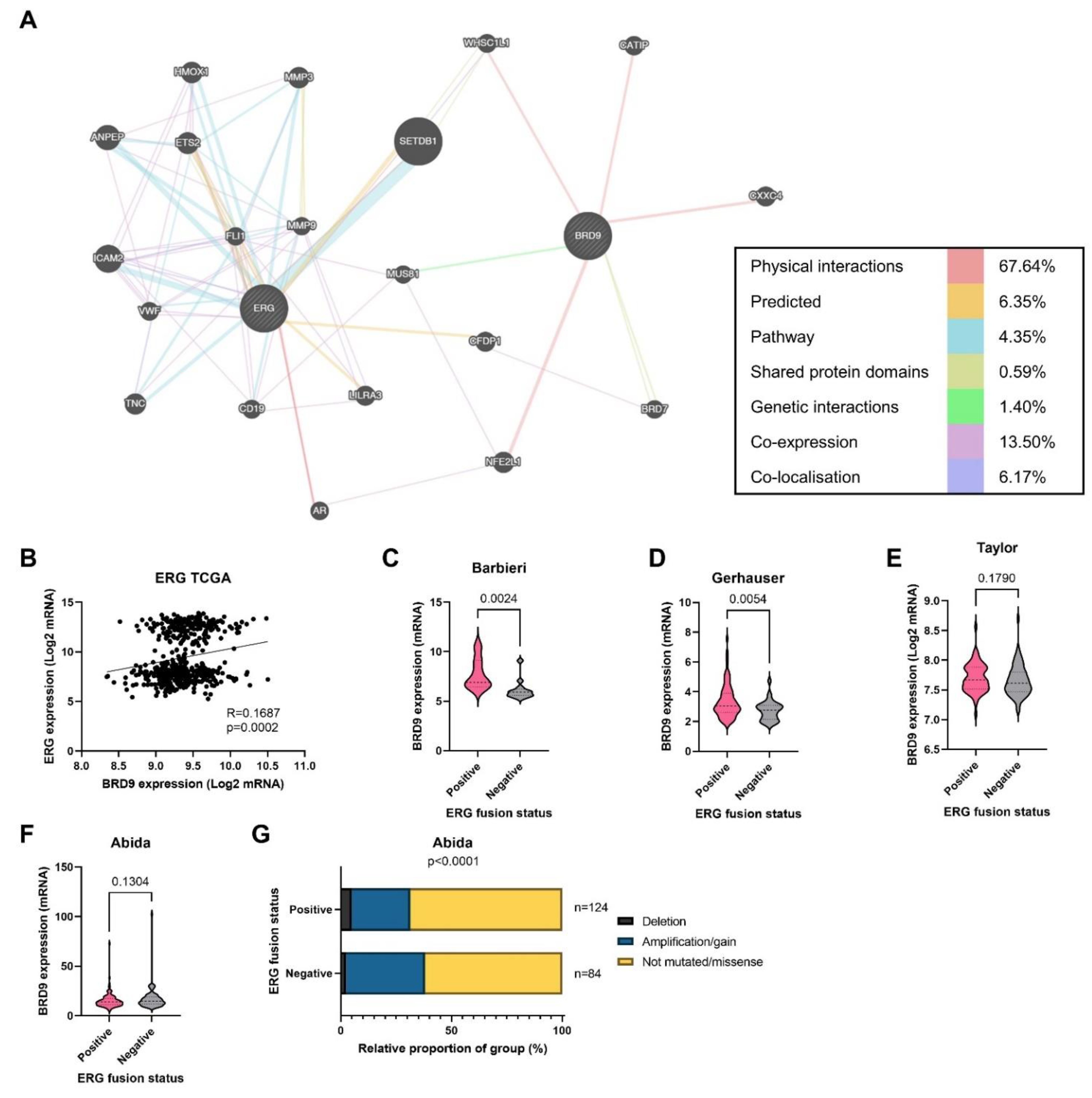
3.7. BRD9 Could Play a Role as a Therapeutic Co-Target Alongside ERG
4. Discussion
4.1. BRD9 as a Biomarker
4.2. BRD9 and CRPC
4.3. Co-Targeting BRD9
4.4. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prostate Cancer UK. About Prostate Cancer. Available online: https://prostatecanceruk.org/prostate-information/about-prostate-cancer (accessed on 19 April 2021).
- Prostate Cancer UK. Prostate Cancer UK’s Best Practice Pathway TREATMENT. Prostate Cancer UK, 2019. Available online: https://prostatecanceruk.org/media/1fknrpz5/treatmentpathwaypathwaycommentaryupdatenov2018-2-1.pdf (accessed on 19 April 2021).
- NICE. Information about PSA Testing. Available online: https://cks.nice.org.uk/topics/prostate-cancer/diagnosis/psa-testing/#:~:text=If%20the%20prostate%2Dspecific%20antigen,2%20weeks)%20to%20a%20specialist (accessed on 19 April 2021).
- Ilic, D.; Djulbegovic, M.; Jung, J.H.; Hwang, E.C.; Zhou, Q.; Cleves, A.; Agoritsas, T.; Dahm, P. Prostate cancer screening with prostate-specific antigen (PSA) test: A systematic review and meta-analysis. BMJ 2018, 362, k3519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filella, X.; Fernández-Galan, E.; Fernández Bonifacio, R.; Foj, L. Emerging biomarkers in the diagnosis of prostate cancer. Pharmgenom. Pers. Med. 2018, 11, 83–94. [Google Scholar] [CrossRef] [Green Version]
- Möller, A.; Olsson, H.; Grönberg, H.; Eklund, M.; Aly, M.; Nordström, T. The Stockholm3 blood-test predicts clinically-significant cancer on biopsy: Independent validation in a multi-center community cohort. Prostate Cancer Prostatic Dis. 2019, 22, 137–142. [Google Scholar] [CrossRef]
- Grönberg, H. Stockholm3 Validation Study in a Multi-Ethnic Cohort (SEPTA). Available online: https://clinicaltrials.gov/ct2/show/NCT04583072 (accessed on 1 May 2021).
- Siow, Z.R.; de Boer, R.H.; Lindeman, G.J.; Mann, G.B. Spotlight on the utility of the Oncotype DX(®) breast cancer assay. Int. J. Womens Health 2018, 10, 89–100. [Google Scholar] [CrossRef] [Green Version]
- CRUK. Survival. Available online: https://www.cancerresearchuk.org/about-cancer/prostate-cancer/survival (accessed on 20 April 2021).
- Fridriksson, J.; Folkvaljon, Y.; Nilsson, P.; Robinson, D.; Franck-Lissbrant, I.; Ehdaie, B.; Eastham, J.A.; Widmark, A.; Karlsson, C.T.; Stattin, P. Long-term adverse effects after curative radiotherapy and radical prostatectomy: Population-based nationwide register study. Scand. J. Urol. 2016, 50, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Litwin, M.S.; Tan, H.J. The Diagnosis and Treatment of Prostate Cancer: A Review. JAMA 2017, 317, 2532–2542. [Google Scholar] [CrossRef]
- NCI Staff. With Two FDA Approvals, Prostate Cancer Treatment Enters the PARP Era 2020. 2021. Available online: https://www.cancer.gov/news-events/cancer-currents-blog/2020/fda-olaparib-rucaparib-prostate-cancer (accessed on 19 April 2021).
- Centore, R.C.; Sandoval, G.J.; Soares, L.M.M.; Kadoch, C.; Chan, H.M. Mammalian SWI/SNF Chromatin Remodeling Complexes: Emerging Mechanisms and Therapeutic Strategies. Trends Genet. 2020, 36, 936–950. [Google Scholar] [CrossRef]
- Shi, J.; Vakoc, C.R. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell 2014, 54, 728–736. [Google Scholar] [CrossRef] [Green Version]
- Wyce, A.; Degenhardt, Y.; Bai, Y.; Le, B.; Korenchuk, S.; Crouthame, M.C.; McHugh, C.F.; Vessella, R.; Creasy, C.L.; Tummino, P.J.; et al. Inhibition of BET bromodomain proteins as a therapeutic approach in prostate cancer. Oncotarget 2013, 4, 2419–2429. [Google Scholar] [CrossRef] [Green Version]
- Gatchalian, J.; Malik, S.; Ho, J.; Lee, D.S.; Kelso, T.W.R.; Shokhirev, M.N.; Dixon, J.R.; Hargreaves, D.C. A non-canonical BRD9-containing BAF chromatin remodeling complex regulates naive pluripotency in mouse embryonic stem cells. Nat. Commun. 2018, 9, 5139. [Google Scholar] [CrossRef] [Green Version]
- Alpsoy, A.; Utturkar, S.M.; Carter, B.C.; Dhiman, A.; Torregrosa-Allen, S.E.; Currie, M.P.; Elzey, B.D.; Dykhuizen, E.C. BRD9 Is a Critical Regulator of Androgen Receptor Signaling and Prostate Cancer Progression. Cancer Res. 2021, 81, 820–833. [Google Scholar] [CrossRef]
- Canesin, G.; Krzyzanowska, A.; Hellsten, R.; Bjartell, A. Cytokines and Janus kinase/signal transducer and activator of transcription signaling in prostate cancer: Overview and therapeutic opportunities. Curr. Opin. Endocr. Metab. Res. 2020, 10, 36–42. [Google Scholar] [CrossRef]
- Haddad, B.R.; Gu, L.; Mirtti, T.; Dagvadorj, A.; Vogiatzi, P.; Hoang, D.T.; Bajaj, R.; Leiby, B.; Ellsworth, E.; Blackmon, S.; et al. STAT5A/B gene locus undergoes amplification during human prostate cancer progression. Am. J. Pathol. 2013, 182, 2264–2275. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Berriguete, G.; Fraile, B.; Martínez-Onsurbe, P.; Olmedilla, G.; Paniagua, R.; Royuela, M. MAP Kinases and Prostate Cancer. J. Signal Transduct. 2012, 2012, 169170. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, R.; McGuinness, D.H.; McCall, P.; Underwood, M.A.; Seywright, M.; Orange, C.; Edwards, J. Upregulation of MAPK pathway is associated with survival in castrate-resistant prostate cancer. Br. J. Cancer 2011, 104, 1920–1928. [Google Scholar] [CrossRef]
- Jonsson Comprehensive Cancer Center. Trametinib in Treating Patients with Progressive Metastatic Hormone-Resistant Prostate Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT02881242 (accessed on 1 May 2021).
- Edlind, M.P.; Hsieh, A.C. PI3K-AKT-mTOR signaling in prostate cancer progression and androgen deprivation therapy resistance. Asian J. Androl. 2014, 16, 378–386. [Google Scholar] [CrossRef]
- De Bono, J.S.; de Giorgi, U.; Rodrigues, D.N.; Massard, C.; Bracarda, S.; Font, A.; Arranz Arija, J.A.; Shih, K.C.; Radavoi, G.D.; Xu, N.; et al. Randomized Phase II Study Evaluating Akt Blockade with Ipatasertib, in Combination with Abiraterone, in Patients with Metastatic Prostate Cancer with and without PTEN Loss. Clin. Cancer Res. 2019, 25, 928–936. [Google Scholar] [CrossRef] [Green Version]
- Roche, H.-L. Ipatasertib Plus Abiraterone Plus Prednisone/Prednisolone, Relative to Placebo Plus Abiraterone Plus Prednisone/Prednisolone in Adult Male Patients with Metastatic Castrate-Resistant Prostate Cancer (IPATential150). Available online: https://clinicaltrials.gov/ct2/show/study/NCT03072238 (accessed on 1 May 2021).
- Bono, J.D. ESMO Virtual Congress 2020: IPATential150: Phase III Study of Ipatasertib plus Abiraterone vs Placebo plus Abiraterone in Metastatic Castration-Resistant Prostate Cancer. In Proceedings of the ESMO Virtual Congress, virtual, 19–20 September 2020. [Google Scholar]
- Tomlins, S.A.; Laxman, B.; Varambally, S.; Cao, X.; Yu, J.; Helgeson, B.E.; Cao, Q.; Prensner, J.R.; Rubin, M.A.; Shah, R.B.; et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia 2008, 10, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Demichelis, F.; Fall, K.; Perner, S.; Andrén, O.; Schmidt, F.; Setlur, S.R.; Hoshida, Y.; Mosquera, J.M.; Pawitan, Y.; Lee, C.; et al. TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene 2007, 26, 4596–4599. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, A.A.; Xavier, C.P.; Sukumar, G.; Tan, S.H.; Ravindranath, L.; Seraj, N.; Kumar, V.; Sreenath, T.; McLeod, D.G.; Petrovics, G.; et al. Identification of a Small Molecule That Selectively Inhibits ERG-Positive Cancer Cell Growth. Cancer Res. 2018, 78, 3659–3671. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Liao, Y.; Tang, L. Targeting BRD9 for Cancer Treatment: A New Strategy. Onco. Targets Ther. 2020, 13, 13191–13200. [Google Scholar] [CrossRef]
- Brien, G.L.; Remillard, D.; Shi, J.; Hemming, M.L.; Chabon, J.; Wynne, K.; Dillon, E.T.; Cagney, G.; van Mierlo, G.; Baltissen, M.P.; et al. Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. eLife 2018, 7. [Google Scholar] [CrossRef]
- Del Gaudio, N.; Di Costanzo, A.; Liu, N.Q.; Conte, L.; Migliaccio, A.; Vermeulen, M.; Martens, J.H.A.; Stunnenberg, H.G.; Nebbioso, A.; Altucci, L. BRD9 binds cell type-specific chromatin regions regulating leukemic cell survival via STAT5 inhibition. Cell Death Dis. 2019, 10, 338. [Google Scholar] [CrossRef]
- Hartley, A.; Leung, H.Y.; Ahmad, I. Targeting the BAF complex in advanced prostate cancer. Expert Opin. Drug Discov. 2021, 16, 173–181. [Google Scholar] [CrossRef]
- Dou, C.; Sun, L.; Wang, L.; Cheng, J.; Wu, W.; Zhang, C.; Xu, Q.; Tu, K.; Liu, J. Bromodomain-containing protein 9 promotes the growth and metastasis of human hepatocellular carcinoma by activating the TUFT1/AKT pathway. Cell Death Dis. 2020, 11, 730. [Google Scholar] [CrossRef]
- Alpsoy, A.; Dykhuizen, E.C. Glioma tumor suppressor candidate region gene 1 (GLTSCR1) and its paralog GLTSCR1-like form SWI/SNF chromatin remodeling subcomplexes. J. Biol. Chem. 2018, 293, 3892–3903. [Google Scholar] [CrossRef] [Green Version]
- Kirby, M.; Hirst, C.; Crawford, E.D. Characterising the castration-resistant prostate cancer population: A systematic review. Int. J. Clin. Pract. 2011, 65, 1180–1192. [Google Scholar] [CrossRef]
- Cortazar, A.R.; Torrano, V.; Martín-Martín, N.; Caro-Maldonado, A.; Camacho, L.; Hermanova, I.; Guruceaga, E.; Lorenzo-Martín, L.F.; Caloto, R.; Gomis, R.R.; et al. CANCERTOOL: A Visualization and Representation Interface to Exploit Cancer Datasets. Cancer Res. 2018, 78, 6320–6328. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, l1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic. Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [PubMed]
- The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [CrossRef] [Green Version]
- Glinsky, G.V.; Glinskii, A.B.; Stephenson, A.J.; Hoffman, R.M.; Gerald, W.L. Gene expression profiling predicts clinical outcome of prostate cancer. J. Clin. Investig. 2004, 113, 913–923. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Varambally, S.; Yu, J.; Laxman, B.; Rhodes, D.R.; Mehra, R.; Tomlins, S.A.; Shah, R.B.; Chandran, U.; Monzon, F.A.; Becich, M.J.; et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell 2005, 8, 393–406. [Google Scholar] [CrossRef] [Green Version]
- Gerhauser, C.; Favero, F.; Risch, T.; Simon, R.; Feuerbach, L.; Assenov, Y.; Heckmann, D.; Sidiropoulos, N.; Waszak, S.M.; Hübschmann, D.; et al. Molecular Evolution of Early-Onset Prostate Cancer Identifies Molecular Risk Markers and Clinical Trajectories. Cancer Cell 2018, 34, 996–1011. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef] [Green Version]
- Ren, S.; Wei, G.-H.; Liu, D.; Wang, L.; Hou, Y.; Zhu, S.; Peng, L.; Zhang, Q.; Cheng, Y.; Su, H.; et al. Whole-genome and Transcriptome Sequencing of Prostate Cancer Identify New Genetic Alterations Driving Disease Progression. Eur. Urol. 2018, 73, 322–339. [Google Scholar] [CrossRef]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [Green Version]
- Dan, R.; van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Coleman, I.; Morrissey, C.; Zhang, X.; True, L.D.; Gulati, R.; Etzioni, R.; Bolouri, H.; Montgomery, B.; White, T.; et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med. 2016, 22, 369–378. [Google Scholar] [CrossRef]
- Weissgerber, T.L.; Winham, S.J.; Heinzen, E.P.; Milin-Lazovic, J.S.; Garcia-Valencia, O.; Bukumiric, Z.; Savic, M.D.; Garovic, V.D.; Milic, N.M. Reveal, Don’t Conceal: Transforming Data Visualization to Improve Transparency. Circulation 2019, 140, 1506–1518. [Google Scholar] [CrossRef]
- BMJ. Correlation and Regression. Available online: https://www.bmj.com/about-bmj/resources-readers/publications/statistics-square-one/11-correlation-and-regression (accessed on 23 April 2021).
- Luszczak, S.; Kumar, C.; Sathyadevan, V.K.; Simpson, B.S.; Gately, K.A.; Whitaker, H.C.; Heavey, S. PIM kinase inhibition: Co-targeted therapeutic approaches in prostate cancer. Signal Transduct. Target. Ther. 2020, 5, 7. [Google Scholar] [CrossRef]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef]
- Guo, J.; Yang, J.; Zhang, X.; Feng, X.; Zhang, H.; Chen, L.; Johnson, H.; Persson, J.L.; Xiao, K. A Panel of Biomarkers for Diagnosis of Prostate Cancer Using Urine Samples. Anticancer Res. 2018, 38, 1471–1477. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Huang, J.; Zhang, C.; Zhao, F.; Kim, W.; Tu, X.; Zhang, Y.; Nowsheen, S.; Zhu, Q.; Deng, M.; et al. The bromodomain containing protein BRD-9 orchestrates RAD51–RAD54 complex formation and regulates homologous recombination-mediated repair. Nat. Commun. 2020, 11, 2639. [Google Scholar] [CrossRef]
- Ghallab, A. In vitro test systems and their limitations. Excli J. 2013, 12, 1024–1026. [Google Scholar]
- Geraghty, R.J.; Capes-Davis, A.; Davis, J.M.; Downward, J.; Freshney, R.I.; Knezevic, I.; Lovell-Badge, R.; Masters, J.R.W.; Meredith, J.; Stacey, G.N.; et al. Guidelines for the use of cell lines in biomedical research. Br. J. Cancer 2014, 111, 1021–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olarerin-George, A.O.; Hogenesch, J.B. Assessing the prevalence of mycoplasma contamination in cell culture via a survey of NCBI’s RNA-seq archive. Nucleic. Acids Res. 2015, 43, 2535–2542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Formaggio, N.; Rubin, M.A.; Theurillat, J.-P. Loss and revival of androgen receptor signaling in advanced prostate cancer. Oncogene 2021, 40, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Bluemn, E.G.; Coleman, I.M.; Lucas, J.M.; Coleman, R.T.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.F.; Kaipainen, A.; Corella, A.N.; et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 2017, 32, 474–489.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzger, A.L.; Abel, S.; Wegner, R.E.; Fuhrer, R.; Mao, S.; Miller, R.; Beriwal, S.; Horne, Z.D. Patterns of care and outcomes in small cell carcinoma of the prostate: A national cancer database analysis. Prostate 2019, 79, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, S.; Troisi, E.C.; Howard, T.P.; Haswell, J.R.; Wolf, B.K.; Hawk, W.H.; Ramos, P.; Oberlick, E.M.; Tzvetkov, E.P.; et al. BRD9 defines a SWI/SNF sub-complex and constitutes a specific vulnerability in malignant rhabdoid tumors. Nat. Commun. 2019, 10, 1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, C.M.; Raffeiner, P.; Hart, J.R.; Vogt, P.K. PIK3CA Cooperates with KRAS to Promote MYC Activity and Tumorigenesis via the Bromodomain Protein BRD9. Cancers 2019, 11, 1634. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Wang, Y.; Li, Q.; Fei, X.; Ma, H.; Hu, R. miR-140-3p functions as a tumor suppressor in squamous cell lung cancer by regulating BRD9. Cancer Lett. 2019, 446, 81–89. [Google Scholar] [CrossRef]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef]
- Sandoval, G.J.; Pulice, J.L.; Pakula, H.; Schenone, M.; Takeda, D.Y.; Pop, M.; Boulay, G.; Williamson, K.E.; McBride, M.J.; Pan, J.; et al. Binding of TMPRSS2-ERG to BAF Chromatin Remodeling Complexes Mediates Prostate Oncogenesis. Mol. Cell 2018, 71, 554–566.e7. [Google Scholar] [CrossRef] [Green Version]
- Miah, S.; Ahmed, H.U.; Freeman, A.; Emberton, M. Does true Gleason pattern 3 merit its cancer descriptor? Nat. Rev. Urol. 2016, 13, 541–548. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TCGA [43] | Glinsky [44] | Grasso [45] | Taylor [46] | Varambally [47] | Gerhauser [48] | Barbieri [49] | Ren [50] | Abida [51] | Dan [52] | Kumar [53] | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cancertool name | TCGA | Glinsky | Grasso | Taylor | Varambally | N/A | N/A | N/A | N/A | N/A | N/A |
| cBioPortal name | TCGA Firehose Legacy | N/A | Metastatic Prostate Adenocarcinoma (MCTP, Nature 2012) | Prostate Adenocarcinoma (MSKCC, Cancer Cell 2010) | N/A | Prostate Cancer (DKFZ, Cancer Cell 2018) | Prostate Adenocarcinoma (Broad/Cornell, Nat Genet 2012) | Prostate Adenocarcinoma (SMMU, Eur Urol 2017) | Metastatic Prostate Adenocarcinoma (SU2C/PCF Dream Team, PNAS 2019) | Metastatic Prostate Cancer SU2C/PCF Dream Team, Cell 2015) | Prostate Adenocarcinoma (Fred Hutchinson CRC, Nat Med 2016) |
| Number of patients (N) | 499 | 79 | 61 | 218 | 13 | 251 | 112 | 65 | 429 | 150 | 176 |
| Cancer type | Primary prostate adenocarcinoma | Recurrent (n = 37) and nonrecurrent (n = 42) disease | Heavily penetrated CRPC (n = 50), high grade localised PCa (n = 11) | Primary tumours (n = 81), metastatic disease (n = 37) | Primary tumours (n = 7), metastatic disease (n = 6) | Primary prostate adenocarcinoma | Prostate adenocarcinoma | Prostate adenocarcinoma | Metastatic CRPC | Metastatic CRPC | Primary and metastatic—mCRPC (n = 63) |
| Treatment (prior to specimen collection) | Naïve | Undergoing routine treatment | Prostatectomy, chemotherapy, hormone therapy, radiotherapy, palliative radiotherapy. Localised cancers were treatment naïve. | No information | No information | Treatment naïve (except for two patients receiving neoadjuvant hormone therapy) | Treatment-naïve | Treatment-naïve | Standard of care (including second generation antiandrogens)/enrolled in a clinical trial (e.g., targeted therapy) | Various—second generation antiandrogens, clinical trial, taxane chemotherapy | ADT followed by second-generation antiandrogens (following disease progression) and docetaxel chemotherapy |
| Method of sample collection | Radical prostatectomy | During therapeutic/diagnostic procedures | Rapid autopsy (CRPC) and radical prostatectomy (localised) | Radical prostatectomy | Radical prostatectomy and rapid autopsy | Radical prostatectomy | Radical prostatectomy | Radical prostatectomy | Radiographic-guided biopsy | Radiographic-guided biopsy | Rapid autopsy |
| Gleason grade | 5: - | 5: - | 5: - | 5: 2 (1%) | - | 5: - | 5: - | 5: - | 5: - | - | - |
| 6: 45 (9%) | 6: 15 (19%) | 6: - | 6: 101 (47%) | - | 6: 13 (11%) | 6: 4 (20%) | 6: 5 (8%) | 6: 15 (9%) | - | - | |
| 7: 244 (50%) | 7: 45 (57%) | 7: 2 (18%) | 7: 77 (36%) | - | 7: 86 (74%) | 7: 13 (65%) 7.5: 1 (5%) | 7: 37 (57%) | 7: 49 (29%) | - | - | |
| 8: 63 (13%) | 8: 10 (13%) | 8: 4 (36%) | 8: 19 (9%) | - | 8: 1 (1%) | 8: 2 (10%) | 8: 9 (14%) | 8: 23 (14%) | - | - | |
| 9: 135 (27%) | 9: 9 (11%) | 9: 5 (46%) | 9: 15 (7%) | - | 9: 15 (13%) | 9: - | 9: 12 (18%) | 9: 67 (40%) | - | - | |
| 10: 4 (1%) | 10: - | 10: - | 10: - | - | 10: 1 (1% | 10: - | 10: 2 (3%) | 10: 13 (8%) | - | - | |
| 11: - | 11: - | 11: - | 11: - | - | 11: - | 11: - | 11: - | 11: 1 (1%) | - | - | |
| Total patients (n) for whom a Gleason grade was available | 491 (100%) | 79 (100%) | 11 (100%) (High grade localised PCas only) | 214 (100%) | No data available | 116 (100%) | 20 (100%) | 65 (100%) | 168 (101%) | No data available | No data available |
| Spearman’s R (Positive) | Spearman’s R (Negative) | |
|---|---|---|
| Very weak | 0–0.19 | 0–−0.19 |
| Weak | 0.2–0.39 | −0.2–−0.39 |
| Moderate | 0.4–0.59 | −0.4–−0.59 |
| Strong | 0.6–0.79 | −0.6–−0.79 |
| Very strong | 0.8–1 | −0.8–−1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barma, N.; Stone, T.C.; Carmona Echeverria, L.M.; Heavey, S. Exploring the Value of BRD9 as a Biomarker, Therapeutic Target and Co-Target in Prostate Cancer. Biomolecules 2021, 11, 1794. https://doi.org/10.3390/biom11121794
Barma N, Stone TC, Carmona Echeverria LM, Heavey S. Exploring the Value of BRD9 as a Biomarker, Therapeutic Target and Co-Target in Prostate Cancer. Biomolecules. 2021; 11(12):1794. https://doi.org/10.3390/biom11121794
Chicago/Turabian StyleBarma, Nafisa, Timothy C. Stone, Lina Maria Carmona Echeverria, and Susan Heavey. 2021. "Exploring the Value of BRD9 as a Biomarker, Therapeutic Target and Co-Target in Prostate Cancer" Biomolecules 11, no. 12: 1794. https://doi.org/10.3390/biom11121794
APA StyleBarma, N., Stone, T. C., Carmona Echeverria, L. M., & Heavey, S. (2021). Exploring the Value of BRD9 as a Biomarker, Therapeutic Target and Co-Target in Prostate Cancer. Biomolecules, 11(12), 1794. https://doi.org/10.3390/biom11121794