
β-Synuclein: An Enigmatic Protein with Diverse Functionality


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Intrinsically Disordered Proteins
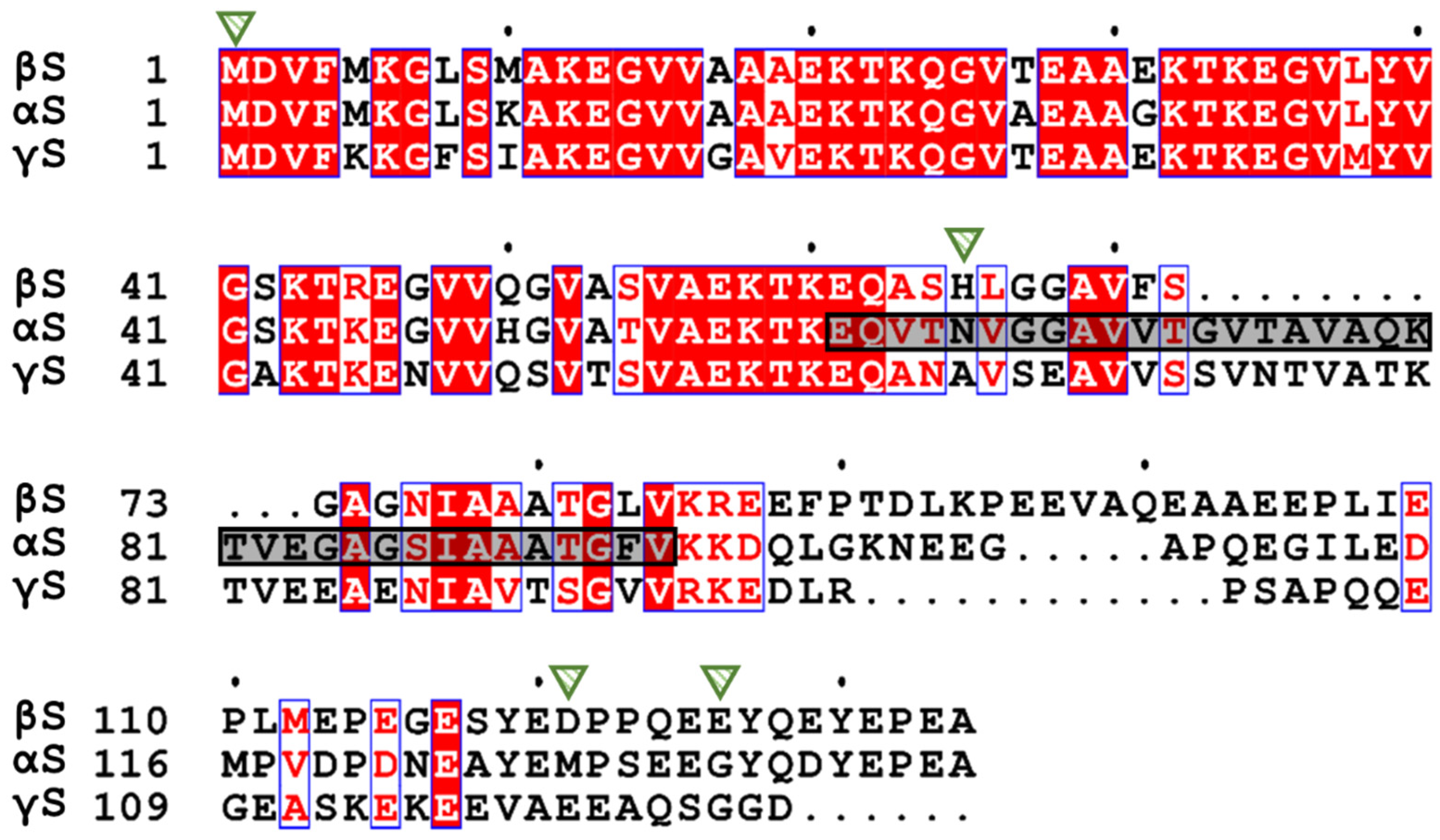
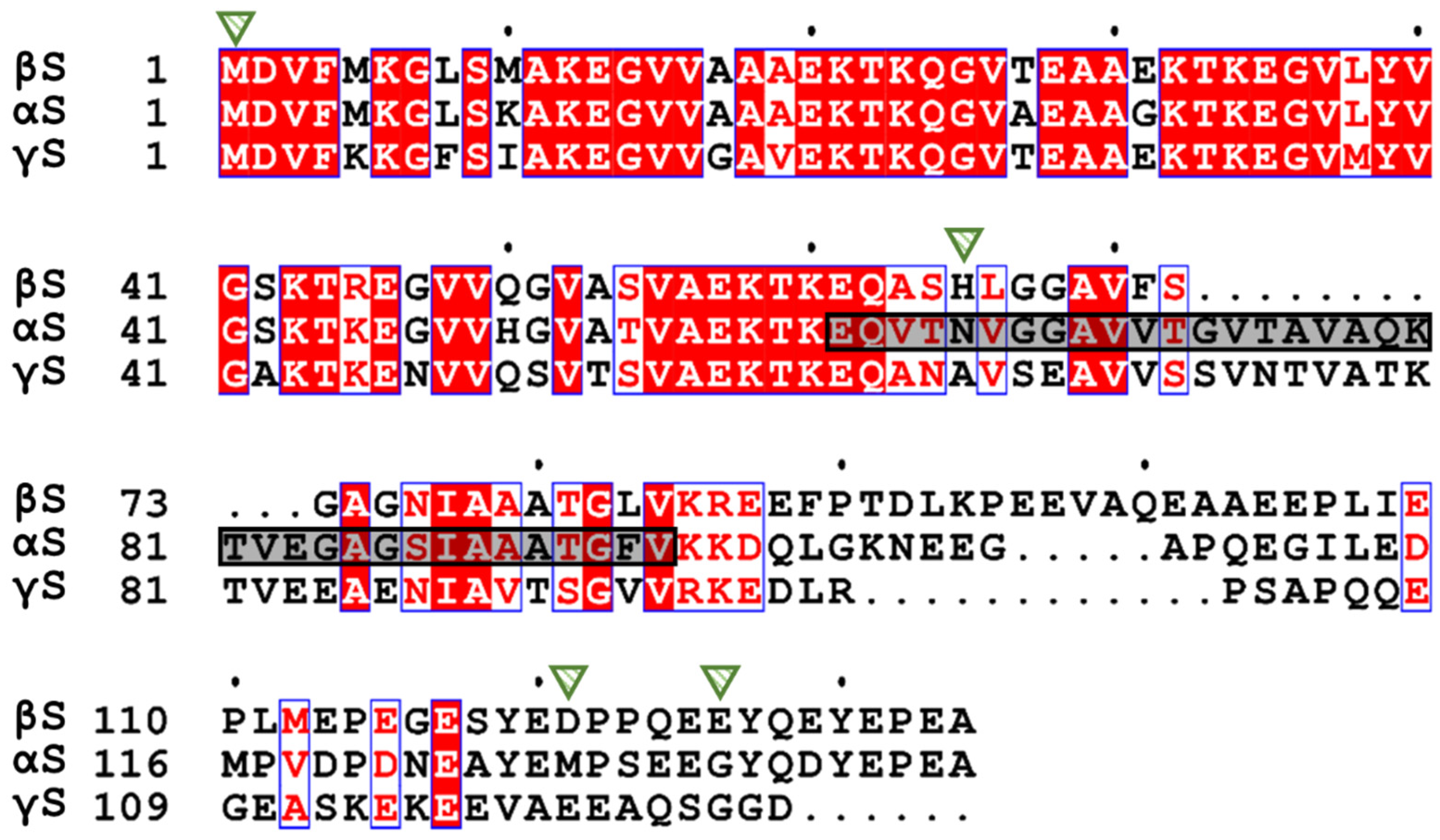
2. The Synuclein Family: α-, β-, and γ-Synuclein
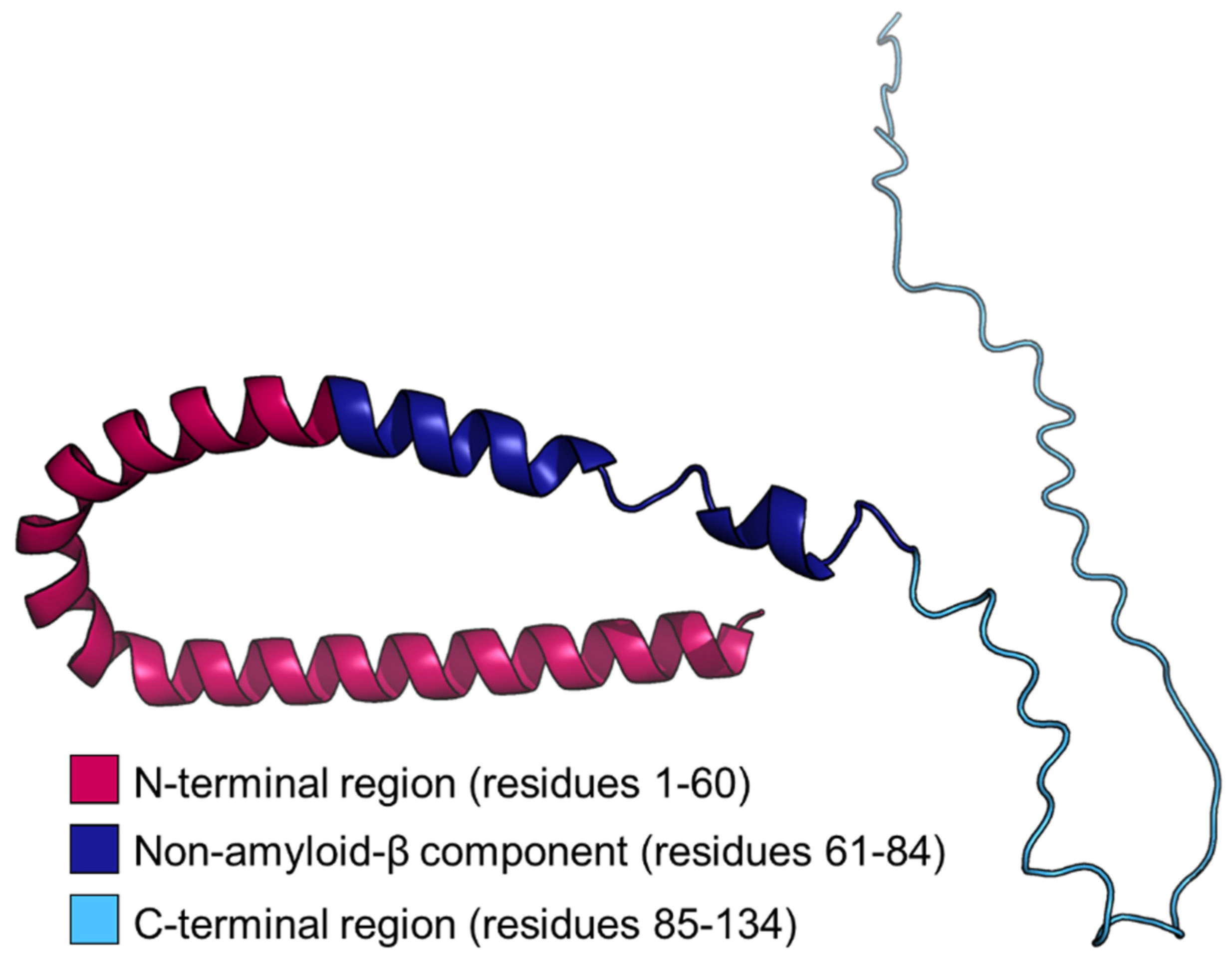
3. The Structure of β-Synuclein
4. The Function of β-Synuclein
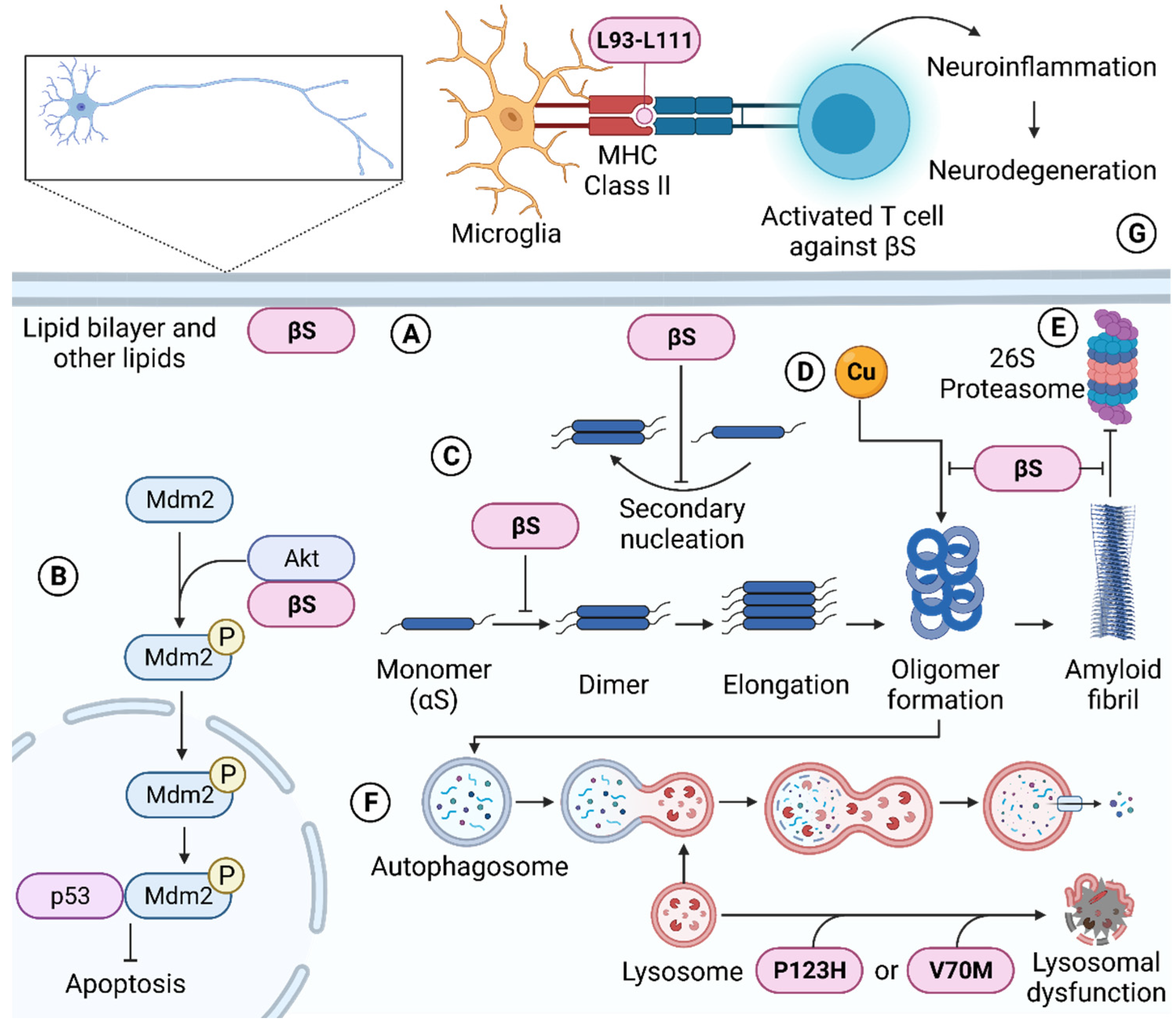
4.1. Molecular Chaperone Ability of β-Synuclein
4.2. β-Synuclein Regulates Synaptic Function, Lipid Binding, and Dopamine Neurotransmission
4.2.1. Structural Changes to β-Synuclein upon Lipid Binding
4.2.2. Structural Changes to the Lipid upon Binding of β-Synuclein
4.2.3. β-Synuclein Regulates the Nigrostriatal Dopaminergic System
4.3. β-Synuclein Regulates Cellular Metal Levels
4.4. β-Synuclein Regulates Apoptosis
4.5. β-Synuclein Regulates Protein Degradation Pathways
4.6. β-Synuclein Promotes Cellular Toxicity and Protein Aggregation
4.6.1. Changes in β-Synuclein Expression in Pathology
4.6.2. β-Synuclein Can Induce Neurotoxicity
5. Pathological Mutations of β-Synuclein
6. Post-Translational Modifications of β-Synuclein
7. Future Directions and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2014, 16, 18–29. [Google Scholar] [CrossRef]
- Deiana, A.; Forcelloni, S.; Porrello, A.; Giansanti, A. Intrinsically disordered proteins and structured proteins with intrinsically disordered regions have different functional roles in the cell. PLoS ONE 2019, 14, e0217889. [Google Scholar] [CrossRef] [Green Version]
- Berlow, R.B.; Dyson, H.J.; Wright, P.E. Functional advantages of dynamic protein disorder. FEBS Lett. 2015, 589, 2433–2440. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, A.; Brookes, D.H.; Yost, S.R.; Dyson, H.J.; Forman-Kay, J.D.; Gunter, D.; Head-Gordon, M.; Hura, G.L.; Pande, V.S.; Wemmer, D.E.; et al. Finding our way in the dark proteome. J. Am. Chem. Soc. 2016, 138, 9730–9742. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the D2 concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef]
- Uyar, B.; Weatheritt, R.J.; Dinkel, H.; Davey, N.E.; Gibson, T.J. Proteome-wide analysis of human disease mutations in short linear motifs: Neglected players in cancer? Mol. Biosyst. 2014, 10, 2626–2642. [Google Scholar] [CrossRef] [Green Version]
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [CrossRef] [Green Version]
- Galvin, J.E.; Schuck, T.M.; Lee, V.M.Y.; Trojanowski, J.Q. Differential expression and distribution of α-, β-, and γ-synuclein in the developing human substantia nigra. Exp. Neurol. 2001, 168, 347–355. [Google Scholar] [CrossRef]
- Connor-Robson, N.; Peters, O.P.; Millership, S.; Ninkina, N.; Buchman, V.L. Combinational losses of synucleins reveal their differential requirements for compensating age-dependent alterations in motor behavior and dopamine metabolism. Neurobiol. Aging 2016, 46, 107–112. [Google Scholar] [CrossRef] [Green Version]
- Chandra, S.; Fornai, F.; Kwon, H.B.; Yazdani, U.; Atasoy, D.; Liu, X.; Hammer, R.E.; Battaglia, G.; German, D.C.; Castillo, P.E.; et al. Double-knockout mice for α- and β-Synucleins: Effect on synaptic functions. Proc. Natl. Acad. Sci. USA 2004, 101, 14966–14971. [Google Scholar] [CrossRef] [Green Version]
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.L.; Etherton, M.R.; Sudhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.; Higgins, D.; Gibson, T. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [Green Version]
- Duda, J.E.; Shah, U.; Arnold, S.E.; Lee, V.M.Y.; Trojanowski, J.Q. The expression of α-, β-, and γ-synucleins in olfactory mucosa from patients with and without neurodegenerative diseases. Exp. Neurol. 1999, 160, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Jakes, R.; Spillantini, M.G.; Goedert, M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994, 345, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Tanji, K.; Mori, F.; Nakajo, S.; Imaizumi, T.; Yoshida, H.; Hirabayashi, T.; Yoshimoto, M.; Satoh, K.; Takahashi, H.; Wakabayashi, K. Expression of β-Synuclein in normal human astrocytes. Neuroreport 2001, 12, 2845–2848. [Google Scholar] [CrossRef]
- Mor, F.; Quintana, F.; Mimran, A.; Cohen, I.R. Autoimmune encephalomyelitis and uveitis induced by T Cell immunity to self β-Synuclein. J. Immunol. 2003, 170, 628–634. [Google Scholar] [CrossRef] [Green Version]
- Oort, P.J.; Knotts, T.A.; Grino, M.; Naour, N.; Bastard, J.-P.; Clément, K.; Ninkina, N.; Buchman, V.L.; Permana, P.A.; Luo, X.; et al. γ-Synuclein is an adipocyte-neuron gene coordinately expressed with leptin and increased in human obesity. J. Nutr. 2008, 138, 841–848. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, M.; Attoub, S.; Singh, M.N.; Martin, F.L.; El-Agnaf, O.M.A. γ-Synuclein and the progression of cancer. FASEB J. 2007, 21, 3419–3430. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Souillac, P.; Millett, I.S.; Doniach, S.; Jakes, R.; Goedert, M.; Fink, A.L. Biophysical properties of the synucleins and their propensities to fibrillate: Inhibition of α-synuclein assembly by β- and γ-synucleins. J. Biol. Chem. 2002, 277, 11970–11978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertoncini, C.W.; Rasia, R.M.; Lamberto, G.R.; Binolfi, A.; Zweckstetter, M.; Griesinger, C.; Fernandez, C.O. Structural characterization of the intrinsically unfolded protein β-Synuclein, a natural negative regulator of α-synuclein aggregation. J. Mol. Biol. 2007, 372, 708–722. [Google Scholar] [CrossRef]
- Jain, M.K.; Singh, P.; Roy, S.; Bhat, R. Comparative analysis of the conformation, aggregation, interaction, and fibril morphologies of human α-, β-, and γ-synuclein proteins. Biochemistry 2018, 57, 3830–3848. [Google Scholar] [CrossRef] [PubMed]
- Syme, C.D.; Blanch, E.W.; Holt, C.; Jakes, R.; Goedert, M.; Hecht, L.; Barron, L.D. A Raman optical activity study of rheomorphism in caseins, synucleins and tau: New insight into the structure and behaviour of natively unfolded proteins. Eur. J. Biochem. 2002, 269, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, U.; Newman, A.J.; Luth, E.S.; Bartels, T.; Selkoe, D. In vivo cross-linking reveals principally oligomeric forms of α-synuclein and β-Synuclein in neurons and non-neural cells. J. Biol. Chem. 2013, 288, 6371–6385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.W.P.; Buell, A.K.; Michaels, T.C.T.; Meisl, G.; Carozza, J.; Flagmeier, P.; Vendruscolo, M.; Knowles, T.P.J.; Dobson, C.M.; Galvagnion, C. β-Synuclein suppresses both the initiation and amplification steps of α-synuclein aggregation via competitive binding to surfaces. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Bar-On, P.; Ho, G.; Takenouchi, T.; Rockenstein, E.; Crews, L.; Masliah, E. β-Synuclein regulates Akt activity in neuronal cells: A possible mechanism for neuroprotection in Parkinson’s disease. J. Biol. Chem. 2004, 279, 23622–23629. [Google Scholar] [CrossRef] [Green Version]
- Janowska, M.K.; Wu, K.P.; Baum, J. Unveiling transient protein-protein interactions that modulate inhibition of alpha-synuclein aggregation by beta-synuclein, a pre-synaptic protein that co-localizes with alpha-synuclein. Sci. Rep. 2015, 5, 1–10. [Google Scholar] [CrossRef]
- Van de Vondel, E.; Baatsen, P.; Van Elzen, R.; Lambeir, A.-M.; Keiderling, T.A.; Herrebout, W.A.; Johannessen, C. Vibrational circular dichroism sheds new light on the competitive effects of crowding and β-Synuclein on the fibrillation process of α-synuclein. Biochemistry 2018, 57, 5989–5995. [Google Scholar] [CrossRef]
- Tsigelny, I.F.; Bar-On, P.; Sharikov, Y.; Crews, L.; Hashimoto, M.; Miller, M.A.; Keller, S.H.; Platoshyn, O.; Yuan, J.X.J.; Masliah, E. Dynamics of α-synuclein aggregation and inhibition of pore-like oligomer development by β-Synuclein. FEBS J. 2007, 274, 1862–1877. [Google Scholar] [CrossRef] [PubMed]
- Leitao, A.; Bhumkar, A.; Hunter, D.; Gambin, Y.; Sierecki, E. Unveiling a selective mechanism for the inhibition of α-synuclein aggregation by β-Synuclein. Int. J. Mol. Sci. 2018, 19, 334. [Google Scholar] [CrossRef] [Green Version]
- Treweek, T.M.; Meehan, S.; Ecroyd, H.; Carver, J.A. Small heat-shock proteins: Important players in regulating cellular proteostasis. Cell. Mol. Life Sci. 2015, 72, 429–451. [Google Scholar] [CrossRef]
- Hayashi, J.; Carver, J.A. The multifaceted nature of αB-crystallin. Cell Stress Chaperones 2020, 25, 639–654. [Google Scholar] [CrossRef]
- Doherty, C.P.A.; Ulamec, S.M.; Maya-Martinez, R.; Good, S.C.; Makepeace, J.; Khan, G.N.; van Oosten-Hawle, P.; Radford, S.E.; Brockwell, D.J. A short motif in the N-terminal region of α-synuclein is critical for both aggregation and function. Nat. Struct. Mol. Biol. 2020, 27, 249–259. [Google Scholar] [CrossRef]
- Angelova, D.M.; Jones, H.B.L.; Brown, D.R. Levels of α- and β-Synuclein regulate cellular susceptibility to toxicity from α-synuclein oligomers. FASEB J. 2018, 32, 995–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, G.; Morse, S.; Ararat, M.; Graham, F.L. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. 2002, 16, 869–871. [Google Scholar] [CrossRef]
- Fan, Y.; Limprasert, P.; Murray, I.V.J.; Smith, A.C.; Lee, V.M.Y.; Trojanowski, J.Q.; Sopher, B.L.; La Spada, A.R. β-Synuclein modulates α-synuclein neurotoxicity by reducing α-synuclein protein expression. Hum. Mol. Genet. 2006, 15, 3002–3011. [Google Scholar] [CrossRef]
- McNaught, K.S.P.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2001, 297, 191–194. [Google Scholar] [CrossRef]
- Snyder, H.; Mensah, K.; Hsu, C.; Hashimoto, M.; Surgucheva, I.G.; Festoff, B.; Surguchov, A.; Masliah, E.; Matouschek, A.; Wolozin, B. β-Synuclein reduces proteasomal inhibition by α-synuclein but not γ-synuclein. J. Biol. Chem. 2005, 280, 7562–7569. [Google Scholar] [CrossRef] [Green Version]
- Windisch, M.; Hutter-Paier, B.; Rockenstein, E.; Hashimoto, M.; Mallory, M.; Masliah, E. Development of a new treatment for Alzheimer’s disease and Parkinson’s disease using anti-aggregatory β-Synuclein-derived peptides. J. Mol. Neurosci. 2002, 19, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Rockenstein, E.; Mante, M.; Mallory, M.; Masliah, E. β-Synuclein inhibits α-synuclein aggregation: A possible role as an anti-Parkinsonian factor. Neuron 2001, 32, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, M.; Rockenstein, E.; Mante, M.; Crews, L.; Bar-On, P.; Gage, F.H.; Marr, R.; Masliah, E. An antiaggregation gene therapy strategy for Lewy body disease utilizing β-Synuclein lentivirus in a transgenic model. Gene Ther. 2004, 11, 1713–1723. [Google Scholar] [CrossRef]
- Shaltiel-Karyo, R.; Frenkel-Pinter, M.; Egoz-Matia, N.; Frydman-Marom, A.; Shalev, D.E.; Segal, D.; Gazit, E. Inhibiting α-synuclein oligomerization by stable cell-penetrating β-Synuclein fragments recovers phenotype of Parkinson’s disease model flies. PLoS ONE 2010, 5, e13863. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.; Paik, S.R.; Choi, K.Y. β-Synuclein exhibits chaperone activity more efficiently than α-synuclein. FEBS Lett. 2004, 576, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Jensen, P.H.; Højrup, P.; Hager, H.; Nielsen, M.S.; Jacobsen, L.; Olesen, O.F.; Gliemann, J.; Jakes, R. Binding of Aβ to α- and β-Synucleins: Identification of segments in α-synuclein/NAC precursor that bind Aβ and NAC. Biochem. J. 1997, 323, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Rekas, A.; Adda, C.G.; Andrew Aquilina, J.; Barnham, K.J.; Sunde, M.; Galatis, D.; Williamson, N.A.; Masters, C.L.; Anders, R.F.; Robinson, C.V.; et al. Interaction of the molecular chaperone αB-crystallin with α-synuclein: Effects on amyloid fibril formation and chaperone activity. J. Mol. Biol. 2004, 340, 1167–1183. [Google Scholar] [CrossRef]
- Sharon, R.; Goldberg, M.S.; Bar-Josef, I.; Betensky, R.A.; Shen, J.; Selkoe, D.J. α-synuclein occurs in lipid-rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid-binding proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 9110–9115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westphal, C.H.; Chandra, S.S. Monomeric synucleins generate membrane curvature. J. Biol. Chem. 2013, 288, 1829–1840. [Google Scholar] [CrossRef] [Green Version]
- Sharon, R.; Bar-Joseph, I.; Mirick, G.E.; Serhan, C.N.; Selkoe, D.J. Altered fatty acid composition of dopaminergic neurons expressing α-synuclein and human brains with α-synucleinopathies. J. Biol. Chem. 2003, 278, 49874–49881. [Google Scholar] [CrossRef] [Green Version]
- Gai, W.P.; Yuan, H.X.; Li, X.Q.; Power, J.T.H.; Blumbergs, P.C.; Jensen, P.H. In situ and in vitro study of colocalization and segregation of α-synuclein, ubiquitin, and lipids in Lewy bodies. Exp. Neurol. 2000, 166, 324–333. [Google Scholar] [CrossRef]
- Israeli, E.; Sharon, R. β-Synuclein occurs in vivo in lipid-associated oligomers and forms hetero-oligomers with α-synuclein. J. Neurochem. 2009, 108, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Salem, S.A.; Allsop, D.; Mann, D.M.A.; Tokuda, T.; El-Agnaf, O.M.A. An investigation into the lipid-binding properties of α-, β- and γ-synucleins in human brain and cerebrospinal fluid. Brain Res. 2007, 1170, 103–111. [Google Scholar] [CrossRef]
- Ducas, V.C.; Rhoades, E. Quantifying interactions of β-Synuclein and γ-synuclein with model membranes. J. Mol. Biol. 2012, 423, 528–539. [Google Scholar] [CrossRef] [Green Version]
- Carnazza, K.E.; Komer, L.; Pineda, A.; Na, Y.; Ramlall, T.; Buchman, V.L.; Eliezer, D.; Sharma, M.; Burre, J. Beta- and gamma-synucleins modulate synaptic vesicle-binding of alpha-synuclein. bioRxiv 2020. [Google Scholar] [CrossRef]
- Rivers, R.C.; Kumita, J.R.; Tartaglia, G.G.; Dedmon, M.M.; Pawar, A.; Vendruscolo, M.; Dobson, C.M.; Christodoulou, J. Molecular determinants of the aggregation behavior of α- and β-Synuclein. Protein Sci. 2008, 17, 887–898. [Google Scholar] [CrossRef]
- Sung, Y.; Eliezer, D. Secondary structure and dynamics of micelle bound β- and γ-synuclein. Protein Sci. 2006, 15, 1162–1174. [Google Scholar] [CrossRef] [Green Version]
- Varkey, J.; Isas, J.M.; Mizuno, N.; Jensen, M.B.; Bhatia, V.K.; Jao, C.C.; Petrlova, J.; Voss, J.C.; Stamou, D.G.; Steven, A.C.; et al. Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins. J. Biol. Chem. 2010, 285, 32486–32493. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Park, J.Y.; Lansbury, P.T. β-Synuclein inhibits formation of α-synuclein protofibrils: A possible therapeutic strategy against Parkinson’s disease. Biochemistry 2003, 42, 3696–3700. [Google Scholar] [CrossRef]
- Hayashi, J.; Ton, J.; Negi, S.; Stephens, D.E.; Pountney, D.L.; Preiss, T.; Carver, J.A. The effect of oxidized dopamine on the structure and molecular chaperone function of the small heat-shock proteins, αB-crystallin and Hsp27. Int. J. Mol. Sci. 2021, 22, 3700. [Google Scholar] [CrossRef]
- LaVoie, M.J.; Ostaszewski, B.L.; Weihofen, A.; Schlossmacher, M.G.; Selkoe, D.J. Dopamine covalently modifies and functionally inactivates parkin. Nat. Med. 2005, 11, 1214–1221. [Google Scholar] [CrossRef]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef] [Green Version]
- Ninkina, N.; Millership, S.J.; Peters, O.M.; Connor-Robson, N.; Chaprov, K.; Kopylov, A.T.; Montoya, A.; Kramer, H.; Withers, D.J.; Buchman, V.L. β-Synuclein potentiates synaptic vesicle dopamine uptake and rescues dopaminergic neurons from MPTP-induced death in the absence of other synucleins. J. Biol. Chem. 2021, 297, 101375. [Google Scholar] [CrossRef]
- De Ricco, R.; Valensin, D.; Dellacqua, S.; Casella, L.; Gaggelli, E.; Valensin, G.; Bubacco, L.; Mangani, S. Differences in the binding of copper(I) to α- and β-Synuclein. Inorg. Chem. 2015, 54, 265–272. [Google Scholar] [CrossRef]
- Pall, H.S.; Blake, D.R.; Gutteridge, J.M.; Williams, A.C.; Lunec, J.; Hall, M.; Taylor, A. Raised cerebrospinal-fluid copper concentration in Parkinson’s disease. Lancet 1987, 330, 238–241. [Google Scholar] [CrossRef]
- Gorell, J.M.; Johnson, C.C.; Rybicki, B.A.; Peterson, E.L.; Kortsha, G.X.; Brown, G.G.; Richardson, R.J. Occupational exposure to manganese, copper, lead, iron, mercury and zinc and the risk of Parkinson’s disease. Neurotoxicology 1999, 20, 239–248. [Google Scholar]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Wright, J.A.; Wang, X.; Brown, D.R. Unique copper-induced oligomers mediate alpha-synuclein toxicity. FASEB J. 2009, 23, 2384–2393. [Google Scholar] [CrossRef]
- Binolfi, A.; Lamberto, G.R.; Duran, R.; Quintanar, L.; Bertoncini, C.W.; Souza, J.M.; Cerveñansky, C.; Zweckstetter, M.; Griesinger, C.; Fernández, C.O. Site-specific interactions of Cu(II) with α and β-Synuclein: Bridging the molecular gap between metal binding and aggregation. J. Am. Chem. Soc. 2008, 130, 11801–11812. [Google Scholar] [CrossRef]
- Davies, P.; Moualla, D.; Brown, D.R. Alpha-synuclein is a cellular ferrireductase. PLoS ONE 2011, 6, 15814. [Google Scholar] [CrossRef]
- Yamin, G.; Munishkina, L.A.; Karymov, M.A.; Lyubchenko, Y.L.; Uversky, V.N.; Fink, A.L. Forcing nonamyloidogenic β-Synuclein to fibrillate. Biochemistry 2005, 44, 9096–9107. [Google Scholar] [CrossRef]
- McHugh, P.C.; Wright, J.A.; Brown, D.R. Transcriptional regulation of the beta-synuclein 5′-promoter metal response element by metal transcription factor-1. PLoS ONE 2011, 6, e17354. [Google Scholar] [CrossRef] [Green Version]
- Da Costa, C.A.; Masliah, E.; Checler, F. β-Synuclein displays an antiapoptotic p53-dependent phenotype and protects neurons from 6-hydroxydopamine-induced caspase 3 activation: Cross-talk with α-synuclein and implication for Parkinson’s disease. J. Biol. Chem. 2003, 278, 37330–37335. [Google Scholar] [CrossRef] [Green Version]
- Brockhaus, K.; Böhm, M.R.R.R.; Melkonyan, H.; Thanos, S. Age-related beta-synuclein alters the p53/Mdm2 pathway and induces the apoptosis of brain microvascular endothelial cells in vitro. Cell Transplant. 2018, 27, 796–813. [Google Scholar] [CrossRef]
- Finkbeiner, S. The autophagy lysosomal pathway and neurodegeneration. Cold Spring Harb. Perspect. Biol. 2020, 12, a033993. [Google Scholar] [CrossRef]
- Evans, T.; Kok, W.L.; Cowan, K.; Hefford, M.; Anichtchik, O. Accumulation of beta-synuclein in cortical neurons is associated with autophagy attenuation in the brains of dementia with Lewy body patients. Brain Res. 2018, 1681, 1–13. [Google Scholar] [CrossRef]
- Popova, B.; Kleinknecht, A.; Arendarski, P.; Mischke, J.; Wang, D.; Braus, G.H. Sumoylation protects against β-Synuclein toxicity in yeast. Front. Mol. Neurosci. 2018, 11, 94. [Google Scholar] [CrossRef]
- Moriarty, G.M.; Olson, M.P.; Atieh, T.B.; Janowska, M.K.; Khare, S.D.; Baum, J. A pH-dependent switch promotes β-Synuclein fibril formation via glutamate residues. J. Biol. Chem. 2017, 292, 16368–16379. [Google Scholar] [CrossRef] [Green Version]
- Galvin, J.E.; Uryu, K.; Lee, V.M.-Y.; Trojanowski, J.Q.; Crowther, R.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. Axon pathology in Parkinson’s disease and Lewy body dementia hippocampus contains alpha -, beta -, and gamma -synuclein. Proc. Natl. Acad. Sci. USA 1999, 96, 13450–13455. [Google Scholar] [CrossRef] [Green Version]
- Oeckl, P.; Metzger, F.; Nagl, M.; Von Arnim, C.A.F.; Halbgebauer, S.; Steinacker, P.; Ludolph, A.C.; Otto, M. Alpha-, beta-, and gamma-synuclein quantification in cerebrospinal fluid by multiple reaction monitoring reveals increased concentrations in Alzheimer’s and Creutzfeldt-Jakob disease but no alteration in synucleinopathies. Mol. Cell. Proteom. 2016, 15, 3126–3138. [Google Scholar] [CrossRef] [Green Version]
- Mori, F.; Nishie, M.; Yoshimoto, M.; Takahashi, H.; Wakabayashi, K. Reciprocal accumulation of β-Synuclein in α-synuclein lesions in multiple system atrophy. Neuroreport 2003, 14, 1783–1786. [Google Scholar] [CrossRef]
- Castaño, E.M.; Maarouf, C.L.; Wu, T.; Leal, M.C.; Whiteside, C.M.; Lue, L.F.; Kokjohn, T.A.; Sabbagh, M.N.; Beach, T.G.; Roher, A.E. Alzheimer disease periventricular white matter lesions exhibit specific proteomic profile alterations. Neurochem. Int. 2013, 62, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Galvin, J.E.; Giasson, B.; Hurtig, H.I.; Lee, V.M.Y.; Trojanowski, J.Q. Neurodegeneration with brain iron accumulation, type 1 is characterized by α-, β-, and γ-synuclein neuropathology. Am. J. Pathol. 2000, 157, 361–368. [Google Scholar] [CrossRef]
- Sánchez, E.; Azcona, L.J.; Paisán-Ruiz, C. Pla2g6 deficiency in zebrafish leads to dopaminergic cell death, axonal degeneration, increased β-Synuclein expression, and defects in brain functions and pathways. Mol. Neurobiol. 2018, 55, 6734–6754. [Google Scholar] [CrossRef]
- Chabas, D.; Baranzini, S.E.; Mitchell, D.; Bernard, C.C.A.; Rittling, S.R.; Denhardt, D.T.; Sobel, R.A.; Lock, C.; Karpuj, M.; Pedotti, R.; et al. The influence of the proinflammatory cytokine, osteopontin, on autoimmue demyelinating desease. Science 2001, 294, 1731–1735. [Google Scholar] [CrossRef]
- Suzuki, K.; Iseki, E.; Katsuse, O.; Yamaguchi, A.; Katsuyama, K.; Aoki, I.; Yamanaka, S.; Kosaka, K. Neuronal accumulation of α- and β-Synucleins in the brain of a GM2 gangliosidosis mouse model. Neuroreport 2003, 14, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Iseki, E.; Togo, T.; Yamaguchi, A.; Katsuse, O.; Katsuyama, K.; Kanzaki, S.; Shiozaki, K.; Kawanishi, C.; Yamashita, S.; et al. Neuronal and glial accumulation of α- and β-Synucleins in human lipidoses. Acta Neuropathol. 2007, 114, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Sriwimol, W.; Limprasert, P. Significant changes in plasma alpha-synuclein and beta-synuclein levels in male children with autism spectrum disorder. Biomed Res. Int. 2018, 2018, 4503871. [Google Scholar] [CrossRef] [Green Version]
- Fung, K.M.; Rorke, L.B.; Giasson, B.; Lee, V.M.Y.; Trojanowski, J.Q. Expression of α-, β-, and γ-synuclein in glial tumor and medulloblastomas. Acta Neuropathol. 2003, 106, 167–175. [Google Scholar] [CrossRef]
- Rockenstein, E.; Hansen, L.A.; Mallory, M.; Trojanowski, J.Q.; Galasko, D.; Masliah, E. Altered expression of the synuclein family mRNA in Lewy body and Alzheimer’s disease. Brain Res. 2001, 914, 48–56. [Google Scholar] [CrossRef]
- Noori-Daloii, M.R.; Kheirollahi, M.; Mahbod, P.; Mohammadi, F.; Astaneh, A.N.; Zarindast, M.R.; Azimi, C.; Mohammadi, M.R. Alpha-and beta-synucleins mRNA expression in lymphocytes of schizophrenia patients. Genet. Test. Mol. Biomarkers 2010, 14, 725–729. [Google Scholar] [CrossRef]
- Brighina, L.; Okubadejo, N.U.; Schneider, N.K.; Lesnick, T.G.; de Andrade, M.; Cunningham, J.M.; Farrer, M.J.; Lincoln, S.J.; Rocca, W.A.; Maraganore, D.M. Beta-synuclein gene variants and Parkinson’s disease: A preliminary case-control study. Neurosci. Lett. 2007, 420, 229–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishioka, K.; Wider, C.; Vilariño-Güell, C.; Soto-Ortolaza, A.I.; Lincoln, S.J.; Kachergus, J.M.; Jasinska-Myga, B.; Ross, O.A.; Rajput, A.; Robinson, C.A.; et al. Association of α-, β-, and γ-synuclein with diffuse lewy body disease. Arch. Neurol. 2010, 67, 970–975. [Google Scholar] [CrossRef] [Green Version]
- Taschenberger, G.; Toloe, J.; Tereshchenko, J.; Akerboom, J.; Wales, P.; Benz, R.; Becker, S.; Outeiro, T.F.; Looger, L.L.; Bähr, M.; et al. β-Synuclein aggregates and induces neurodegeneration in dopaminergic neurons. Ann. Neurol. 2013, 74, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Tenreiro, S.; Rosado-Ramos, R.; Gerhardt, E.; Favretto, F.; Magalhães, F.; Popova, B.; Becker, S.; Zweckstetter, M.; Braus, G.H.; Outeiro, T.F. Yeast reveals similar molecular mechanisms underlying alpha- and beta-synuclein toxicity. Hum. Mol. Genet. 2016, 25, 275–290. [Google Scholar] [CrossRef]
- Kela-Madar, N.; de Rosbo, N.K.; Ronen, A.; Mor, F.; Ben-Nun, A. Autoimmune spread to myelin is associated with experimental autoimmune encephalomyelitis induced by a neuronal protein, β-Synuclein. J. Neuroimmunol. 2009, 208, 19–29. [Google Scholar] [CrossRef]
- Lodygin, D.; Hermann, M.; Schweingruber, N.; Flügel-Koch, C.; Watanabe, T.; Schlosser, C.; Merlini, A.; Körner, H.; Chang, H.F.; Fischer, H.J.; et al. β-Synuclein-reactive T cells induce autoimmune CNS grey matter degeneration. Nature 2019, 566, 503–508. [Google Scholar] [CrossRef]
- Ohtake, H.; Limprasert, P.; Fan, Y.; Onodera, O.; Kakita, A.; Takahashi, H.; Bonner, L.T.; Tsuang, D.W.; Murray, I.V.J.; Lee, V.M.Y.; et al. β-Synuclein gene alterations in dementia with Lewy bodies. Neurology 2004, 63, 805–811. [Google Scholar] [CrossRef]
- Fujita, M.; Sugama, S.; Sekiyama, K.; Sekigawa, A.; Tsukui, T.; Nakai, M.; Waragai, M.; Takenouchi, T.; Takamatsu, Y.; Wei, J.; et al. A β-Synuclein mutation linked to dementia produces neurodegeneration when expressed in mouse brain. Nat. Commun. 2010, 1, 110. [Google Scholar] [CrossRef]
- Hagihara, H.; Fujita, M.; Umemori, J.; Hashimoto, M.; Miyakawa, T. Immature-like molecular expression patterns in the hippocampus of a mouse model of dementia with Lewy body-linked mutant β-Synuclein. Mol. Brain 2018, 11, 38. [Google Scholar] [CrossRef] [PubMed]
- Janowska, M.K.; Baum, J. The loss of inhibitory C-terminal conformations in disease associated P123H β-Synuclein. Protein Sci. 2016, 25, 286–294. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Fujita, M.; Nakai, M.; Waragai, M.; Watabe, K.; Akatsu, H.; Rockenstein, E.; Masliah, E.; Hashimoto, M. Enhanced lysosomal pathology caused by β-Synuclein mutants linked to dementia with Lewy bodies. J. Biol. Chem. 2007, 282, 28904–28914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, R.N.; Hart, G.W. Cytosolic O-glycosylation is abundant in nerve terminals. J. Neurochem. 2001, 79, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Vigneswara, V.; Lowenson, J.; Powell, C.; Thakur, M.; Bailey, K.; Clarke, S.; Ray, D.; Carter, W. Proteomic identification of novel substrates of a protein isoaspartyl methyltransferase repair enzyme. J. Biol. Chem. 2006, 281, 32619–32629. [Google Scholar] [CrossRef] [Green Version]
- Vigneswara, V.; Cass, S.; Wayne, D.; Bolt, E.L.; Ray, D.E.; Carter, W.G. Molecular ageing of alpha- and beta-synucleins: Protein damage and repair mechanisms. PLoS ONE 2013, 8, e61442. [Google Scholar] [CrossRef] [Green Version]
- Mbefo, M.; Paleologou, K.; Boucharaba, A.; Oueslati, A.; Schell, H.; Fournier, M.; Olschewski, D.; Yin, G.; Zweckstetter, M.; Masliah, E.; et al. Phosphorylation of synucleins by members of the Polo-like kinase family. J. Biol. Chem. 2010, 285, 2807–2822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pronin, A.; Morris, A.; Surguchov, A.; Benovic, J. Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J. Biol. Chem. 2000, 275, 26515–26522. [Google Scholar] [CrossRef] [Green Version]
- Payton, J.E.; Perrin, R.J.; Woods, W.S.; George, J.M. Structural determinants of PLD2 inhibition by α-synuclein. J. Mol. Biol. 2004, 337, 1001–1009. [Google Scholar] [CrossRef]
- Shigeo, N.; Tsukuda, K.; Omata, K.; Nakamura, Y.; Nakaya, K. A new brain-specific 14-kDa protein is a phosphoprotein. Eur. J. Biochem. 1993, 217, 1057–1063. [Google Scholar] [CrossRef]
- Ia, K.K.; Jeschke, G.R.; Deng, Y.; Kamaruddin, M.A.; Williamson, N.A.; Scanlon, D.B.; Culvenor, J.G.; Hossain, M.I.; Purcell, A.W.; Liu, S.; et al. Defining the substrate specificity determinants recognized by the active site of C-terminal Src kinase-homologous kinase (CHK) and identification of β-Synuclein as a potential CHK physiological substrate. Biochemistry 2011, 50, 6667–6677. [Google Scholar] [CrossRef] [Green Version]
- Lek, S.; Vargas-Medrano, J.; Villanueva, E.; Marcus, B.S.; Godfrey, W.; Perez, R.G. Recombinant α-, β- and γ-synucleins stimulate protein phosphatase 2a catalytic subunit activity in cell free assays. J. Vis. Exp. 2017, 2017, 55361. [Google Scholar] [CrossRef] [Green Version]
- Stark, C.; Breitkreutz, B.-J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef] [Green Version]
- Lam, S.S.; Martell, J.D.; Kamer, K.J.; Deerinck, T.J.; Ellisman, M.H.; Mootha, V.K.; Ting, A.Y. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 2015, 12, 51–54. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayashi, J.; Carver, J.A. β-Synuclein: An Enigmatic Protein with Diverse Functionality. Biomolecules 2022, 12, 142. https://doi.org/10.3390/biom12010142
Hayashi J, Carver JA. β-Synuclein: An Enigmatic Protein with Diverse Functionality. Biomolecules. 2022; 12(1):142. https://doi.org/10.3390/biom12010142
Chicago/Turabian StyleHayashi, Junna, and John A. Carver. 2022. "β-Synuclein: An Enigmatic Protein with Diverse Functionality" Biomolecules 12, no. 1: 142. https://doi.org/10.3390/biom12010142
APA StyleHayashi, J., & Carver, J. A. (2022). β-Synuclein: An Enigmatic Protein with Diverse Functionality. Biomolecules, 12(1), 142. https://doi.org/10.3390/biom12010142