
Extracellular Vesicles as Mediators of Therapy Resistance in the Breast Cancer Microenvironment



Abstract
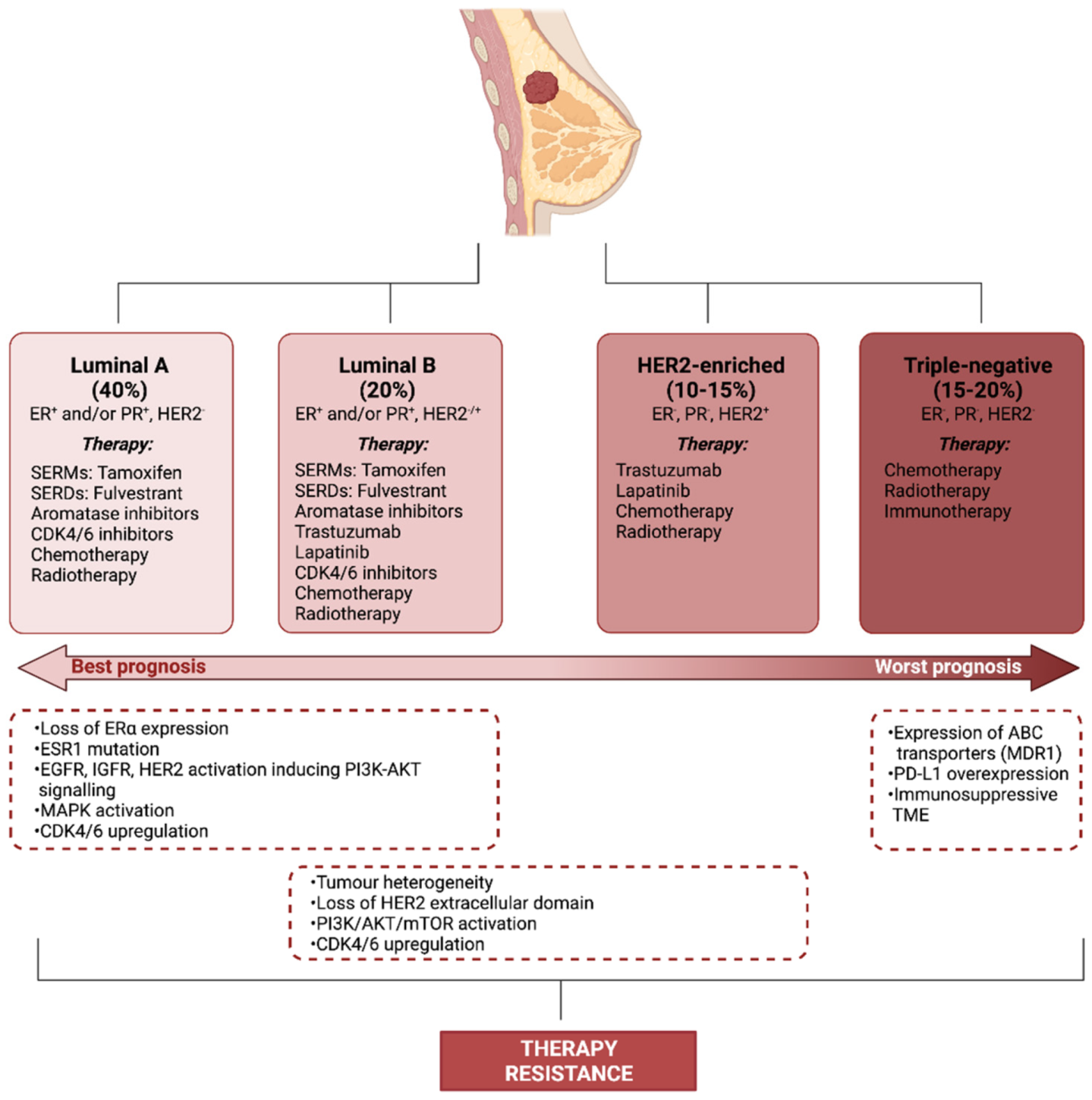
:1. Introduction
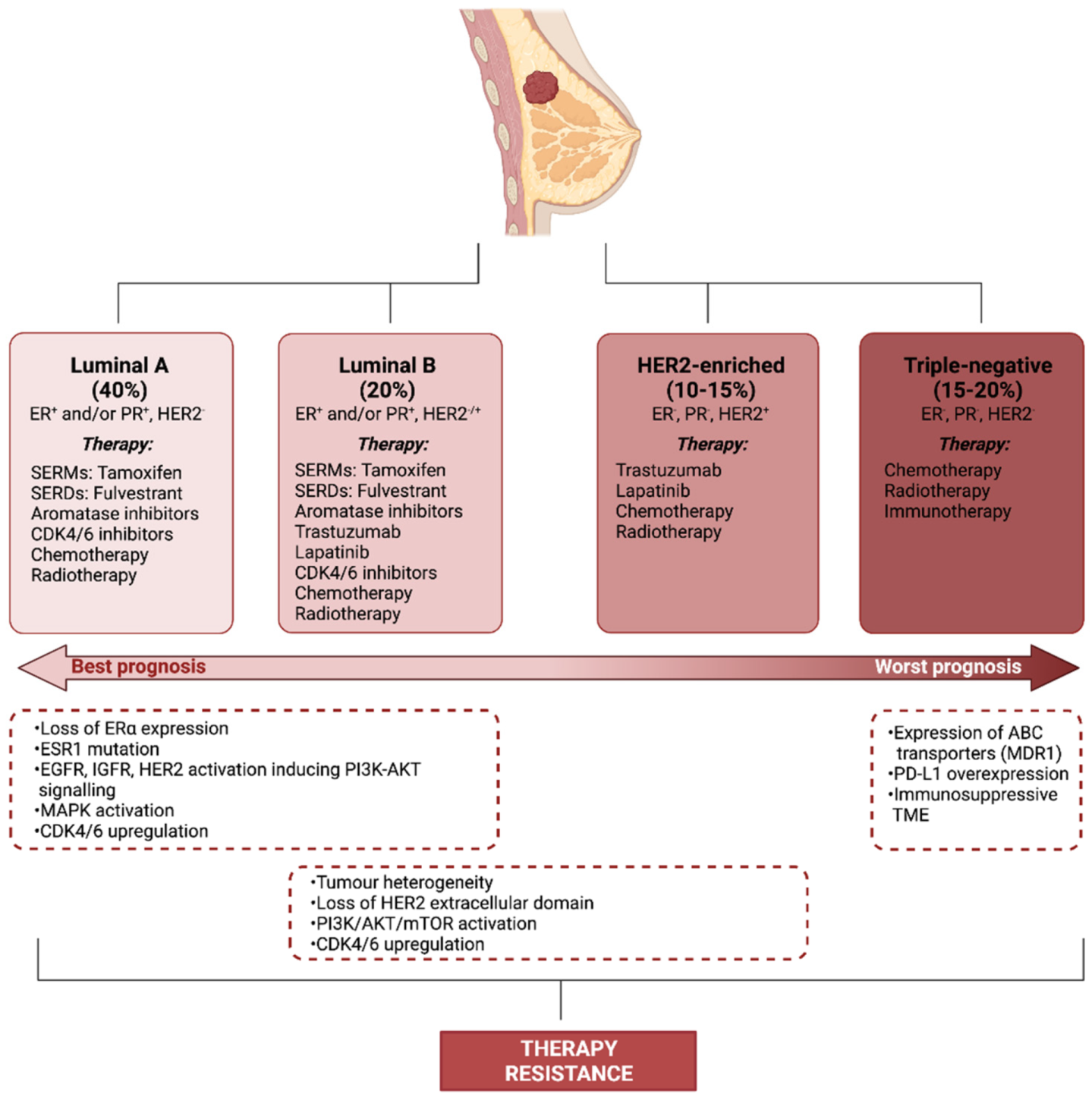
2. Therapy Resistance
3. The Tumour Microenvironment
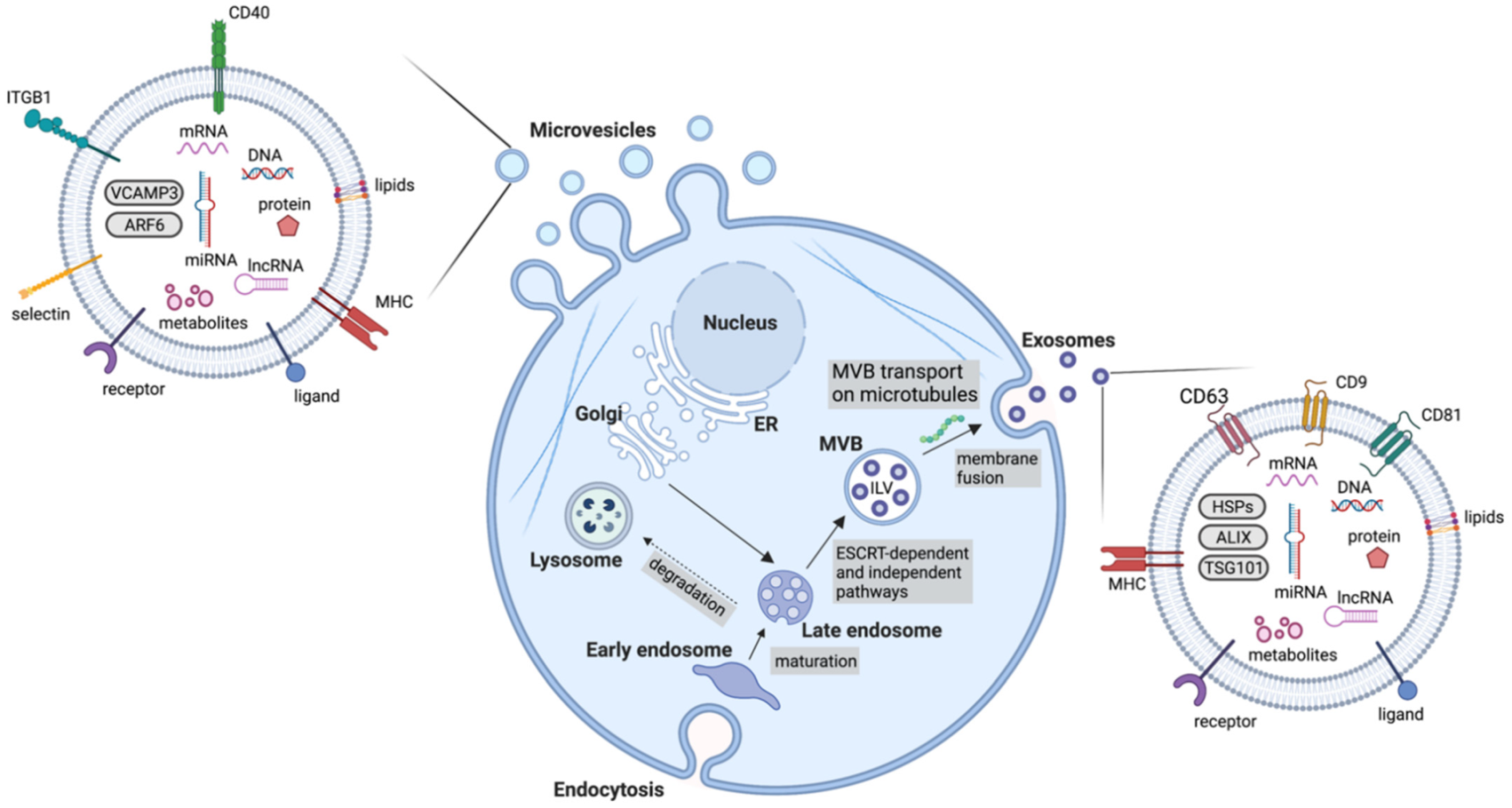
4. Extracellular Vesicles
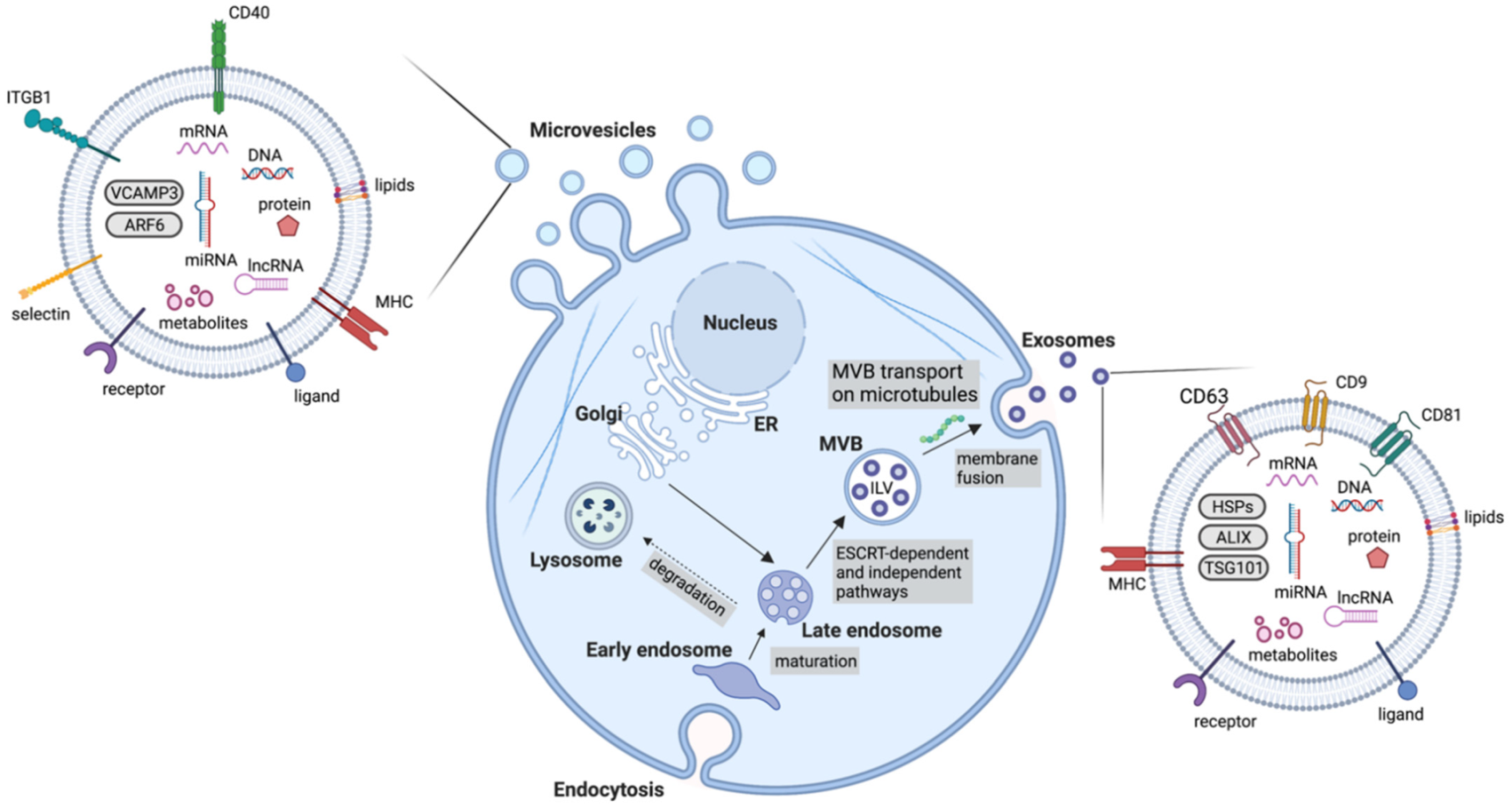
4.1. Exosomes
4.2. Microvesicles
4.3. Extracellular Vesicle Separation
4.4. Extracellular Vesicle Cargo
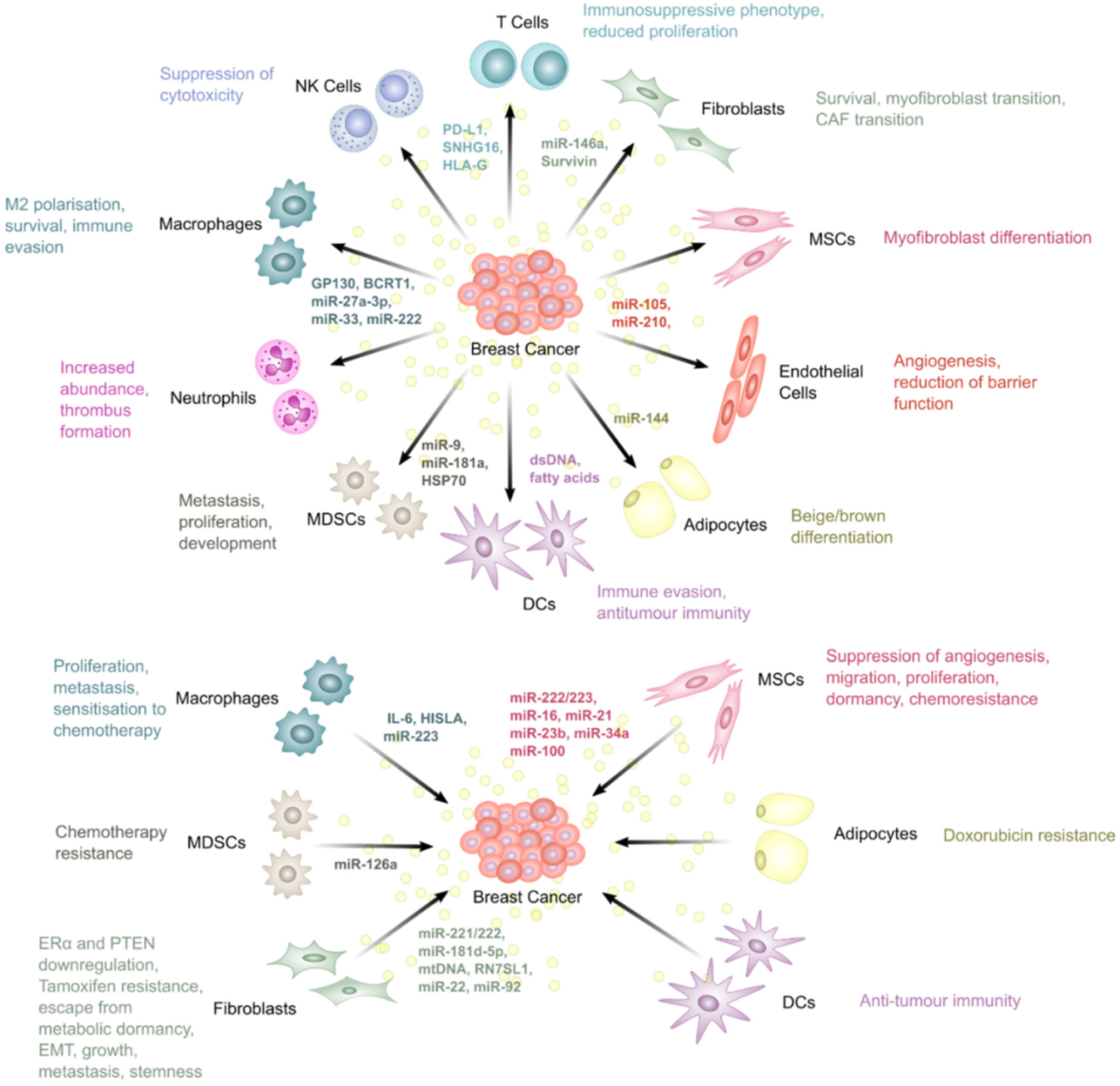
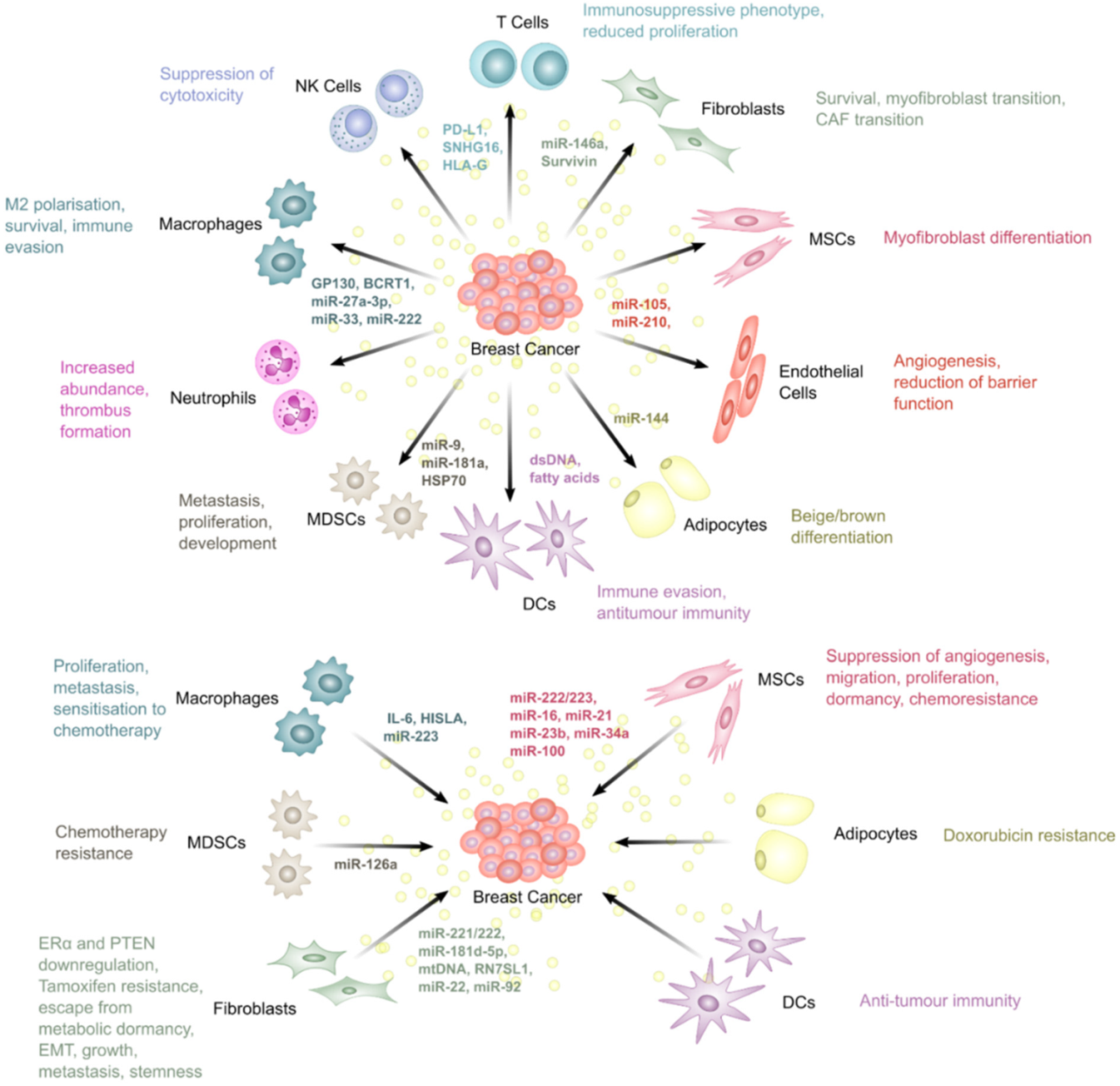
5. The Roles of Extracellular Vesicles in Breast Cancer Therapy Resistance
5.1. Fibroblasts
5.2. Mesenchymal Stem Cells
5.3. Endothelial Cells
5.4. Adipocytes
5.5. Macrophages
5.6. T Lymphocytes
5.7. Dendritic Cells
5.8. Myeloid-Derived Suppressor Cells
5.9. Natural Killer Cells
5.10. Neutrophils
6. Clinical Applications of Extracellular Vesicles in Therapy Resistance
6.1. Extracellular Vesicles as Biomarkers
6.2. Targeting Extracellular Vesicles to Overcome Therapy Resistance
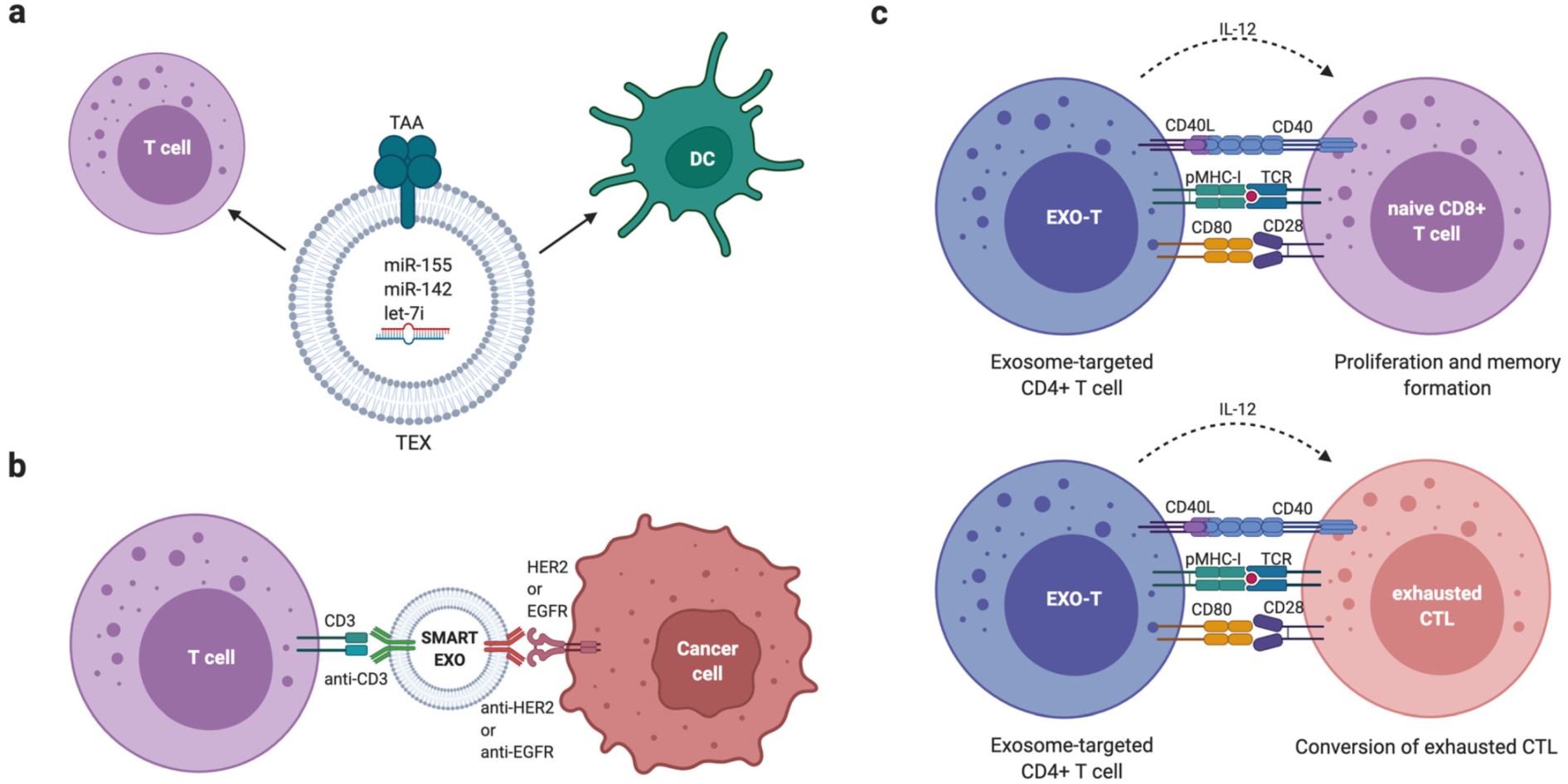
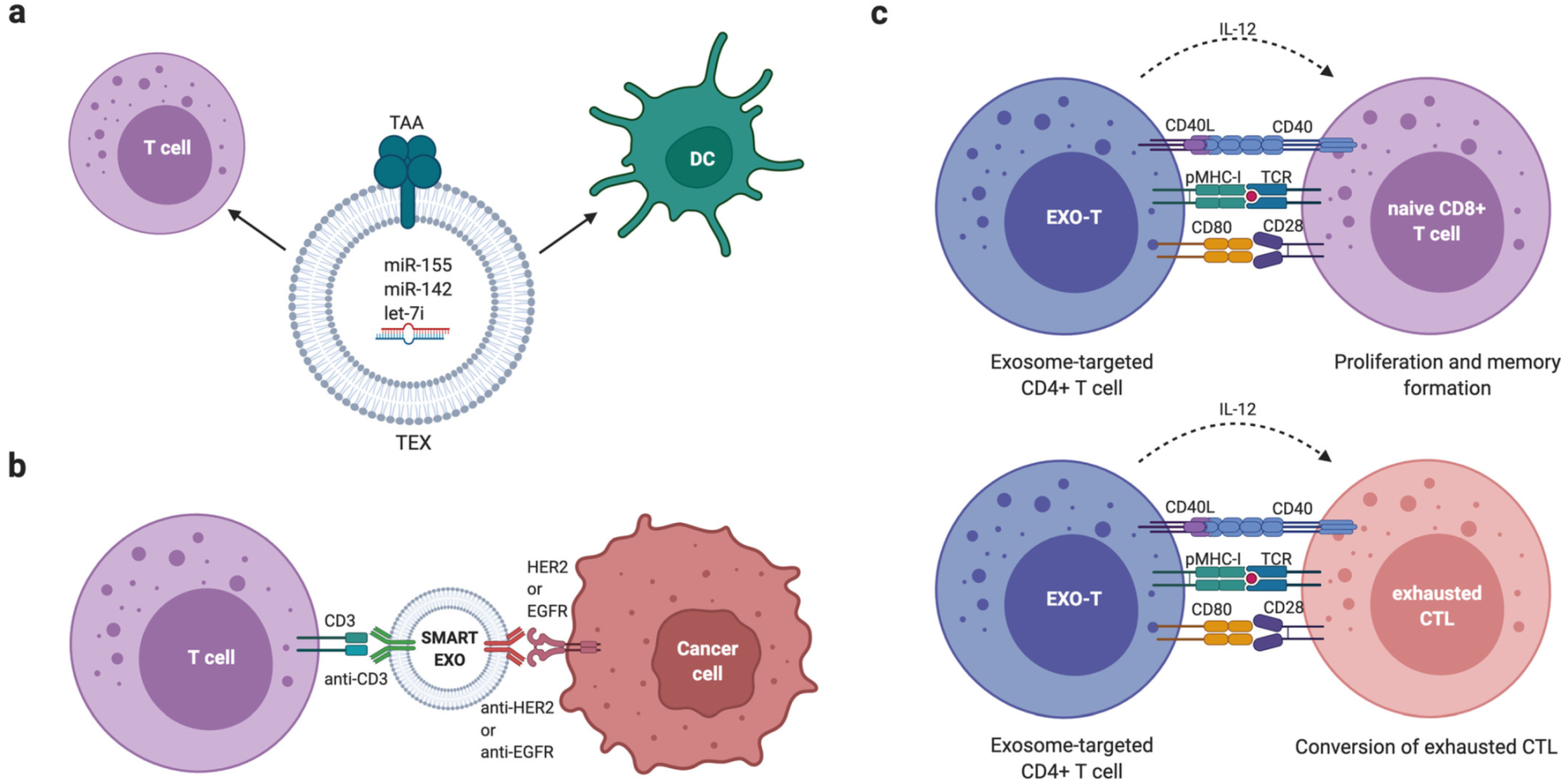
6.3. Extracellular Vesicle-Based Cancer Immunotherapies
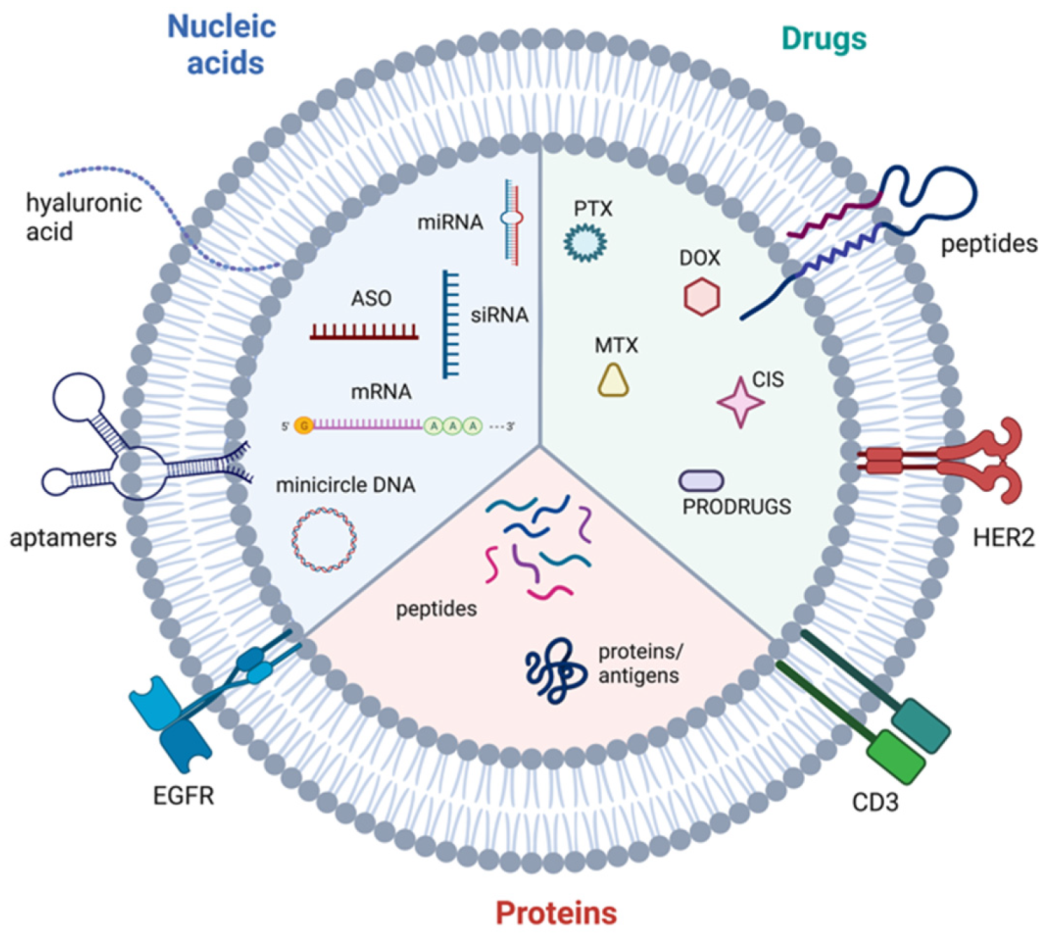
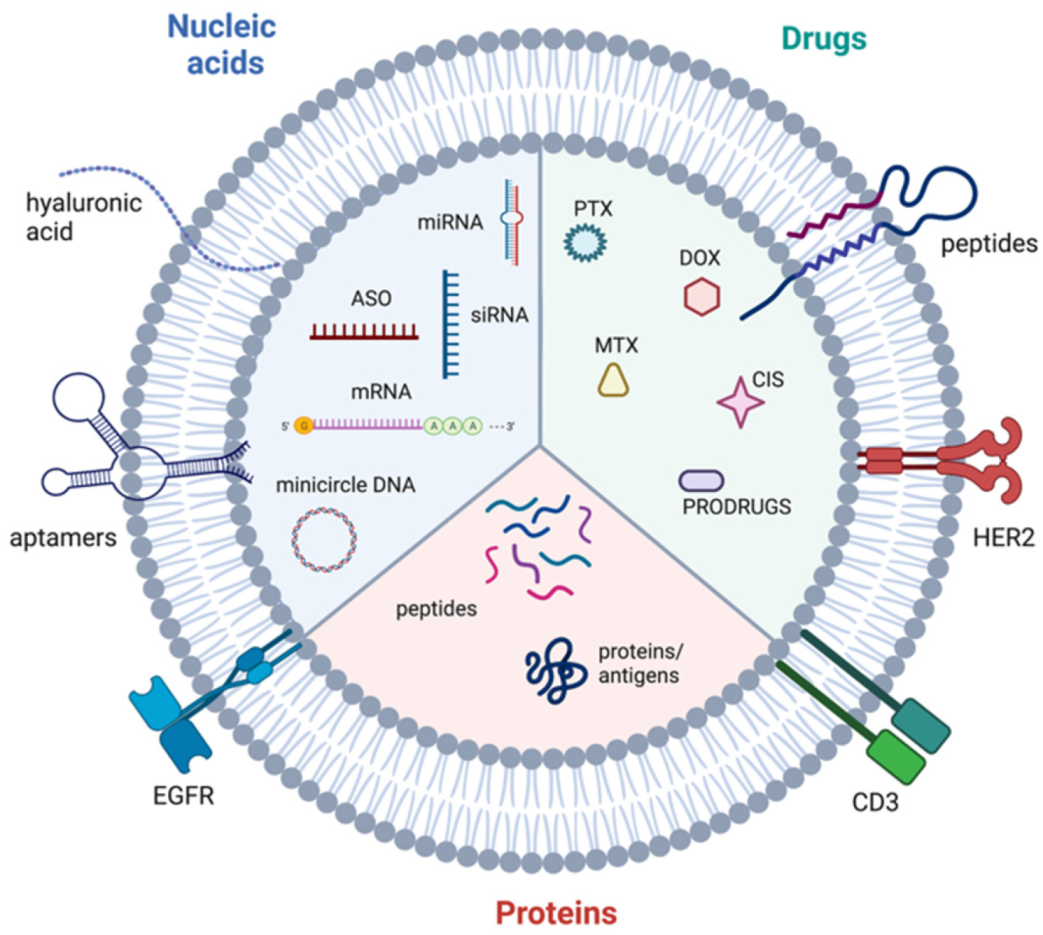
6.4. Extracellular Vesicles as Delivery Vehicles for Therapeutic Agents
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Nordenskjold, A.E.; Fohlin, H.; Arnesson, L.G.; Einbeigi, Z.; Holmberg, E.; Albertsson, P.; Karlsson, P. Breast cancer survival trends in different stages and age groups—A population-based study 1989–2013. Acta Oncol. 2019, 58, 45–51. [Google Scholar] [CrossRef]
- Lafourcade, A.; His, M.; Baglietto, L.; Boutron-Ruault, M.C.; Dossus, L.; Rondeau, V. Factors associated with breast cancer recurrences or mortality and dynamic prediction of death using history of cancer recurrences: The French E3N cohort. BMC Cancer 2018, 18, 171. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar]
- Early Breast Cancer Trialists’ Collaborative Group. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: An overview of the randomised trials. Lancet 2005, 365, 1687–1717. [Google Scholar] [CrossRef]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Geyer, C.E.; Forster, J.; Lindquist, D.; Chan, S.; Romieu, C.G.; Pienkowski, T.; Jagiello-Gruszfeld, A.; Crown, J.; Chan, A.; Kaufman, B.; et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2006, 355, 2733–2743. [Google Scholar] [CrossRef] [Green Version]
- Mehanna, J.; Haddad, F.G.; Eid, R.; Lambertini, M.; Kourie, H.R. Triple-negative breast cancer: Current perspective on the evolving therapeutic landscape. Int. J. Womens Health 2019, 11, 431–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Gao, J.; Wang, M.; Li, M. Potential Prospect of CDK4/6 Inhibitors in Triple-Negative Breast Cancer. Cancer Manag. Res. 2021, 13, 5223–5237. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosentino, G.; Plantamura, I.; Tagliabue, E.; Iorio, M.V.; Cataldo, A. Breast Cancer Drug Resistance: Overcoming the Challenge by Capitalizing on MicroRNA and Tumor Microenvironment Interplay. Cancers 2021, 13, 3691. [Google Scholar] [CrossRef]
- Fan, W.; Chang, J.; Fu, P. Endocrine therapy resistance in breast cancer: Current status, possible mechanisms and overcoming strategies. Future Med. Chem. 2015, 7, 1511–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, R.A.; Bhat-Nakshatri, P.; Patel, N.M.; Constantinidou, D.; Ali, S.; Nakshatri, H. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: A new model for anti-estrogen resistance. J. Biol. Chem. 2001, 276, 9817–9824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutcheson, I.R.; Knowlden, J.M.; Madden, T.A.; Barrow, D.; Gee, J.M.; Wakeling, A.E.; Nicholson, R.I. Oestrogen receptor-mediated modulation of the EGFR/MAPK pathway in tamoxifen-resistant MCF-7 cells. Breast Cancer Res. Treat. 2003, 81, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Roodi, N.; Bailey, L.R.; Kao, W.Y.; Verrier, C.S.; Yee, C.J.; Dupont, W.D.; Parl, F.F. Estrogen receptor gene analysis in estrogen receptor-positive and receptor-negative primary breast cancer. J. Natl. Cancer Inst. 1995, 87, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.L.; Cobleigh, M.A.; Tripathy, D.; Gutheil, J.C.; Harris, L.N.; Fehrenbacher, L.; Slamon, D.J.; Murphy, M.; Novotny, W.F.; Burchmore, M.; et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J. Clin. Oncol. 2002, 20, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Nitta, H.; Wei, L.; Banks, P.M.; Portier, B.; Parwani, A.V.; Li, Z. HER2 intratumoral heterogeneity is independently associated with incomplete response to anti-HER2 neoadjuvant chemotherapy in HER2-positive breast carcinoma. Breast Cancer Res. Treat. 2017, 166, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.C.; Liu, X.; Li, Y.; Covington, M.; Wynn, R.; Huber, R.; Hillman, M.; Yang, G.; Ellis, D.; Marando, C.; et al. Identification of ADAM10 as a major source of HER2 ectodomain sheddase activity in HER2 overexpressing breast cancer cells. Cancer Biol. Ther. 2006, 5, 657–664. [Google Scholar] [CrossRef] [Green Version]
- Scaltriti, M.; Rojo, F.; Ocana, A.; Anido, J.; Guzman, M.; Cortes, J.; Di Cosimo, S.; Matias-Guiu, X.; Ramon y Cajal, S.; Arribas, J.; et al. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J. Natl. Cancer Inst. 2007, 99, 628–638. [Google Scholar] [CrossRef] [Green Version]
- Berns, K.; Horlings, H.M.; Hennessy, B.T.; Madiredjo, M.; Hijmans, E.M.; Beelen, K.; Linn, S.C.; Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Hauptmann, M.; et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 2007, 12, 395–402. [Google Scholar] [CrossRef] [Green Version]
- Nedeljkovic, M.; Damjanovic, A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer-How We Can Rise to the Challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef] [Green Version]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.; Turnbull, A.K.; Ward, C.; Meehan, J.; Martinez-Perez, C.; Bonello, M.; Pang, L.Y.; Langdon, S.P.; Kunkler, I.H.; Murray, A.; et al. Development and characterisation of acquired radioresistant breast cancer cell lines. Radiat. Oncol. 2019, 14, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, S.; Gatti-Mays, M.E.; Kalinsky, K.; Korde, L.A.; Sharon, E.; Amiri-Kordestani, L.; Bear, H.; McArthur, H.L.; Frank, E.; Perlmutter, J.; et al. Current Landscape of Immunotherapy in Breast Cancer: A Review. JAMA Oncol. 2019, 5, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Criscitiello, C. Tumor-associated antigens in breast cancer. Breast Care 2012, 7, 262–266. [Google Scholar] [CrossRef] [Green Version]
- Gil Del Alcazar, C.R.; Aleckovic, M.; Polyak, K. Immune Escape during Breast Tumor Progression. Cancer Immunol. Res. 2020, 8, 422–427. [Google Scholar] [CrossRef] [Green Version]
- Hanna, A.; Balko, J.M. Breast cancer resistance mechanisms: Challenges to immunotherapy. Breast Cancer Res. Treat. 2021, 190, 5–17. [Google Scholar] [CrossRef]
- Soysal, S.D.; Tzankov, A.; Muenst, S.E. Role of the Tumor Microenvironment in Breast Cancer. Pathobiology 2015, 82, 142–152. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Zarrilli, G.; Businello, G.; Dieci, M.V.; Paccagnella, S.; Carraro, V.; Cappellesso, R.; Miglietta, F.; Griguolo, G.; Guarneri, V.; Lo Mele, M.; et al. The Tumor Microenvironment of Primitive and Metastatic Breast Cancer: Implications for Novel Therapeutic Strategies. Int. J. Mol. Sci. 2020, 21, 8102. [Google Scholar] [CrossRef]
- Psaila, B.; Kaplan, R.N.; Port, E.R.; Lyden, D. Priming the ‘soil’ for breast cancer metastasis: The pre-metastatic niche. Breast Dis. 2006, 26, 65–74. [Google Scholar] [CrossRef]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.Z.; Jin, W.L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef] [PubMed]
- Velaei, K.; Samadi, N.; Barazvan, B.; Soleimani Rad, J. Tumor microenvironment-mediated chemoresistance in breast cancer. Breast 2016, 30, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Giordano, C.; La Camera, G.; Gelsomino, L.; Barone, I.; Bonofiglio, D.; Ando, S.; Catalano, S. The Biology of Exosomes in Breast Cancer Progression: Dissemination, Immune Evasion and Metastatic Colonization. Cancers 2020, 12, 2179. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Tian, F.; Zhang, S.; Liu, C.; Han, Z.; Liu, Y.; Deng, J.; Li, Y.; Wu, X.; Cai, L.; Qin, L.; et al. Protein analysis of extracellular vesicles to monitor and predict therapeutic response in metastatic breast cancer. Nat. Commun. 2021, 12, 2536. [Google Scholar] [CrossRef]
- Anand, S.; Samuel, M.; Mathivanan, S. Exomeres: A New Member of Extracellular Vesicles Family. Subcell. Biochem. 2021, 97, 89–97. [Google Scholar] [CrossRef]
- Tavano, S.; Heisenberg, C.P. Migrasomes take center stage. Nat. Cell Biol. 2019, 21, 918–920. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, biologic function and clinical potential. Cell Biosci. 2019, 9, 19. [Google Scholar] [CrossRef]
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The ESCRT pathway. Dev. Cell 2011, 21, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Prag, G.; Watson, H.; Kim, Y.C.; Beach, B.M.; Ghirlando, R.; Hummer, G.; Bonifacino, J.S.; Hurley, J.H. The Vps27/Hse1 complex is a GAT domain-based scaffold for ubiquitin-dependent sorting. Dev. Cell 2007, 12, 973–986. [Google Scholar] [CrossRef] [Green Version]
- Raiborg, C.; Bremnes, B.; Mehlum, A.; Gillooly, D.J.; D’Arrigo, A.; Stang, E.; Stenmark, H. FYVE and coiled-coil domains determine the specific localisation of Hrs to early endosomes. J. Cell Sci. 2001, 114, 2255–2263. [Google Scholar] [CrossRef]
- Kostelansky, M.S.; Sun, J.; Lee, S.; Kim, J.; Ghirlando, R.; Hierro, A.; Emr, S.D.; Hurley, J.H. Structural and functional organization of the ESCRT-I trafficking complex. Cell 2006, 125, 113–126. [Google Scholar] [CrossRef] [Green Version]
- Langelier, C.; von Schwedler, U.K.; Fisher, R.D.; De Domenico, I.; White, P.L.; Hill, C.P.; Kaplan, J.; Ward, D.; Sundquist, W.I. Human ESCRT-II complex and its role in human immunodeficiency virus type 1 release. J. Virol. 2006, 80, 9465–9480. [Google Scholar] [CrossRef] [Green Version]
- Babst, M.; Katzmann, D.J.; Estepa-Sabal, E.J.; Meerloo, T.; Emr, S.D. Escrt-III: An endosome-associated heterooligomeric protein complex required for mvb sorting. Dev. Cell 2002, 3, 271–282. [Google Scholar] [CrossRef] [Green Version]
- Teo, H.; Perisic, O.; Gonzalez, B.; Williams, R.L. ESCRT-II, an endosome-associated complex required for protein sorting: Crystal structure and interactions with ESCRT-III and membranes. Dev. Cell 2004, 7, 559–569. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, O.; Teis, D. The ESCRT machinery. Curr. Biol. 2012, 22, R116–R120. [Google Scholar] [CrossRef] [Green Version]
- Sogaard, M.; Tani, K.; Ye, R.R.; Geromanos, S.; Tempst, P.; Kirchhausen, T.; Rothman, J.E.; Sollner, T. A rab protein is required for the assembly of SNARE complexes in the docking of transport vesicles. Cell 1994, 78, 937–948. [Google Scholar] [CrossRef]
- Blanc, L.; Vidal, M. New insights into the function of Rab GTPases in the context of exosomal secretion. Small GTPases 2018, 9, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Castro, B.M.; Prieto, M.; Silva, L.C. Ceramide: A simple sphingolipid with unique biophysical properties. Prog. Lipid Res. 2014, 54, 53–67. [Google Scholar] [CrossRef]
- Perez-Hernandez, D.; Gutierrez-Vazquez, C.; Jorge, I.; Lopez-Martin, S.; Ursa, A.; Sanchez-Madrid, F.; Vazquez, J.; Yanez-Mo, M. The intracellular interactome of tetraspanin-enriched microdomains reveals their function as sorting machineries toward exosomes. J. Biol. Chem. 2013, 288, 11649–11661. [Google Scholar] [CrossRef] [Green Version]
- Wei, D.; Zhan, W.; Gao, Y.; Huang, L.; Gong, R.; Wang, W.; Zhang, R.; Wu, Y.; Gao, S.; Kang, T. RAB31 marks and controls an ESCRT-independent exosome pathway. Cell Res. 2021, 31, 157–177. [Google Scholar] [CrossRef]
- Tricarico, C.; Clancy, J.; D’Souza-Schorey, C. Biology and biogenesis of shed microvesicles. Small GTPases 2017, 8, 220–232. [Google Scholar] [CrossRef] [Green Version]
- Muralidharan-Chari, V.; Clancy, J.; Plou, C.; Romao, M.; Chavrier, P.; Raposo, G.; D’Souza-Schorey, C. ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr. Biol. 2009, 19, 1875–1885. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.H.; Catling, A.D.; Webb, D.J.; Sankovic, M.; Walker, L.A.; Somlyo, A.V.; Weber, M.J.; Gonias, S.L. Myosin light chain kinase functions downstream of Ras/ERK to promote migration of urokinase-type plasminogen activator-stimulated cells in an integrin-selective manner. J. Cell Biol. 1999, 146, 149–164. [Google Scholar] [CrossRef]
- Li, B.; Antonyak, M.A.; Zhang, J.; Cerione, R.A. RhoA triggers a specific signaling pathway that generates transforming microvesicles in cancer cells. Oncogene 2012, 31, 4740–4749. [Google Scholar] [CrossRef] [Green Version]
- Liangsupree, T.; Multia, E.; Riekkola, M.L. Modern isolation and separation techniques for extracellular vesicles. J. Chromatogr. A 2021, 1636, 461773. [Google Scholar] [CrossRef]
- Zhou, E.; Li, Y.; Wu, F.; Guo, M.; Xu, J.; Wang, S.; Tan, Q.; Ma, P.; Song, S.; Jin, Y. Circulating extracellular vesicles are effective biomarkers for predicting response to cancer therapy. EBioMedicine 2021, 67, 103365. [Google Scholar] [CrossRef]
- Bergsmedh, A.; Szeles, A.; Henriksson, M.; Bratt, A.; Folkman, M.J.; Spetz, A.L.; Holmgren, L. Horizontal transfer of oncogenes by uptake of apoptotic bodies. Proc. Natl. Acad. Sci. USA 2001, 98, 6407–6411. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. (Lausanne) 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Skotland, T.; Sagini, K.; Sandvig, K.; Llorente, A. An emerging focus on lipids in extracellular vesicles. Adv. Drug Deliv. Rev. 2020, 159, 308–321. [Google Scholar] [CrossRef] [PubMed]
- Subra, C.; Grand, D.; Laulagnier, K.; Stella, A.; Lambeau, G.; Paillasse, M.; De Medina, P.; Monsarrat, B.; Perret, B.; Silvente-Poirot, S.; et al. Exosomes account for vesicle-mediated transcellular transport of activatable phospholipases and prostaglandins. J. Lipid Res. 2010, 51, 2105–2120. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Xu, K.; Zheng, X.; Chen, T.; Wang, J.; Song, Y.; Shao, Y.; Zheng, S. Application of exosomes as liquid biopsy in clinical diagnosis. Signal Transduct. Target. Ther. 2020, 5, 144. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rockey, D.C.; Weymouth, N.; Shi, Z. Smooth muscle alpha actin (Acta2) and myofibroblast function during hepatic wound healing. PLoS ONE 2013, 8, e77166. [Google Scholar] [CrossRef] [Green Version]
- Strell, C.; Paulsson, J.; Jin, S.B.; Tobin, N.P.; Mezheyeuski, A.; Roswall, P.; Mutgan, C.; Mitsios, N.; Johansson, H.; Wickberg, S.M.; et al. Impact of Epithelial-Stromal Interactions on Peritumoral Fibroblasts in Ductal Carcinoma in Situ. J. Natl. Cancer Inst. 2019, 111, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; Martin, J.D.; Stylianopoulos, T. The role of mechanical forces in tumor growth and therapy. Annu. Rev. Biomed. Eng. 2014, 16, 321–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.S.; Ma, S.; Dou, H.; Liu, F.; Zhang, S.Y.; Jiang, C.; Xiao, M.; Huang, Y.X. Breast cancer-derived exosomes regulate cell invasion and metastasis in breast cancer via miR-146a to activate cancer associated fibroblasts in tumor microenvironment. Exp. Cell Res. 2020, 391, 111983. [Google Scholar] [CrossRef] [PubMed]
- Dou, D.; Ren, X.; Han, M.; Xu, X.; Ge, X.; Gu, Y.; Wang, X. Cancer-Associated Fibroblasts-Derived Exosomes Suppress Immune Cell Function in Breast Cancer via the miR-92/PD-L1 Pathway. Front. Immunol. 2020, 11, 2026. [Google Scholar] [CrossRef] [PubMed]
- Kreger, B.T.; Johansen, E.R.; Cerione, R.A.; Antonyak, M.A. The Enrichment of Survivin in Exosomes from Breast Cancer Cells Treated with Paclitaxel Promotes Cell Survival and Chemoresistance. Cancers 2016, 8, 111. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Liu, T.; Chen, J.; Ni, H.; Li, W. Survivin in breast cancer-derived exosomes activates fibroblasts by up-regulating SOD1, whose feedback promotes cancer proliferation and metastasis. J. Biol. Chem. 2020, 295, 13737–13752. [Google Scholar] [CrossRef]
- Wang, H.; Wei, H.; Wang, J.; Li, L.; Chen, A.; Li, Z. MicroRNA-181d-5p-Containing Exosomes Derived from CAFs Promote EMT by Regulating CDX2/HOXA5 in Breast Cancer. Mol. Ther. Nucleic Acids 2020, 19, 654–667. [Google Scholar] [CrossRef] [PubMed]
- Strotbek, M.; Schmid, S.; Sanchez-Gonzalez, I.; Boerries, M.; Busch, H.; Olayioye, M.A. miR-181 elevates Akt signaling by co-targeting PHLPP2 and INPP4B phosphatases in luminal breast cancer. Int. J. Cancer 2017, 140, 2310–2320. [Google Scholar] [CrossRef] [PubMed]
- Luga, V.; Zhang, L.; Viloria-Petit, A.M.; Ogunjimi, A.A.; Inanlou, M.R.; Chiu, E.; Buchanan, M.; Hosein, A.N.; Basik, M.; Wrana, J.L. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell 2012, 151, 1542–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Li, X.; Zeng, C.; Liu, C.; Hao, Q.; Li, W.; Zhang, K.; Zhang, W.; Wang, S.; Zhao, H.; et al. CD63(+) Cancer-Associated Fibroblasts Confer Tamoxifen Resistance to Breast Cancer Cells through Exosomal miR-22. Adv. Sci. (Weinh) 2020, 7, 2002518. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.E.; Ghoshal, K.; Ramaswamy, B.; Roy, S.; Datta, J.; Shapiro, C.L.; Jacob, S.; Majumder, S. MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27Kip1. J. Biol. Chem. 2008, 283, 29897–29903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansone, P.; Berishaj, M.; Rajasekhar, V.K.; Ceccarelli, C.; Chang, Q.; Strillacci, A.; Savini, C.; Shapiro, L.; Bowman, R.L.; Mastroleo, C.; et al. Evolution of Cancer Stem-like Cells in Endocrine-Resistant Metastatic Breast Cancers Is Mediated by Stromal Microvesicles. Cancer Res 2017, 77, 1927–1941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.H.; Miller, P.; Garcia-Contreras, M.; Ao, Z.; Machlin, L.; Issa, E.; El-Ashry, D. Hierarchical paracrine interaction of breast cancer associated fibroblasts with cancer cells via hMAPK-microRNAs to drive ER-negative breast cancer phenotype. Cancer Biol. Ther. 2015, 16, 1671–1681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boelens, M.C.; Wu, T.J.; Nabet, B.Y.; Xu, B.; Qiu, Y.; Yoon, T.; Azzam, D.J.; Twyman-Saint Victor, C.; Wiemann, B.Z.; Ishwaran, H.; et al. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell 2014, 159, 499–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabet, B.Y.; Qiu, Y.; Shabason, J.E.; Wu, T.J.; Yoon, T.; Kim, B.C.; Benci, J.L.; DeMichele, A.M.; Tchou, J.; Marcotrigiano, J.; et al. Exosome RNA Unshielding Couples Stromal Activation to Pattern Recognition Receptor Signaling in Cancer. Cell 2017, 170, 352–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pittenger, M.F.; Discher, D.E.; Peault, B.M.; Phinney, D.G.; Hare, J.M.; Caplan, A.I. Mesenchymal stem cell perspective: Cell biology to clinical progress. npj Regen. Med. 2019, 4, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, S.Y. The Role of MSCs in the Tumor Microenvironment and Tumor Progression. Anticancer Res. 2020, 40, 3039–3047. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.A.; Park, H.; Lim, E.H.; Lee, K.W. Exosomes from breast cancer cells can convert adipose tissue-derived mesenchymal stem cells into myofibroblast-like cells. Int. J. Oncol. 2012, 40, 130–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallabhaneni, K.C.; Penfornis, P.; Dhule, S.; Guillonneau, F.; Adams, K.V.; Mo, Y.Y.; Xu, R.; Liu, Y.; Watabe, K.; Vemuri, M.C.; et al. Extracellular vesicles from bone marrow mesenchymal stem/stromal cells transport tumor regulatory microRNA, proteins, and metabolites. Oncotarget 2015, 6, 4953–4967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.; Wang, S.; Zhao, R.C. Exosomes from human adipose-derived mesenchymal stem cells promote migration through Wnt signaling pathway in a breast cancer cell model. Mol. Cell Biochem. 2013, 383, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Merikhian, P.; Eisavand, M.R.; Farahmand, L. Triple-negative breast cancer: Understanding Wnt signaling in drug resistance. Cancer Cell Int. 2021, 21, 419. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Kosaka, N.; Tominaga, N.; Yoshioka, Y.; Takeshita, F.; Takahashi, R.U.; Yoshida, M.; Tsuda, H.; Tamura, K.; Ochiya, T. Exosomes from bone marrow mesenchymal stem cells contain a microRNA that promotes dormancy in metastatic breast cancer cells. Sci. Signal. 2014, 7, ra63. [Google Scholar] [CrossRef] [PubMed]
- Bliss, S.A.; Sinha, G.; Sandiford, O.A.; Williams, L.M.; Engelberth, D.J.; Guiro, K.; Isenalumhe, L.L.; Greco, S.J.; Ayer, S.; Bryan, M.; et al. Mesenchymal Stem Cell-Derived Exosomes Stimulate Cycling Quiescence and Early Breast Cancer Dormancy in Bone Marrow. Cancer Res. 2016, 76, 5832–5844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.K.; Park, S.R.; Jung, B.K.; Jeon, Y.K.; Lee, Y.S.; Kim, M.K.; Kim, Y.G.; Jang, J.Y.; Kim, C.W. Exosomes derived from mesenchymal stem cells suppress angiogenesis by down-regulating VEGF expression in breast cancer cells. PLoS ONE 2013, 8, e84256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pakravan, K.; Babashah, S.; Sadeghizadeh, M.; Mowla, S.J.; Mossahebi-Mohammadi, M.; Ataei, F.; Dana, N.; Javan, M. MicroRNA-100 shuttled by mesenchymal stem cell-derived exosomes suppresses in vitro angiogenesis through modulating the mTOR/HIF-1alpha/VEGF signaling axis in breast cancer cells. Cell. Oncol. 2017, 40, 457–470. [Google Scholar] [CrossRef]
- Harris, A.L. Hypoxia--a key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Robert, N.J.; Dieras, V.; Glaspy, J.; Brufsky, A.M.; Bondarenko, I.; Lipatov, O.N.; Perez, E.A.; Yardley, D.A.; Chan, S.Y.; Zhou, X.; et al. RIBBON-1: Randomized, double-blind, placebo-controlled, phase III trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2-negative, locally recurrent or metastatic breast cancer. J. Clin. Oncol. 2011, 29, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Madu, C.O.; Wang, S.; Madu, C.O.; Lu, Y. Angiogenesis in Breast Cancer Progression, Diagnosis, and Treatment. J. Cancer 2020, 11, 4474–4494. [Google Scholar] [CrossRef] [PubMed]
- Senger, D.R.; Galli, S.J.; Dvorak, A.M.; Perruzzi, C.A.; Harvey, V.S.; Dvorak, H.F. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983, 219, 983–985. [Google Scholar] [CrossRef]
- Chaudhary, P.; Gibbs, L.D.; Maji, S.; Lewis, C.M.; Suzuki, S.; Vishwanatha, J.K. Serum exosomal-annexin A2 is associated with African-American triple-negative breast cancer and promotes angiogenesis. Breast Cancer Res. 2020, 22, 11. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, N.; Iguchi, H.; Hagiwara, K.; Yoshioka, Y.; Takeshita, F.; Ochiya, T. Neutral sphingomyelinase 2 (nSMase2)-dependent exosomal transfer of angiogenic microRNAs regulate cancer cell metastasis. J. Biol. Chem. 2013, 288, 10849–10859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Modica, M.; Regondi, V.; Sandri, M.; Iorio, M.V.; Zanetti, A.; Tagliabue, E.; Casalini, P.; Triulzi, T. Breast cancer-secreted miR-939 downregulates VE-cadherin and destroys the barrier function of endothelial monolayers. Cancer Lett. 2017, 384, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Chen, X.; Meng, F.; Ogawa, K.; Li, M.; Song, R.; Zhang, S.; Zhang, Z.; Kong, X.; Xu, Q.; et al. ASPH-notch Axis guided Exosomal delivery of Prometastatic Secretome renders breast Cancer multi-organ metastasis. Mol. Cancer 2019, 18, 156. [Google Scholar] [CrossRef]
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’Connor, S.T.; Chin, A.R.; et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 2014, 25, 501–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, D.T.; Phuong, T.N.T.; Tien, N.L.B.; Tran, D.K.; Nguyen, T.T.; Thanh, V.V.; Quang, T.L.; Minh, L.B.; Pham, V.H.; Ngoc, V.T.N.; et al. The Effects of Adipocytes on the Regulation of Breast Cancer in the Tumor Microenvironment: An Update. Cells 2019, 8, 857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Li, B.; Li, Z.; Li, J.; Sun, S.; Sun, S. Cancer-associated adipocytes: Key players in breast cancer progression. J. Hematol. Oncol. 2019, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Li, J.; Li, Z.; Sun, S.; Zhu, S.; Wang, L.; Wu, J.; Yuan, J.; Zhang, Y.; Sun, S.; et al. Exosomes from the tumour-adipocyte interplay stimulate beige/brown differentiation and reprogram metabolism in stromal adipocytes to promote tumour progression. J. Exp. Clin. Cancer Res. 2019, 38, 223. [Google Scholar] [CrossRef] [Green Version]
- Lehuede, C.; Li, X.; Dauvillier, S.; Vaysse, C.; Franchet, C.; Clement, E.; Esteve, D.; Longue, M.; Chaltiel, L.; Le Gonidec, S.; et al. Adipocytes promote breast Cancer Res.istance to chemotherapy, a process amplified by obesity: Role of the major vault protein (MVP). Breast Cancer Res. 2019, 21, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayasingam, S.D.; Citartan, M.; Thang, T.H.; Mat Zin, A.A.; Ang, K.C.; Ch’ng, E.S. Evaluating the Polarization of Tumor-Associated Macrophages Into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 2019, 9, 1512. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Qiu, S.Q.; Waaijer, S.J.H.; Zwager, M.C.; de Vries, E.G.E.; van der Vegt, B.; Schroder, C.P. Tumor-associated macrophages in breast cancer: Innocent bystander or important player? Cancer Treat. Rev. 2018, 70, 178–189. [Google Scholar] [CrossRef] [Green Version]
- Laoui, D.; Movahedi, K.; Van Overmeire, E.; Van den Bossche, J.; Schouppe, E.; Mommer, C.; Nikolaou, A.; Morias, Y.; De Baetselier, P.; Van Ginderachter, J.A. Tumor-associated macrophages in breast cancer: Distinct subsets, distinct functions. Int. J. Dev. Biol. 2011, 55, 861–867. [Google Scholar] [CrossRef]
- Tao, S.; Zhao, Z.; Zhang, X.; Guan, X.; Wei, J.; Yuan, B.; He, S.; Zhao, D.; Zhang, J.; Liu, Q.; et al. The role of macrophages during breast cancer development and response to chemotherapy. Clin. Transl. Oncol. 2020, 22, 1938–1951. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Mandal, G.; Roy Chowdhury, S.; Purohit, S.; Payne, K.K.; Anadon, C.; Gupta, A.; Swanson, P.; Yu, X.; Conejo-Garcia, J.R.; et al. Exosomes Produced by Mesenchymal Stem Cells Drive Differentiation of Myeloid Cells into Immunosuppressive M2-Polarized Macrophages in Breast Cancer. J. Immunol. 2019, 203, 3447–3460. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Song, X.; Li, Y.; Chen, B.; Zhao, W.; Wang, L.; Zhang, H.; Liu, Y.; Han, D.; Zhang, N.; et al. LncRNA BCRT1 promotes breast cancer progression by targeting miR-1303/PTBP3 axis. Mol. Cancer 2020, 19, 85. [Google Scholar] [CrossRef] [PubMed]
- Ham, S.; Lima, L.G.; Chai, E.P.Z.; Muller, A.; Lobb, R.J.; Krumeich, S.; Wen, S.W.; Wiegmans, A.P.; Moller, A. Breast Cancer-Derived Exosomes Alter Macrophage Polarization via gp130/STAT3 Signaling. Front. Immunol. 2018, 9, 871. [Google Scholar] [CrossRef] [Green Version]
- Piao, Y.J.; Kim, H.S.; Hwang, E.H.; Woo, J.; Zhang, M.; Moon, W.K. Breast cancer cell-derived exosomes and macrophage polarization are associated with lymph node metastasis. Oncotarget 2018, 9, 7398–7410. [Google Scholar] [CrossRef]
- Chen, W.X.; Wang, D.D.; Zhu, B.; Zhu, Y.Z.; Zheng, L.; Feng, Z.Q.; Qin, X.H. Exosomal miR-222 from adriamycin-resistant MCF-7 breast cancer cells promote macrophages M2 polarization via PTEN/Akt to induce tumor progression. Aging 2021, 13, 10415–10430. [Google Scholar] [CrossRef]
- Yao, X.; Tu, Y.; Xu, Y.; Guo, Y.; Yao, F.; Zhang, X. Endoplasmic reticulum stress-induced exosomal miR-27a-3p promotes immune escape in breast cancer via regulating PD-L1 expression in macrophages. J. Cell. Mol. Med. 2020, 24, 9560–9573. [Google Scholar] [CrossRef] [PubMed]
- Prose, N.S. HIV infection in children. J. Am. Acad. Dermatol. 1990, 22, 1223–1231. [Google Scholar] [CrossRef]
- Chen, F.; Chen, J.; Yang, L.; Liu, J.; Zhang, X.; Zhang, Y.; Tu, Q.; Yin, D.; Lin, D.; Wong, P.P.; et al. Extracellular vesicle-packaged HIF-1alpha-stabilizing lncRNA from tumour-associated macrophages regulates aerobic glycolysis of breast cancer cells. Nat. Cell Biol. 2019, 21, 498–510. [Google Scholar] [CrossRef]
- Yu, X.; Zhang, Q.; Zhang, X.; Han, Q.; Li, H.; Mao, Y.; Wang, X.; Guo, H.; Irwin, D.M.; Niu, G.; et al. Exosomes from Macrophages Exposed to Apoptotic Breast Cancer Cells Promote Breast Cancer Proliferation and Metastasis. J. Cancer 2019, 10, 2892–2906. [Google Scholar] [CrossRef] [Green Version]
- Walker, N.D.; Elias, M.; Guiro, K.; Bhatia, R.; Greco, S.J.; Bryan, M.; Gergues, M.; Sandiford, O.A.; Ponzio, N.M.; Leibovich, S.J.; et al. Exosomes from differentially activated macrophages influence dormancy or resurgence of breast cancer cells within bone marrow stroma. Cell Death Dis. 2019, 10, 59. [Google Scholar] [CrossRef] [Green Version]
- Moradi-Chaleshtori, M.; Bandehpour, M.; Heidari, N.; Mohammadi-Yeganeh, S.; Mahmoud Hashemi, S. Exosome-mediated miR-33 transfer induces M1 polarization in mouse macrophages and exerts antitumor effect in 4T1 breast cancer cell line. Int. Immunopharmacol. 2021, 90, 107198. [Google Scholar] [CrossRef]
- Stanton, S.E.; Disis, M.L. Clinical significance of tumor-infiltrating lymphocytes in breast cancer. J. Immunother. Cancer 2016, 4, 59. [Google Scholar] [CrossRef] [Green Version]
- Salgado, R.; Loi, S. Tumour infiltrating lymphocytes in breast cancer: Increasing clinical relevance. Lancet Oncol. 2018, 19, 3–5. [Google Scholar] [CrossRef]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gogenur, I. Cytotoxic CD8(+) T cells in cancer and cancer immunotherapy. Br. J. Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Knutson, K.L.; Disis, M.L. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol. Immunother. 2005, 54, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Cilibrasi, C.; Papanastasopoulos, P.; Samuels, M.; Giamas, G. Reconstituting Immune Surveillance in Breast Cancer: Molecular Pathophysiology and Current Immunotherapy Strategies. Int. J. Mol. Sci. 2021, 22, 2015. [Google Scholar] [CrossRef] [PubMed]
- Shang, B.; Liu, Y.; Jiang, S.J.; Liu, Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef] [Green Version]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Yang, Y.; Li, C.W.; Chan, L.C.; Wei, Y.; Hsu, J.M.; Xia, W.; Cha, J.H.; Hou, J.; Hsu, J.L.; Sun, L.; et al. Exosomal PD-L1 harbors active defense function to suppress T cell killing of breast cancer cells and promote tumor growth. Cell Res. 2018, 28, 862–864. [Google Scholar] [CrossRef] [Green Version]
- Monypenny, J.; Milewicz, H.; Flores-Borja, F.; Weitsman, G.; Cheung, A.; Chowdhury, R.; Burgoyne, T.; Arulappu, A.; Lawler, K.; Barber, P.R.; et al. ALIX Regulates Tumor-Mediated Immunosuppression by Controlling EGFR Activity and PD-L1 Presentation. Cell Rep 2018, 24, 630–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Rong, L.; Li, R.; Li, S.; Luo, R. Immunosuppression of breast cancer cells mediated by transforming growth factor-beta in exosomes from cancer cells. Oncol. Lett. 2016, 11, 500–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S.; Chatterjee, A.; Jana, S.; Dey, S.; Roy, H.; Das, M.K.; Alam, J.; Adhikary, A.; Chowdhury, A.; Biswas, A.; et al. Transforming growth factor beta orchestrates PD-L1 enrichment in tumor-derived exosomes and mediates CD8 T-cell dysfunction regulating early phosphorylation of TCR signalome in breast cancer. Carcinogenesis 2021, 42, 38–47. [Google Scholar] [CrossRef]
- Poggio, M.; Hu, T.; Pai, C.C.; Chu, B.; Belair, C.D.; Chang, A.; Montabana, E.; Lang, U.E.; Fu, Q.; Fong, L.; et al. Suppression of Exosomal PD-L1 Induces Systemic Anti-tumor Immunity and Memory. Cell 2019, 177, 414–427. [Google Scholar] [CrossRef] [Green Version]
- Wen, S.W.; Sceneay, J.; Lima, L.G.; Wong, C.S.; Becker, M.; Krumeich, S.; Lobb, R.J.; Castillo, V.; Wong, K.N.; Ellis, S.; et al. The Biodistribution and Immune Suppressive Effects of Breast Cancer-Derived Exosomes. Cancer Res. 2016, 76, 6816–6827. [Google Scholar] [CrossRef] [Green Version]
- Ni, C.; Fang, Q.Q.; Chen, W.Z.; Jiang, J.X.; Jiang, Z.; Ye, J.; Zhang, T.; Yang, L.; Meng, F.B.; Xia, W.J.; et al. Breast cancer-derived exosomes transmit lncRNA SNHG16 to induce CD73+gammadelta1 Treg cells. Signal Transduct. Target. Ther. 2020, 5, 41. [Google Scholar] [CrossRef]
- Ning, Y.; Shen, K.; Wu, Q.; Sun, X.; Bai, Y.; Xie, Y.; Pan, J.; Qi, C. Tumor exosomes block dendritic cells maturation to decrease the T cell immune response. Immunol. Lett. 2018, 199, 36–43. [Google Scholar] [CrossRef]
- Schwich, E.; Ho, G.T.; LeMaoult, J.; Bade-Doding, C.; Carosella, E.D.; Horn, P.A.; Rebmann, V. Soluble HLA-G and HLA-G Bearing Extracellular Vesicles Affect ILT-2 Positive and ILT-2 Negative CD8 T Cells Complementary. Front. Immunol. 2020, 11, 2046. [Google Scholar] [CrossRef]
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 2014, 26, 638–652. [Google Scholar] [CrossRef] [Green Version]
- Palucka, K.; Coussens, L.M.; O’Shaughnessy, J. Dendritic cells, inflammation, and breast cancer. Cancer J. 2013, 19, 511–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudek, A.M.; Martin, S.; Garg, A.D.; Agostinis, P. Immature, Semi-Mature, and Fully Mature Dendritic Cells: Toward a DC-Cancer Cells Interface That Augments Anticancer Immunity. Front. Immunol. 2013, 4, 438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfers, J.; Lozier, A.; Raposo, G.; Regnault, A.; Thery, C.; Masurier, C.; Flament, C.; Pouzieux, S.; Faure, F.; Tursz, T.; et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat. Med. 2001, 7, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Zeng, W.; Wu, B.; Wang, L.; Wang, Z.; Tian, H.; Wang, L.; Jiang, Y.; Clay, R.; Wei, X.; et al. PPARalpha Inhibition Overcomes Tumor-Derived Exosomal Lipid-Induced Dendritic Cell Dysfunction. Cell Rep. 2020, 33, 108278. [Google Scholar] [CrossRef] [PubMed]
- Diamond, J.M.; Vanpouille-Box, C.; Spada, S.; Rudqvist, N.P.; Chapman, J.R.; Ueberheide, B.M.; Pilones, K.A.; Sarfraz, Y.; Formenti, S.C.; Demaria, S. Exosomes Shuttle TREX1-Sensitive IFN-Stimulatory dsDNA from Irradiated Cancer Cells to DCs. Cancer Immunol. Res. 2018, 6, 910–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; Demaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef]
- Zitvogel, L.; Regnault, A.; Lozier, A.; Wolfers, J.; Flament, C.; Tenza, D.; Ricciardi-Castagnoli, P.; Raposo, G.; Amigorena, S. Eradication of established murine tumors using a novel cell-free vaccine: Dendritic cell-derived exosomes. Nat. Med. 1998, 4, 594–600. [Google Scholar] [CrossRef]
- Markowitz, J.; Wesolowski, R.; Papenfuss, T.; Brooks, T.R.; Carson, W.E., 3rd. Myeloid-derived suppressor cells in breast cancer. Breast Cancer Res. Treat. 2013, 140, 13–21. [Google Scholar] [CrossRef]
- Diaz-Montero, C.M.; Salem, M.L.; Nishimura, M.I.; Garrett-Mayer, E.; Cole, D.J.; Montero, A.J. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol. Immunother. 2009, 58, 49–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashid, M.H.; Borin, T.F.; Ara, R.; Piranlioglu, R.; Achyut, B.R.; Korkaya, H.; Liu, Y.; Arbab, A.S. Critical immunosuppressive effect of MDSCderived exosomes in the tumor microenvironment. Oncol. Rep. 2021, 45, 1171–1181. [Google Scholar] [CrossRef]
- Liu, Q.W.; Chen, Y.; Li, J.Y.; Xiao, L.; Zhang, W.J.; Zhao, J.L.; Gu, H.C.; Wu, H.Y.; Zuo, G.S.; Deng, K.Y.; et al. Bone marrow cells are differentiated into MDSCs by BCC-Ex through down-regulating the expression of CXCR4 and activating STAT3 signalling pathway. J. Cell. Mol. Med. 2021, 25, 5497–5510. [Google Scholar] [CrossRef]
- Deng, Z.; Rong, Y.; Teng, Y.; Zhuang, X.; Samykutty, A.; Mu, J.; Zhang, L.; Cao, P.; Yan, J.; Miller, D.; et al. Exosomes miR-126a released from MDSC induced by DOX treatment promotes lung metastasis. Oncogene 2017, 36, 639–651. [Google Scholar] [CrossRef] [Green Version]
- Gu, P.; Sun, M.; Li, L.; Yang, Y.; Jiang, Z.; Ge, Y.; Wang, W.; Mu, W.; Wang, H. Breast Tumor-Derived Exosomal MicroRNA-200b-3p Promotes Specific Organ Metastasis Through Regulating CCL2 Expression in Lung Epithelial Cells. Front. Cell Dev. Biol. 2021, 9, 657158. [Google Scholar] [CrossRef] [PubMed]
- Guerra, N.; Tan, Y.X.; Joncker, N.T.; Choy, A.; Gallardo, F.; Xiong, N.; Knoblaugh, S.; Cado, D.; Greenberg, N.M.; Raulet, D.H. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity 2008, 28, 571–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.Y.; Fu, T.; Jiang, Y.Z.; Shao, Z.M. Natural killer cells in cancer biology and therapy. Mol. Cancer 2020, 19, 120. [Google Scholar] [CrossRef]
- Zhang, H.G.; Kim, H.; Liu, C.; Yu, S.; Wang, J.; Grizzle, W.E.; Kimberly, R.P.; Barnes, S. Curcumin reverses breast tumor exosomes mediated immune suppression of NK cell tumor cytotoxicity. Biochim. Biophys. Acta 2007, 1773, 1116–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosales, C. Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types? Front. Physiol. 2018, 9, 113. [Google Scholar] [CrossRef]
- Gago-Dominguez, M.; Matabuena, M.; Redondo, C.M.; Patel, S.P.; Carracedo, A.; Ponte, S.M.; Martinez, M.E.; Castelao, J.E. Neutrophil to lymphocyte ratio and breast cancer risk: Analysis by subtype and potential interactions. Sci. Rep. 2020, 10, 13203. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leal, A.C.; Mizurini, D.M.; Gomes, T.; Rochael, N.C.; Saraiva, E.M.; Dias, M.S.; Werneck, C.C.; Sielski, M.S.; Vicente, C.P.; Monteiro, R.Q. Tumor-Derived Exosomes Induce the Formation of Neutrophil Extracellular Traps: Implications For The Establishment of Cancer-Associated Thrombosis. Sci. Rep. 2017, 7, 6438. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.H.; Xue, L.; Hsu, C.C.; Paez, J.S.; Pan, L.; Andaluz, H.; Wendt, M.K.; Iliuk, A.B.; Zhu, J.K.; Tao, W.A. Phosphoproteins in extracellular vesicles as candidate markers for breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 3175–3180. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Liu, C.; Pan, W.; Shen, J.; Guo, J.; Luo, T.; Feng, J.; Situ, B.; An, T.; Zhang, Y.; et al. Facile fluorescent aptasensor using aggregation-induced emission luminogens for exosomal proteins profiling towards liquid biopsy. Biosens. Bioelectron. 2020, 168, 112520. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shi, J.; Zhang, H.; Zhu, Y.; Liu, W.; Zhang, K.; Zhang, Z. Localized fluorescent imaging of multiple proteins on individual extracellular vesicles using rolling circle amplification for cancer diagnosis. J. Extracell. Vesicles 2020, 10, e12025. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Tian, H.; Li, X.; Jin, D.; Li, X.; Kong, J.; Yang, C.; Yang, X.; Lu, Y.; Luo, Y.; et al. Clinical application of a microfluidic chip for immunocapture and quantification of circulating exosomes to assist breast cancer diagnosis and molecular classification. PLoS ONE 2017, 12, e0175050. [Google Scholar] [CrossRef] [Green Version]
- Bandini, E.; Rossi, T.; Scarpi, E.; Gallerani, G.; Vannini, I.; Salvi, S.; Azzali, I.; Melloni, M.; Salucci, S.; Battistelli, M.; et al. Early Detection and Investigation of Extracellular Vesicles Biomarkers in Breast Cancer. Front. Mol. Biosci. 2021, 8, 732900. [Google Scholar] [CrossRef] [PubMed]
- Moon, P.G.; Lee, J.E.; Cho, Y.E.; Lee, S.J.; Jung, J.H.; Chae, Y.S.; Bae, H.I.; Kim, Y.B.; Kim, I.S.; Park, H.Y.; et al. Identification of Developmental Endothelial Locus-1 on Circulating Extracellular Vesicles as a Novel Biomarker for Early Breast Cancer Detection. Clin. Cancer Res. 2016, 22, 1757–1766. [Google Scholar] [CrossRef] [Green Version]
- Ortega, F.G.; Regiart, M.D.; Rodriguez-Martinez, A.; de Miguel-Perez, D.; Serrano, M.J.; Lorente, J.A.; Tortella, G.; Rubilar, O.; Sapag, K.; Bertotti, M.; et al. Sandwich-Type Electrochemical Paper-Based Immunosensor for Claudin 7 and CD81 Dual Determination on Extracellular Vesicles from Breast Cancer Patients. Anal. Chem. 2021, 93, 1143–1153. [Google Scholar] [CrossRef] [PubMed]
- Ando, W.; Kikuchi, K.; Uematsu, T.; Yokomori, H.; Takaki, T.; Sogabe, M.; Kohgo, Y.; Otori, K.; Ishikawa, S.; Okazaki, I. Novel breast cancer screening: Combined expression of miR-21 and MMP-1 in urinary exosomes detects 95% of breast cancer without metastasis. Sci. Rep. 2019, 9, 13595. [Google Scholar] [CrossRef] [PubMed]
- Zhong, G.; Wang, K.; Li, J.; Xiao, S.; Wei, W.; Liu, J. Determination of Serum Exosomal H19 as a Noninvasive Biomarker for Breast Cancer Diagnosis. Onco Targets Ther. 2020, 13, 2563–2571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Mao, J.H.; Wang, B.Y.; Wang, L.X.; Wen, H.Y.; Xu, L.J.; Fu, J.X.; Yang, H. Exosomal miR-1910-3p promotes proliferation, metastasis, and autophagy of breast cancer cells by targeting MTMR3 and activating the NF-kappaB signaling pathway. Cancer Lett. 2020, 489, 87–99. [Google Scholar] [CrossRef]
- Rodriguez-Martinez, A.; de Miguel-Perez, D.; Ortega, F.G.; Garcia-Puche, J.L.; Robles-Fernandez, I.; Exposito, J.; Martorell-Marugan, J.; Carmona-Saez, P.; Garrido-Navas, M.D.C.; Rolfo, C.; et al. Exosomal miRNA profile as complementary tool in the diagnostic and prediction of treatment response in localized breast cancer under neoadjuvant chemotherapy. Breast Cancer Res. 2019, 21, 21. [Google Scholar] [CrossRef]
- Vinik, Y.; Ortega, F.G.; Mills, G.B.; Lu, Y.; Jurkowicz, M.; Halperin, S.; Aharoni, M.; Gutman, M.; Lev, S. Proteomic analysis of circulating extracellular vesicles identifies potential markers of breast cancer progression, recurrence, and response. Sci. Adv. 2020, 6, eaba5714. [Google Scholar] [CrossRef] [PubMed]
- Konig, L.; Kasimir-Bauer, S.; Bittner, A.K.; Hoffmann, O.; Wagner, B.; Santos Manvailer, L.F.; Kimmig, R.; Horn, P.A.; Rebmann, V. Elevated levels of extracellular vesicles are associated with therapy failure and disease progression in breast cancer patients undergoing neoadjuvant chemotherapy. Oncoimmunology 2017, 7, e1376153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia Garre, E.; Luengo Gil, G.; Montoro Garcia, S.; Gonzalez Billalabeitia, E.; Zafra Poves, M.; Garcia Martinez, E.; Roldan Schilling, V.; Navarro Manzano, E.; Ivars Rubio, A.; Lip, G.Y.H.; et al. Circulating small-sized endothelial microparticles as predictors of clinical outcome after chemotherapy for breast cancer: An exploratory analysis. Breast Cancer Res. Treat. 2018, 169, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Stevic, I.; Muller, V.; Weber, K.; Fasching, P.A.; Karn, T.; Marme, F.; Schem, C.; Stickeler, E.; Denkert, C.; van Mackelenbergh, M.; et al. Specific microRNA signatures in exosomes of triple-negative and HER2-positive breast cancer patients undergoing neoadjuvant therapy within the GeparSixto trial. BMC Med. 2018, 16, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvador-Coloma, C.; Santaballa, A.; Sanmartin, E.; Calvo, D.; Garcia, A.; Hervas, D.; Cordon, L.; Quintas, G.; Ripoll, F.; Panadero, J.; et al. Immunosuppressive profiles in liquid biopsy at diagnosis predict response to neoadjuvant chemotherapy in triple-negative breast cancer. Eur. J. Cancer 2020, 139, 119–134. [Google Scholar] [CrossRef]
- Dong, H.; Wang, W.; Chen, R.; Zhang, Y.; Zou, K.; Ye, M.; He, X.; Zhang, F.; Han, J. Exosome-mediated transfer of lncRNASNHG14 promotes trastuzumab chemoresistance in breast cancer. Int. J. Oncol. 2018, 53, 1013–1026. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Zheng, K.; Tang, Y.; Li, Z.; Zou, T.; Liu, D. Overexpression of serum exosomal HOTAIR is correlated with poor survival and poor response to chemotherapy in breast cancer patients. J. Biosci. 2019, 44, 37. [Google Scholar] [CrossRef]
- Wang, T.; Ning, K.; Lu, T.X.; Sun, X.; Jin, L.; Qi, X.; Jin, J.; Hua, D. Increasing circulating exosomes-carrying TRPC5 predicts chemoresistance in metastatic breast cancer patients. Cancer Sci. 2017, 108, 448–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, K.; Wang, T.; Sun, X.; Zhang, P.; Chen, Y.; Jin, J.; Hua, D. UCH-L1-containing exosomes mediate chemotherapeutic resistance transfer in breast cancer. J. Surg. Oncol. 2017, 115, 932–940. [Google Scholar] [CrossRef]
- Martinez, V.G.; O’Neill, S.; Salimu, J.; Breslin, S.; Clayton, A.; Crown, J.; O’Driscoll, L. Resistance to HER2-targeted anti-cancer drugs is associated with immune evasion in cancer cells and their derived extracellular vesicles. Oncoimmunology 2017, 6, e1362530. [Google Scholar] [CrossRef] [PubMed]
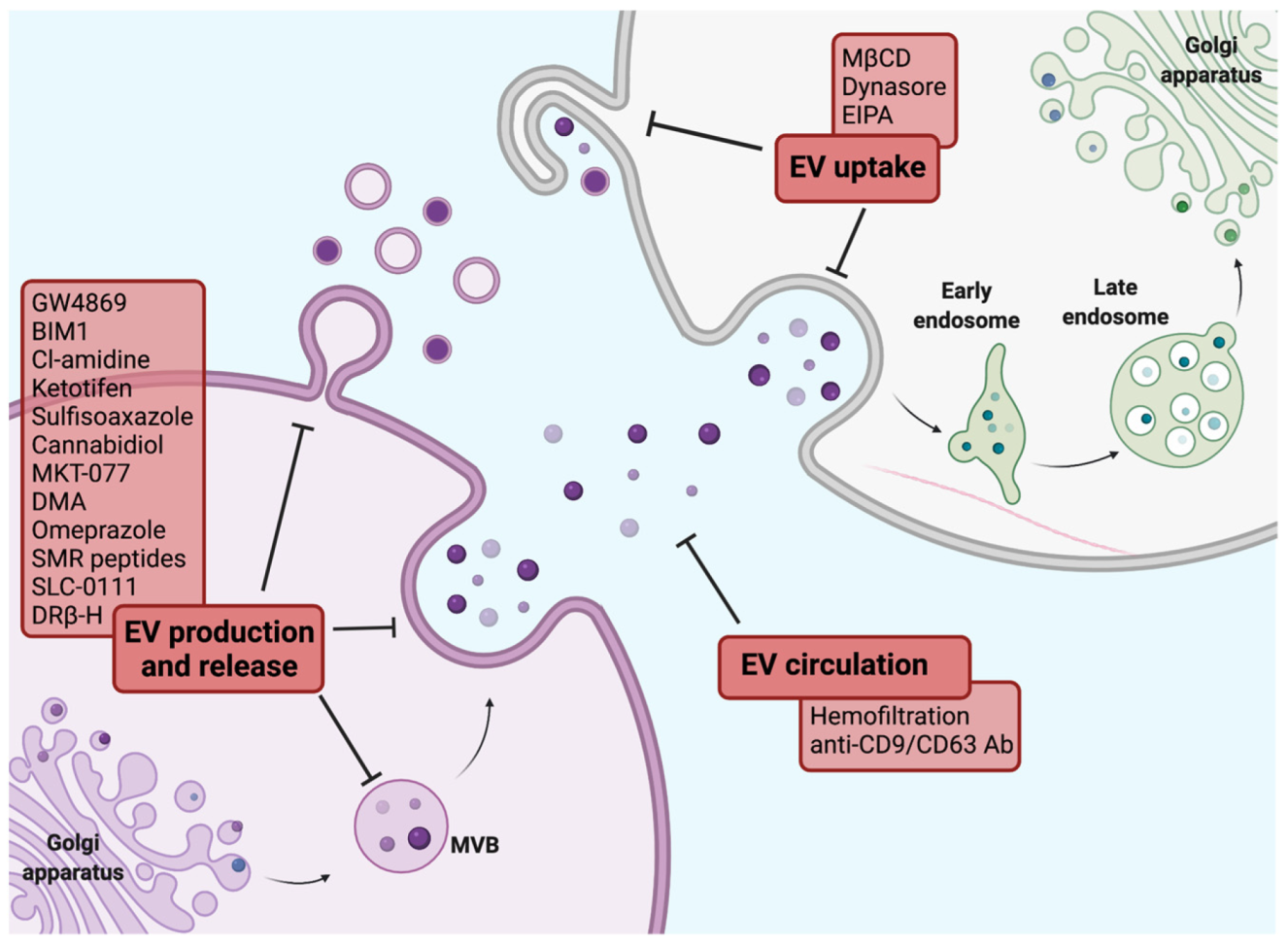
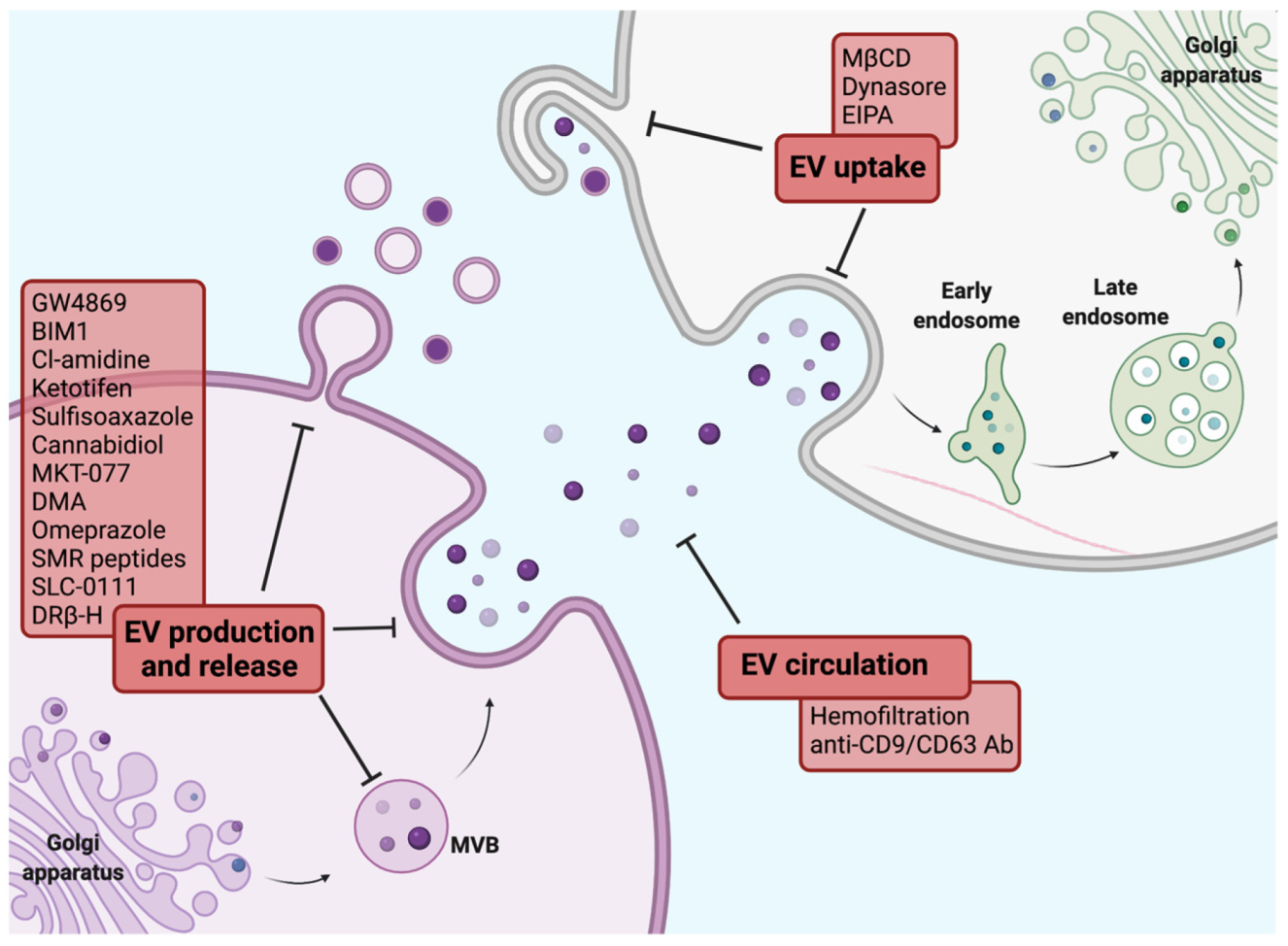
- Hayatudin, R.; Fong, Z.; Ming, L.C.; Goh, B.H.; Lee, W.L.; Kifli, N. Overcoming Chemoresistance via Extracellular Vesicle Inhibition. Front. Mol. Biosci. 2021, 8, 629874. [Google Scholar] [CrossRef] [PubMed]
- Catalano, M.; O’Driscoll, L. Inhibiting extracellular vesicles formation and release: A review of EV inhibitors. J. Extracell. Vesicles 2020, 9, 1703244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kavanagh, E.L.; Lindsay, S.; Halasz, M.; Gubbins, L.C.; Weiner-Gorzel, K.; Guang, M.H.Z.; McGoldrick, A.; Collins, E.; Henry, M.; Blanco-Fernandez, A.; et al. Protein and chemotherapy profiling of extracellular vesicles harvested from therapeutic induced senescent triple negative breast cancer cells. Oncogenesis 2017, 6, e388. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Li, Y.; Peng, S.; Li, J.; Qin, X. Exosomal-mediated transfer of OIP5-AS1 enhanced cell chemoresistance to trastuzumab in breast cancer via up-regulating HMGB3 by sponging miR-381-3p. Open Med. 2021, 16, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Gu, Y.; Lu, P.; Li, J.; Cao, H.; Li, X.; Qian, X.; Yu, C.; Yang, Y.; Yang, X.; et al. Exosome-mediated lncRNA AFAP1-AS1 promotes trastuzumab resistance through binding with AUF1 and activating ERBB2 translation. Mol. Cancer 2020, 19, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosgodage, U.S.; Trindade, R.P.; Thompson, P.R.; Inal, J.M.; Lange, S. Chloramidine/Bisindolylmaleimide-I-Mediated Inhibition of Exosome and Microvesicle Release and Enhanced Efficacy of Cancer Chemotherapy. Int. J. Mol. Sci. 2017, 18, 1007. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Berger, S.A. Ketotifen reverses MDR1-mediated multidrug resistance in human breast cancer cells in vitro and alleviates cardiotoxicity induced by doxorubicin in vivo. Cancer Chemother. Pharmacol. 2003, 51, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Park, M.K.; Kim, S.Y.; Lee, C.H. Novel Suppressive Effects of Ketotifen on Migration and Invasion of MDA-MB-231 and HT-1080 Cancer Cells. Biomol. Ther. 2014, 22, 540–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Crump, M.; Berge, S.A. Purging of contaminating breast cancer cells from hematopoietic progenitor cell preparations using activation enhanced cell death. Breast Cancer Res. Treat. 2002, 72, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Khan, F.M.; Saleh, E.; Alawadhi, H.; Harati, R.; Zimmermann, W.H.; El-Awady, R. Inhibition of exosome release by ketotifen enhances sensitivity of cancer cells to doxorubicin. Cancer Biol. Ther. 2018, 19, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Im, E.J.; Lee, C.H.; Moon, P.G.; Rangaswamy, G.G.; Lee, B.; Lee, J.M.; Lee, J.C.; Jee, J.G.; Bae, J.S.; Kwon, T.K.; et al. Sulfisoxazole inhibits the secretion of small extracellular vesicles by targeting the endothelin receptor A. Nat. Commun. 2019, 10, 1387. [Google Scholar] [CrossRef] [PubMed]
- Pisanti, S.; Malfitano, A.M.; Ciaglia, E.; Lamberti, A.; Ranieri, R.; Cuomo, G.; Abate, M.; Faggiana, G.; Proto, M.C.; Fiore, D.; et al. Cannabidiol: State of the art and new challenges for therapeutic applications. Pharmacol. Ther. 2017, 175, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Kosgodage, U.S.; Mould, R.; Henley, A.B.; Nunn, A.V.; Guy, G.W.; Thomas, E.L.; Inal, J.M.; Bell, J.D.; Lange, S. Cannabidiol (CBD) Is a Novel Inhibitor for Exosome and Microvesicle (EMV) Release in Cancer. Front. Pharmacol. 2018, 9, 889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, Q.; Wadhwa, R.; Kawai, R.; Kaul, S.C.; Sifers, R.N.; Bick, R.J.; Smith, J.R.; Pereira-Smith, O.M. Extramitochondrial localization of mortalin/mthsp70/PBP74/GRP75. Biochem. Biophys. Res. Commun. 2000, 275, 174–179. [Google Scholar] [CrossRef]
- Huang, M.B.; Gonzalez, R.R.; Lillard, J.; Bond, V.C. Secretion modification region-derived peptide blocks exosome release and mediates cell cycle arrest in breast cancer cells. Oncotarget 2017, 8, 11302–11315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.B.; Wu, J.Y.; Lillard, J.; Bond, V.C. SMR peptide antagonizes mortalin promoted release of extracellular vesicles and affects mortalin protection from complement-dependent cytotoxicity in breast cancer cells and leukemia cells. Oncotarget 2019, 10, 5419–5438. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Knowlton, A.A. HSP60 trafficking in adult cardiac myocytes: Role of the exosomal pathway. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H3052–H3056. [Google Scholar] [CrossRef] [Green Version]
- Ghiringhelli, F.; Larmonier, N.; Schmitt, E.; Parcellier, A.; Cathelin, D.; Garrido, C.; Chauffert, B.; Solary, E.; Bonnotte, B.; Martin, F. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur. J. Immunol. 2004, 34, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Chalmin, F.; Ladoire, S.; Mignot, G.; Vincent, J.; Bruchard, M.; Remy-Martin, J.P.; Boireau, W.; Rouleau, A.; Simon, B.; Lanneau, D.; et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J. Clin. Investig. 2010, 120, 457–471. [Google Scholar] [CrossRef]
- Bobrie, A.; Krumeich, S.; Reyal, F.; Recchi, C.; Moita, L.F.; Seabra, M.C.; Ostrowski, M.; Thery, C. Rab27a supports exosome-dependent and -independent mechanisms that modify the tumor microenvironment and can promote tumor progression. Cancer Res. 2012, 72, 4920–4930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.X.; Xu, L.Y.; Qian, Q.; He, X.; Peng, W.T.; Fan, W.Q.; Zhu, Y.L.; Tang, J.H.; Cheng, L. d Rhamnose beta-hederin reverses chemoresistance of breast cancer cells by regulating exosome-mediated resistance transmission. BioSci. Rep. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef] [Green Version]
- Logozzi, M.; Mizzoni, D.; Angelini, D.F.; Di Raimo, R.; Falchi, M.; Battistini, L.; Fais, S. Microenvironmental pH and Exosome Levels Interplay in Human Cancer Cell Lines of Different Histotypes. Cancers 2018, 10, 370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, P.C.; Chia, S.; Bedard, P.L.; Chu, Q.; Lyle, M.; Tang, L.; Singh, M.; Zhang, Z.; Supuran, C.T.; Renouf, D.J.; et al. A Phase 1 Study of SLC-0111, a Novel Inhibitor of Carbonic Anhydrase IX, in Patients With Advanced Solid Tumors. Am. J. Clin. Oncol. 2020, 43, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.; McDonald, P.C.; Oloumi, A.; Chia, S.; Ostlund, C.; Ahmadi, A.; Kyle, A.; Auf dem Keller, U.; Leung, S.; Huntsman, D.; et al. Targeting tumor hypoxia: Suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res. 2011, 71, 3364–3376. [Google Scholar] [CrossRef] [Green Version]
- Lock, F.E.; McDonald, P.C.; Lou, Y.; Serrano, I.; Chafe, S.C.; Ostlund, C.; Aparicio, S.; Winum, J.Y.; Supuran, C.T.; Dedhar, S. Targeting carbonic anhydrase IX depletes breast cancer stem cells within the hypoxic niche. Oncogene 2013, 32, 5210–5219. [Google Scholar] [CrossRef]
- Hedlund, E.E.; McDonald, P.C.; Nemirovsky, O.; Awrey, S.; Jensen, L.D.E.; Dedhar, S. Harnessing Induced Essentiality: Targeting Carbonic Anhydrase IX and Angiogenesis Reduces Lung Metastasis of Triple Negative Breast Cancer Xenografts. Cancers 2019, 11, 1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chafe, S.C.; Lou, Y.; Sceneay, J.; Vallejo, M.; Hamilton, M.J.; McDonald, P.C.; Bennewith, K.L.; Moller, A.; Dedhar, S. Carbonic anhydrase IX promotes myeloid-derived suppressor cell mobilization and establishment of a metastatic niche by stimulating G-CSF production. Cancer Res. 2015, 75, 996–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreucci, E.; Ruzzolini, J.; Peppicelli, S.; Bianchini, F.; Laurenzana, A.; Carta, F.; Supuran, C.T.; Calorini, L. The carbonic anhydrase IX inhibitor SLC-0111 sensitises cancer cells to conventional chemotherapy. J. Enzyme Inhib. Med. Chem. 2019, 34, 117–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koumangoye, R.B.; Sakwe, A.M.; Goodwin, J.S.; Patel, T.; Ochieng, J. Detachment of breast tumor cells induces rapid secretion of exosomes which subsequently mediate cellular adhesion and spreading. PLoS ONE 2011, 6, e24234. [Google Scholar] [CrossRef] [Green Version]
- Menck, K.; Scharf, C.; Bleckmann, A.; Dyck, L.; Rost, U.; Wenzel, D.; Dhople, V.M.; Siam, L.; Pukrop, T.; Binder, C.; et al. Tumor-derived microvesicles mediate human breast cancer invasion through differentially glycosylated EMMPRIN. J. Mol. Cell Biol. 2015, 7, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, A.I. Pharmacological inhibition of endocytic pathways: Is it specific enough to be useful? Methods Mol. Biol. 2008, 440, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Jayashankar, V.; Edinger, A.L. Macropinocytosis confers resistance to therapies targeting cancer anabolism. Nat. Commun. 2020, 11, 1121. [Google Scholar] [CrossRef]
- Marleau, A.M.; Chen, C.S.; Joyce, J.A.; Tullis, R.H. Exosome removal as a therapeutic adjuvant in cancer. J. Transl. Med. 2012, 10, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida-Aoki, N.; Tominaga, N.; Takeshita, F.; Sonoda, H.; Yoshioka, Y.; Ochiya, T. Disruption of Circulating Extracellular Vesicles as a Novel Therapeutic Strategy against Cancer Metastasis. Mol. Ther. 2017, 25, 181–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gobbo, J.; Marcion, G.; Cordonnier, M.; Dias, A.M.M.; Pernet, N.; Hammann, A.; Richaud, S.; Mjahed, H.; Isambert, N.; Clausse, V.; et al. Restoring Anticancer Immune Response by Targeting Tumor-Derived Exosomes With a HSP70 Peptide Aptamer. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Pikina, E.S.; Ostrovskii, B.I. Nucleation and growth of droplets in the overheated free-standing smectic films. Eur. Phys. J. E Soft Matter 2017, 40, 24. [Google Scholar] [CrossRef]
- Shi, X.; Cheng, Q.; Hou, T.; Han, M.; Smbatyan, G.; Lang, J.E.; Epstein, A.L.; Lenz, H.J.; Zhang, Y. Genetically Engineered Cell-Derived Nanoparticles for Targeted Breast Cancer Immunotherapy. Mol. Ther. 2020, 28, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Shi, X.; Han, M.; Smbatyan, G.; Lenz, H.J.; Zhang, Y. Reprogramming Exosomes as Nanoscale Controllers of Cellular Immunity. J. Am. Chem. Soc. 2018, 140, 16413–16417. [Google Scholar] [CrossRef] [PubMed]
- Taghikhani, A.; Hassan, Z.M.; Ebrahimi, M.; Moazzeni, S.M. microRNA modified tumor-derived exosomes as novel tools for maturation of dendritic cells. J. Cell. Physiol. 2019, 234, 9417–9427. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Dorval, T.; Chaput, N.; Andre, F.; Caby, M.P.; Novault, S.; Flament, C.; Leboulaire, C.; Borg, C.; Amigorena, S.; et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: Results of thefirst phase I clinical trial. J. Transl. Med. 2005, 3, 10. [Google Scholar] [CrossRef] [Green Version]
- Morse, M.A.; Garst, J.; Osada, T.; Khan, S.; Hobeika, A.; Clay, T.M.; Valente, N.; Shreeniwas, R.; Sutton, M.A.; Delcayre, A.; et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J. Transl. Med. 2005, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Bu, N.; Wu, H.; Sun, B.; Zhang, G.; Zhan, S.; Zhang, R.; Zhou, L. Exosome-loaded dendritic cells elicit tumor-specific CD8+ cytotoxic T cells in patients with glioma. J. Neurooncol. 2011, 104, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Chibbar, R.; Xiang, J. Novel EXO-T vaccine using polyclonal CD4(+) T cells armed with HER2-specific exosomes for HER2-positive breast cancer. Onco Targets Ther. 2018, 11, 7089–7093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Wu, J.; Xu, A.; Ahmeqd, S.; Sami, A.; Chibbar, R.; Freywald, A.; Zheng, C.; Xiang, J. Heterologous human/rat HER2-specific exosome-targeted T cell vaccine stimulates potent humoral and CTL responses leading to enhanced circumvention of HER2 tolerance in double transgenic HLA-A2/HER2 mice. Vaccine 2018, 36, 1414–1422. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Bai, X.; Ni, J.; Zhang, H.; Duan, W.; Graham, P.; Li, Y. Exosomes and breast cancer drug resistance. Cell Death Dis. 2020, 11, 987. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Oller, L.; Seras-Franzoso, J.; Andrade, F.; Rafael, D.; Abasolo, I.; Gener, P.; Schwartz, S., Jr. Extracellular Vesicles as Drug Delivery Systems in Cancer. Pharmaceutics 2020, 12, 1146. [Google Scholar] [CrossRef]
- Zhao, X.; Wu, D.; Ma, X.; Wang, J.; Hou, W.; Zhang, W. Exosomes as drug carriers for cancer therapy and challenges regarding exosome uptake. Biomed. Pharmacother. 2020, 128, 110237. [Google Scholar] [CrossRef] [PubMed]
- Luan, X.; Sansanaphongpricha, K.; Myers, I.; Chen, H.; Yuan, H.; Sun, D. Engineering exosomes as refined biological nanoplatforms for drug delivery. Acta Pharmacol. Sin. 2017, 38, 754–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ye, Z.; Xiang, M.; Chang, B.; Cui, J.; Ji, T.; Zhao, L.; Li, Q.; Deng, Y.; Xu, L.; et al. Functional extracellular vesicles engineered with lipid-grafted hyaluronic acid effectively reverse cancer drug resistance. Biomaterials 2019, 223, 119475. [Google Scholar] [CrossRef] [PubMed]
- Pi, F.; Binzel, D.W.; Lee, T.J.; Li, Z.; Sun, M.; Rychahou, P.; Li, H.; Haque, F.; Wang, S.; Croce, C.M.; et al. Nanoparticle orientation to control RNA loading and ligand display on extracellular vesicles for cancer regression. Nat. Nanotechnol. 2018, 13, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T.; et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther. 2013, 21, 185–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Chen, X.; Tian, B.; Liu, J.; Yang, L.; Zeng, L.; Chen, T.; Hong, A.; Wang, X. Nucleolin-targeted Extracellular Vesicles as a Versatile Platform for Biologics Delivery to Breast Cancer. Theranostics 2017, 7, 1360–1372. [Google Scholar] [CrossRef]
- O’Brien, K.P.; Khan, S.; Gilligan, K.E.; Zafar, H.; Lalor, P.; Glynn, C.; O’Flatharta, C.; Ingoldsby, H.; Dockery, P.; De Bhulbh, A.; et al. Employing mesenchymal stem cells to support tumor-targeted delivery of extracellular vesicle (EV)-encapsulated microRNA-379. Oncogene 2018, 37, 2137–2149. [Google Scholar] [CrossRef] [Green Version]
- Naseri, Z.; Oskuee, R.K.; Forouzandeh-Moghadam, M.; Jaafari, M.R. Delivery of LNA-antimiR-142-3p by Mesenchymal Stem Cells-Derived Exosomes to Breast Cancer Stem Cells Reduces Tumorigenicity. Stem Cell Rev. Rep. 2020, 16, 541–556. [Google Scholar] [CrossRef]
- Yuan, Z.; Kolluri, K.K.; Gowers, K.H.; Janes, S.M. TRAIL delivery by MSC-derived extracellular vesicles is an effective anticancer therapy. J. Extracell. Vesicles 2017, 6, 1265291. [Google Scholar] [CrossRef]
- Wang, P.; Wang, H.; Huang, Q.; Peng, C.; Yao, L.; Chen, H.; Qiu, Z.; Wu, Y.; Wang, L.; Chen, W. Exosomes from M1-Polarized Macrophages Enhance Paclitaxel Antitumor Activity by Activating Macrophages-Mediated Inflammation. Theranostics 2019, 9, 1714–1727. [Google Scholar] [CrossRef]
- Fan, Z.; Xiao, K.; Lin, J.; Liao, Y.; Huang, X. Functionalized DNA Enables Programming Exosomes/Vesicles for Tumor Imaging and Therapy. Small 2019, 15, e1903761. [Google Scholar] [CrossRef]
- Li, S.; Wu, Y.; Ding, F.; Yang, J.; Li, J.; Gao, X.; Zhang, C.; Feng, J. Engineering macrophage-derived exosomes for targeted chemotherapy of triple-negative breast cancer. Nanoscale 2020, 12, 10854–10862. [Google Scholar] [CrossRef]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef]
- Gong, C.; Tian, J.; Wang, Z.; Gao, Y.; Wu, X.; Ding, X.; Qiang, L.; Li, G.; Han, Z.; Yuan, Y.; et al. Functional exosome-mediated co-delivery of doxorubicin and hydrophobically modified microRNA 159 for triple-negative breast cancer therapy. J. Nanobiotechnol. 2019, 17, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, R.; Wang, Z.; Niu, R.; Wang, H.; Guan, W.; Chang, J. Tumor Exosome Mimicking Nanoparticles for Tumor Combinatorial Chemo-Photothermal Therapy. Front. Bioeng. Biotechnol. 2020, 8, 1010. [Google Scholar] [CrossRef] [PubMed]
- Kanada, M.; Kim, B.D.; Hardy, J.W.; Ronald, J.A.; Bachmann, M.H.; Bernard, M.P.; Perez, G.I.; Zarea, A.A.; Ge, T.J.; Withrow, A.; et al. Microvesicle-Mediated Delivery of Minicircle DNA Results in Effective Gene-Directed Enzyme Prodrug Cancer Therapy. Mol. Cancer Ther. 2019, 18, 2331–2342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Zhang, Y.; Tang, K.; Zhang, H.; Yin, X.; Li, Y.; Xu, P.; Sun, Y.; Ma, R.; Ji, T.; et al. Reversing drug resistance of soft tumor-repopulating cells by tumor cell-derived chemotherapeutic microparticles. Cell Res. 2016, 26, 713–727. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Gu, C.; Gan, Y.; Shao, L.; Chen, H.; Zhu, H. Exosome-mediated siRNA delivery to suppress postoperative breast cancer metastasis. J. Control. Release 2020, 318, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Usman, W.M.; Pham, T.C.; Kwok, Y.Y.; Vu, L.T.; Ma, V.; Peng, B.; Chan, Y.S.; Wei, L.; Chin, S.M.; Azad, A.; et al. Efficient RNA drug delivery using red blood cell extracellular vesicles. Nat. Commun. 2018, 9, 2359. [Google Scholar] [CrossRef]
- Aqil, F.; Munagala, R.; Jeyabalan, J.; Agrawal, A.K.; Gupta, R. Exosomes for the Enhanced Tissue Bioavailability and Efficacy of Curcumin. AAPS J. 2017, 19, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Wu, J.Y.; Wang, J.M.; Hu, X.B.; Cai, J.X.; Xiang, D.X. Gemcitabine loaded autologous exosomes for effective and safe chemotherapy of pancreatic cancer. Acta Biomater. 2020, 101, 519–530. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of EV | Size (nm) | Biogenesis | Main Components |
|---|---|---|---|
| Exosomes | 30–150 | Early endosomes mature into late endosomes. Through the action of ESCRT machinery, MVBs containing ILVs are formed and fuse with the plasma membrane for release | Tetraspanins (CD9, CD63, CD81), HSPs, MVB biogenesis components (ALIX, TSG101) |
| Microvesicles | 100–1000 | Outward budding of the membrane followed by fission through contractile machinery | Cell adhesion molecules (integrins, selectins), Death receptors (CD40), VCAMP3, ARF6 |
| Apoptotic bodies | >1000 | Cytoplasmic fragmentation during programmed cell death | Histones, Annexin V, Caspase 3, Phosphatidylserine |
| Oncosomes | >1000 | Cleavage of large cytoplasmic extensions from cell body | Cytoskeleton components (cytokeratin 18), Tetraspanins (CD9, CD81), Cell adhesion molecules (integrins, ICAM, CD44) |
| Biological Fluid | Biomarkers | Therapeutic Approaches Involved | Changes Detected | Potential Clinical Application | Refs |
|---|---|---|---|---|---|
| Plasma | 144 phosphoproteins | n.a. | Increased in BC patients | BC diagnosis | [165] |
| Plasma | EGFR, EpCAM, HER2 | n.a. | Increased in BC patients | BC diagnosis | [166] |
| Plasma | CD63/EpCAM/mucin 1 | n.a. | Increased in BC patients | BC diagnosis | [167] |
| Plasma | EpCAM | n.a. | Increased in BC patients | BC diagnosis | [168] |
| Plasma | CD3, CD56, CD2, CD25, CD9, CD44, CD326, CD133/1, CD142, CD45, CD14 | Surgery + adjuvant therapy | Increased in BC patients. CD146 and CD45 downregulated 1 month after surgery | BC diagnosis and monitoring after surgical resection and during adjuvant therapy | [169] |
| Plasma | Del-1 | Surgery | Increased in BC patients and returned to almost normal after tumour removal | BC diagnosis and monitoring after surgical resection | [170] |
| Claudin 7 | n.a. | Increased in BC patients | BC diagnosis | [171] | |
| Urine | miR-21, MMP-1 | n.a. | miR-21: decreased in BC patients; MMP1: increased in BC patients | BC diagnosis | [172] |
| Serum | H19 | Surgery | H19 levels increased in BC patients. Median serum EV H19 levels were significantly lower in post-operative than that in the pre-operative setting. EV H19 expression levels were associated with lymph node metastasis, distant metastasis, TNM stages, ER, PR, and HER2 expression | BC diagnosis and monitoring after surgical resection | [173] |
| Serum | miR-1910-3p | n.a. | Increased in BC patients | BC diagnosis | [174] |
| Serum | miRNA-21, miRNA-105 miRNA-222 | Neoadjuvant therapy | miRNA-21 and 105: increased in metastatic vs non-metastatic patients and healthy controls; miRNA-222 increased in basal-like and in luminal B versus luminal A tumour subtypes. Correlated with clinical and pathological variables such as PR status and Ki67. miRNA-21: during neoadjuvant treatment expression levels directly correlated with tumour size and inversely with Ki67 expression. | BC diagnosis and monitoring disease during neoadjuvant therapy | [175] |
| Plasma | Ca15-3, CEA, Ca125, HER2, EGFR, PSMA, EpCAM, VEGF | Surgery, chemotherapy | Increased in BC patients. A higher level was significantly associated with PFS | BC diagnosis and monitoring during therapy | [39] |
| Serum | AnxA2 | n.a. | Significantly elevated in TNBC in comparison to ER+, HER2+. Associated with tumour grade poor overall survival and poor disease-free survival | BC diagnosis | [100] |
| Plasma | FAK, Fibronectin, P-cadherin, TAZ, IGFRβ, HSP70 | Chemotherapy, Radiotherapy, Surgery | FAK, Fibronectin: differentiated BC patients from healthy individuals; P-cadherin, TAZ: reduced in stage IIA; IGFRβ: overexpressed in stage IIA; HSP70: associated with a high risk of relapse | BC diagnosis and monitoring of relapse | [176] |
| Plasma | EV concentration | Neoadjuvant therapy | Higher in BC patients. EV concentration increased during therapy, an increased EV concentration before neoadjuvant chemotherapy was associated with therapy failure and an elevated EV concentration after neoadjuvant chemotherapy was associated with a reduced three-year progression-free and overall survival | BC diagnosis and monitoring during neoadjuvant therapy | [177] |
| Plasma | sEMP concentration | Neoadjuvant or first line chemotherapy | Median levels of sEMP decreased after chemotherapy and was associated with better overall survival and progression free survival | BC monitoring during therapy | [178] |
| Plasma | miR-155, miR-301 | Neoadjuvant therapy | predicted pathological complete response | BC monitoring during neoadjuvant therapy | [179] |
| Blood | miR-185, miR-4283, miR-5008, miR-3613 miR-1302, miR-4715, miR-3144 | Neoadjuvant therapy | miR-185, miR-4283, miR-5008, miR-3613: lower expression in non-responsive patients; miR-1302, miR-4715, miR-3144: higher expression in non-responsive patients | TNBC monitoring during neoadjuvant therapy | [180] |
| Serum | SNHG14 | Trastuzumab | upregulated in HER2+ patients who exhibited resistance to Trastuzumab | HER2+ BC monitoring during Trastuzumab treatment | [181] |
| Serum | HOTAIR | Surgery, neoadjuvant therapy | Decreased after surgery | BC monitoring during therapy | [182] |
| Plasma | TRPC | Chemotherapy | Increased cirExo-TRPC5 level after chemotherapy preceded PD based on imaging examination and strongly predicted acquired chemoresistance | BC monitoring during therapy | [183] |
| Serum | UCH-L | Chemotherapy | Highly expressed in BC patients with poor prognosis due to chemo-resistance | BC monitoring during therapy | [184] |
| Serum | TGFβ1 | Neoadjuvant therapy | Increased in patients with HER2-overexpressing BC who did not respond to neoadjuvant HER2-targeted drug treatment | BC monitoring during neoadjuvant therapy | [185] |
| Source of EVs | Cargo | Outcomes | Refs |
|---|---|---|---|
| HEK293T | Doxorubicin | Inhibited MDR tumour growth and extended animal survival times | [236] |
| EGFR aptamer + Survivin siRNA | Inhibited BC growth in mice | [237] | |
| Transmembrane domain of PDGFR fused to the GE11 peptide + let-7a miRNA | Inhibited tumour development in vitro and in vivo | [238] | |
| DC | AS1411 aptamer + let-7 and VEGF miRNA | High anti-tumour activity in vitro and in vivo | [239] |
| MSC | miR-379 | Therapeutic effect in vitro and in vivo, mediated, in part, through regulation of COX-2 | [240] |
| LNA-antimiR-142-3p | Reduced miR-142-3p and miR-150 expression levels and inhibited clonogenicity and tumorigenicity of BC stem-like cells | [241] | |
| TRAIL | Induced pronounced apoptosis and overcame TRAIL resistance in BC cells | [242] | |
| M1-type RAW264.7 | Paclitaxel | Enhanced anticancer efficiency of paclitaxel in BC in vitro and in vivo | [243] |
| Doxorubicin | Enhanced anti-tumour activity in a BC mouse model | [244] | |
| poly(lactic-co-glycolic acid) + anti-c-Met peptide | Improved the cellular uptake efficiency and the anti-tumour efficacy of doxorubicin in TNBC | [245] | |
| imDCs | Lamp2b fused to CRGDKGPDC peptide | Inhibited tumour growth without over toxicity | [246] |
| THP1 | Doxorubicin + Cho-miR159 | Improved anticancer results in vivo and in vitro in TNBC | [247] |
| 4T1 | Indocyanine + Doxorubicin | Showed synergistic effects of chemotherapy and photothermal therapy against BC | [248] |
| TK/NTR minicircle DNA | Effective killing of BC cells | [249] | |
| MCF7 | Cisplatin + Methotrexate + Doxorubicin | Reverted BC resistance to chemotherapeutic drugs | [250] |
| Autologous BC cells | siS200A4 | Targeted lung premetastatic niche in a TNBC model | [251] |
| RBCs | ASOs against miR-125b | Efficient genome editing in BC cells in vitro and in vivo, without any observable cytotoxicity | [252] |
| Milk | Curcumin | Enhanced antiproliferative, anti-inflammatory, and anti-tumour activities | [253] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samuels, M.; Cilibrasi, C.; Papanastasopoulos, P.; Giamas, G. Extracellular Vesicles as Mediators of Therapy Resistance in the Breast Cancer Microenvironment. Biomolecules 2022, 12, 132. https://doi.org/10.3390/biom12010132
Samuels M, Cilibrasi C, Papanastasopoulos P, Giamas G. Extracellular Vesicles as Mediators of Therapy Resistance in the Breast Cancer Microenvironment. Biomolecules. 2022; 12(1):132. https://doi.org/10.3390/biom12010132
Chicago/Turabian StyleSamuels, Mark, Chiara Cilibrasi, Panagiotis Papanastasopoulos, and Georgios Giamas. 2022. "Extracellular Vesicles as Mediators of Therapy Resistance in the Breast Cancer Microenvironment" Biomolecules 12, no. 1: 132. https://doi.org/10.3390/biom12010132
APA StyleSamuels, M., Cilibrasi, C., Papanastasopoulos, P., & Giamas, G. (2022). Extracellular Vesicles as Mediators of Therapy Resistance in the Breast Cancer Microenvironment. Biomolecules, 12(1), 132. https://doi.org/10.3390/biom12010132