
Mechanistic Link between Vitamin B12 and Alzheimer’s Disease

 ,
, 
Abstract

1. Introduction
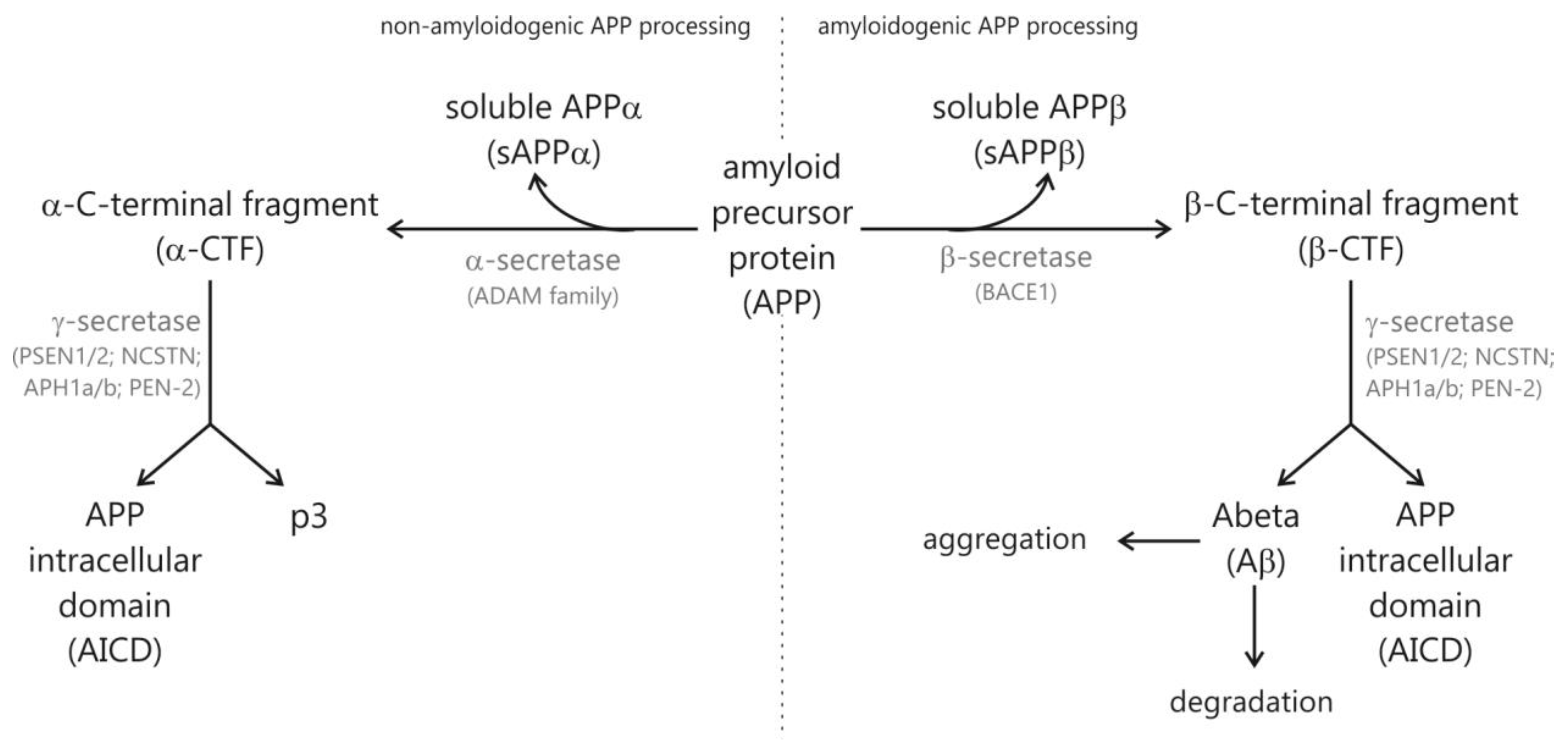
1.1. Hallmarks of Alzheimer’s Disease
1.2. Risk Factors for Sporadic AD
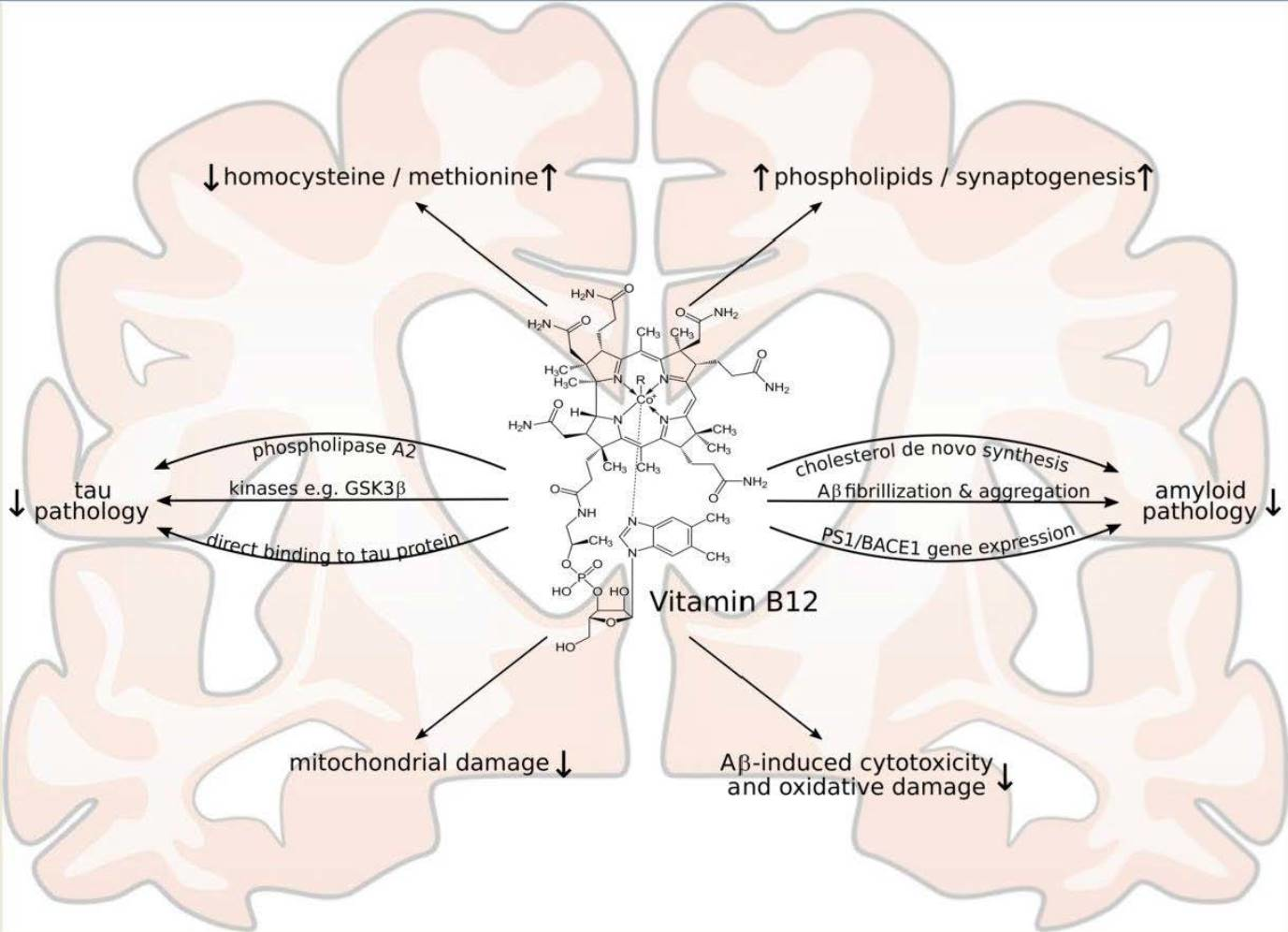
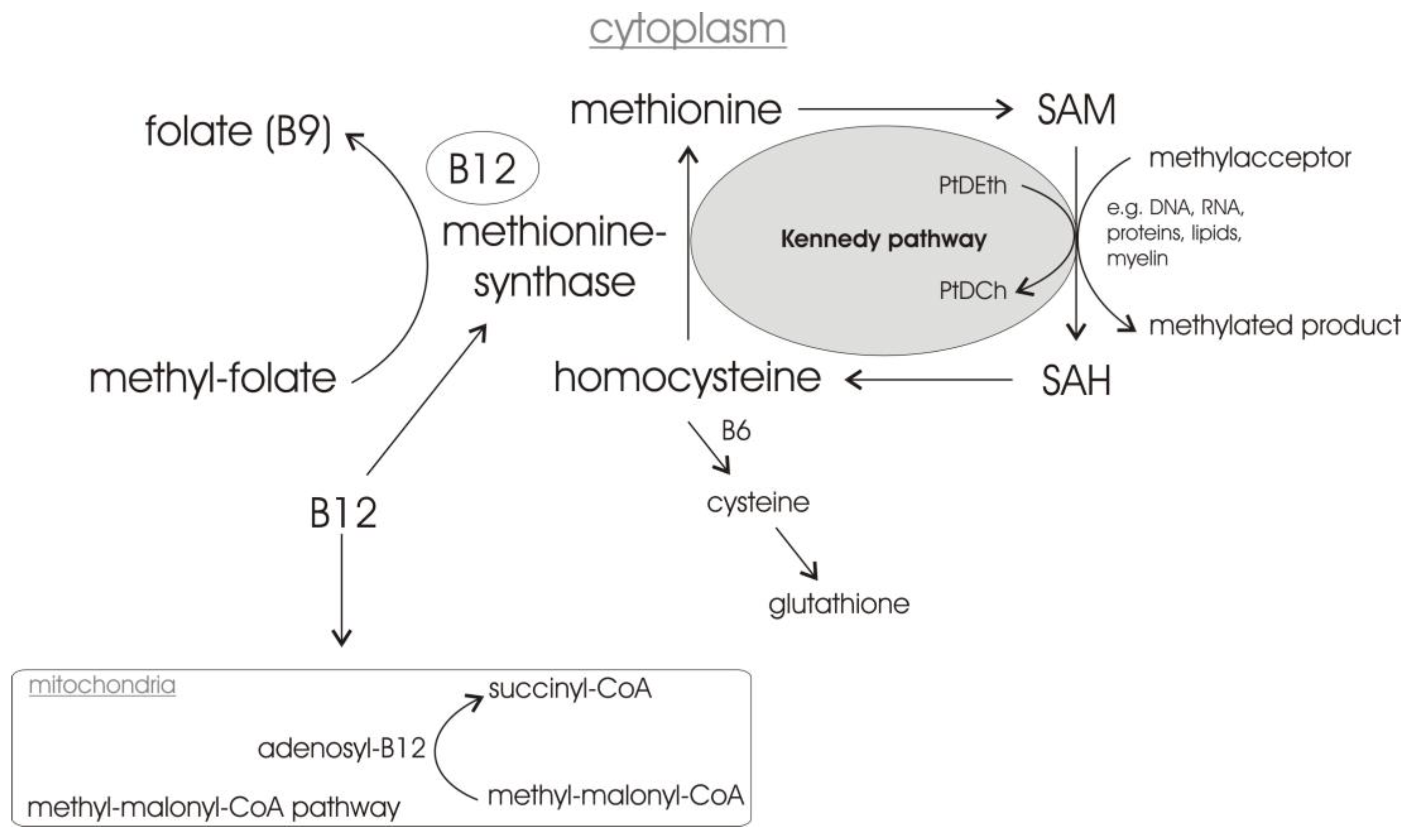
1.3. Vitamin B12
2. Vitamin B12 Cell Culture and Animal Studies Related to the Molecular Mechanisms of AD and AD Pathology
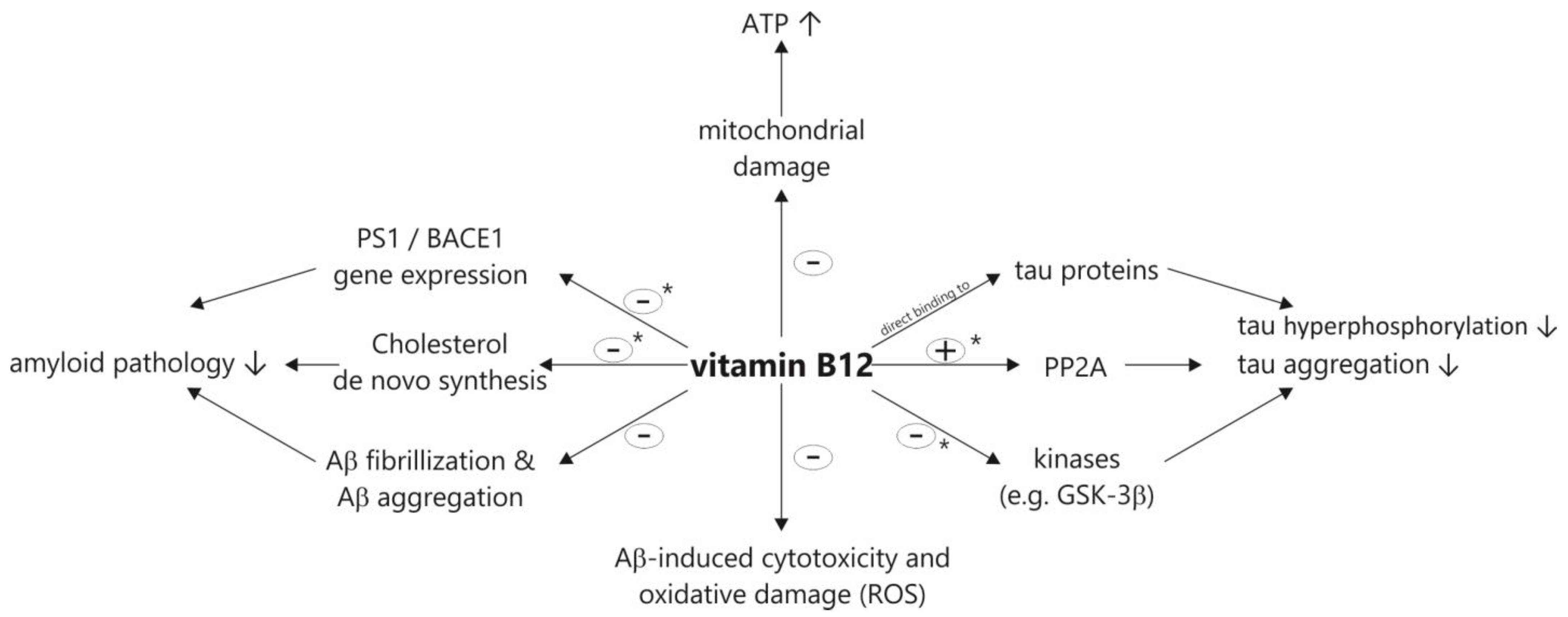
2.1. Effect of Vitamin B12 Deficiency on the Aβ Peptide Level and Aβ Deposition in AD Mice Models
2.2. Reduced Gene Expression of the Vitamin B12 Transporter Cubulin in the Intestinal Epithelium of Pre-Symptomatic Young AD Mice Models
2.3. Vitamin B12 Supplementation Antagonizes Homocysteine Induced Changes in APP Processing and Tau Phosphorylation in Wildtype Animals
2.4. Effect of Vitamin B12 on Amyloid Toxicity in Aβ-Expressing C. elegans as an AD Animal Model
2.5. Vitamin B12 Inhibits Aβ Aggregation In Vitro
2.6. Vitamin B12 Protects Cells from Cytotoxicity and Aβ-Induced Oxidative Damage
2.7. Vitamin B12 Deficiency Increases the Aβ Level in Neuroblastoma Cell Lines by an Elevation in the PS1 and BACE1 Protein Level
2.8. Vitamin B12 Inhibits Tau Polymerization by Direct Binding to Tau Proteins
2.9. Vitamin B12 Deficiency Increases the Cholesterol Level in Human Adipocyte Cell Cultures
3. Clinical Studies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author | Year | Type of Study/Duration/n | Main Finding |
|---|---|---|---|
| Perla-Kaján et al. [164] | 2021 | RCT/2 years/intervention group (n = 95) and placebo group (n = 101) | A daily dose of folic acid, vitamin B12 and B6 ameliorates detrimental effects of paraoxonase 1 (PON1) on cognition in individuals with mild cognitive impairment |
| Li et al. [167] | 2021 | Meta-Analysis/until 1 December 2019/21 RCTs (7571 participants) | Vitamin B supplements (vitamin B12, B6, folic acid alone or in combination) show preventive efficacy on cognitive decline of elderly adults |
| Zhang et al. [166] | 2020 | Meta-Analysis/until 8 August 2019/21 observational studies (sample sizes: 155–7030) | Higher levels of vitamin B12 concentration were associated with better cognition in cross-sectional studies |
| Ma et al. [168] | 2019 | RCT/6 months/240 participants with MCI (four treatment groups) | Daily oral uptake of vitamin B12 (25 µg) in combination with folic acid (800 µg) significantly improved cognitive performance and reduced inflammatory cytokine levels in peripheral blood in MCI elderly |
| Oulhaj et al. [173] | 2016 | RCT/2 years/266 participants with MCI aged ≥70 years | The effect of vitamin B treatment on cognitive decline in MCI depends on the omega-3 fatty acid concentrations |
| Remington et al. [174] | 2015 | RCT/6 months nutraceutical formulation (NF) and placebo + 6 months extension with NF for all participants/34 individuals with MCI | Intervention with nutraceutical formulation (400 µg folic acid, 6 µg B12, 30 I.U. alpha-tocopherol, 400 mg SAM, 600 mg NAC, and 500 mg ALCAR) improved cognitive performance |
| Jernerén et al. [172] | 2015 | RCT/2 years/intervention group (n = 85) and placebo groups (n = 83) | High plasma long-chain omega-3 fatty acids are important for the beneficial effect of vitamin B treatment (folic acid, vitamin B6 and B12) on brain atrophy in MCI patients |
| Douaud et al. [171] | 2013 | RCT/2 years/intervention group (n = 80) and placebo group (n = 76) | High-dose vitamin B treatment (folic acid, vitamin B6 and B12) slow the atrophy of specific brain regions related to AD and cognitive decline in MCI patients |
| A de Jager et al. [170] | 2012 | RCT/2 years/intervention group (n = 133) and placebo group (n = 133) | Vitamins B (folic acid, vitamin B6 and B12) appear to slow cognitive and clinical decline in MCI patients, especially among participants with elevated baseline homocysteine levels |
| Ford et al. [165] | 2010 | RCT/2–8 years/299 hypertensive men ≥ 75 years | No beneficial effect of supplementation with B vitamins (B12, B6, folic acid) on cognitive function (2 years outcome) or the risk of cognitive impairment or dementia (8 years outcome) |
| Smit et al. [169] | 2010 | RCT/2 years/intervention group (n = 85) and placebo group (n = 83) | Accelerated brain atrophy in MCI patients can be slowed by treatment with B vitamins (folic acid, vitamin B6 and B12) |
| Flicker et al. [175] | 2008 | RCT/2 years/intervention group (n = 150) and placebo group (n = 149) | Reduced increase of plasma Aβ40 levels in older men treated with a combination of folate, vitamin B6 and B12 compared to placebo group |
| Frick et al. [176] | 2006 | Clinical Trial/1 month/58 patients (AD, n = 30; vascular dementia, n = 12; MCI, n = 16) | Daily supplementation of B vitamins (vitamins B1, B6, B12, folic acid) declines concentrations of homocysteine but not of neopterin in demented patients |
| An et al. [177] | 2019 | Clinical trial/2.3 years/2533 participants for longitudinal study + a subgroup of 109 MCI patients and 73 controls for DNA methylation and biochemical analyses | Significant association between inadequate dietary intake of vitamin B12 and accelerated cognitive decline, which may be mediated by affected methylation levels of specific redox-related genes |
| Van Dyck et al. [178] | 2009 | Controlled clinical trial/16 weeks/replacement group with low serum B12 levels (n = 28) and control group with normal serum B12 levels (n = 28) | Vitamin B12 replacement in dementia with low serum B12 levels resulted in significant improvements in hematologic and metabolic parameters but is unlikely to benefit cognitive or psychiatric symptoms |
| Author | Year | Type of Study/Duration/n | Main Finding |
|---|---|---|---|
| Chen et al. [197] | 2021 | RCT/6 months/intervention group (n = 51) and placebo group (n = 50) | Supplementation of folic acid and vitamin B12 had a beneficial therapeutic effect in AD patients who were not on a folic acid-fortified diet |
| Guzman-Martinez et al. [196] | 2021 | RCT/24 weeks/82 mild to moderate AD patients | The nutraceutical BrainUp-10®, containing vitamin B12, produces a significant improvement in apathy, ameliorating neuropsychiatric distress of patients |
| Rasmussen [193] | 2019 | RCT/24 + 12 months/311 patients with prodromal AD | Fortasyn Connect, a multi-nutrient combination containing vitamin B12, may show benefit on domains of cognition affected by AD |
| Vakilian et al. [191] | 2017 | Clinical trial | Vitamin B12 in combination with antipsychotic drugs is able to reduce and induce the expression of pro- and anti-inflammatory cytokines in AD patients |
| Zhang et al. [185] | 2017 | Meta-Analysis/until 7 May 2015/4 studies included | Data on vitamin B-induced improvement in cognition by reducing homocysteine levels are conflicting and should be addressed in further studies |
| Remington et al. [189] | 2016 | RCT/12 months/24 individuals diagnosed with AD | Over the duration of nutraceutical formulation (folate, alpha-tocopherol, vitamin B12, SAM, NAC, ALCAR) supplementation behavioral and psychological symptoms of dementia as well as baseline cognitive performance were maintained |
| Remington et al. [188] | 2015 | Clinical trial/3- or 6-months intervention + 6 months open-label extension/106 individuals with AD | The results of this trial extended phase I studies showing maintained or improved cognitive performance and mood/behavior after supplementation of nutraceutical formulation (folate, alpha-tocopherol, vitamin B12, SAM, NAC, ALCAR) in AD patients |
| Rommer et al. [192] | 2016 | Clinical trial/3 months/healthy control (n = 15), AD or MCI (n = 16), supplemented AD or MCI (n = 17) | Supplementation of vitamins B1, B6, B12 and folic acid for three months resulted in decreased levels of carbonyl proteins, which negatively correlated with MMSE in AD/MCI patients |
| Shen et al. [179] | 2015 | Meta-Analysis/up to January 2014/68 studies included | Higher homocysteine and lower folic acid and vitamin B12 levels in AD patients than healthy individuals |
| Lopes da Silva et al. [180] | 2014 | Meta-Analysis/literature published after 1990/more than five publications for a specific nutrient | Significantly lower plasma levels of vitamin B12 were found in AD patients. |
| Cornelli [190] | 2010 | Clinical trial/6 months/52 moderate AD patients already being treated with 5 mg donepezil per day for at least two months | Treatment with formula F (Carnosine, vitamins B1, B2, B3, B6, B9, B12, C, E, Coenzyme Q10, β-carotene, selenium, L-cysteine, Ginkgo biloba) decreased oxidative stress and homocysteine levels and improved MMSE II scores significantly |
| Remington et al. [187] | 2009 | RCT/9 months/12 institutionalized patients diagnosed with moderate-stage to later-stage AD | Supplementation of a vitamin/nutraceutical formulation containing folate, vitamin B12, alpha-tocopherol, S-adenosyl methionine (SAM), N-acetyl cysteine (NAC), acetyl-L-carnitine (ALCAR) seems to delay the decline in cognition, mood, and daily function |
| Chan et al. [186] | 2008 | Clinical trial/12 months/14 community-dwelling individuals with early-stage AD | Treatment with a vitamin/nutraceutical formulation (folate, vitamin B12, alpha-tocopherol, SAM, NAC, ALCAR) resulted in improved cognitive performance |
| Aisen et al. [184] | 2008 | RCT/18 months/intervention group (n = 202) and placebo group (n = 138) of AD patients | Daily supplementation of folate, vitamin B6 and B12 for 18 months was effective in reducing homocysteine levels, but not in slowing cognitive decline in individuals with mild to moderate AD |
| Sun et al. [183] | 2007 | RCT/26 weeks/89 patients with mild to moderate AD and normal folic acid and vitamin B12 concentrations | Multivitamin supplement including vitamins B12, B6 and folic acid reduced concentrations of homocysteine but had no statistically significant beneficial effects on cognition compared to placebo treatment |
| Aisen et al. [182] | 2003 | Clinical trial/8 weeks/69 subjects with AD, including 33 with standard multivitamin supplements | This open-label trial shows high-dose, combined vitamin B12 and B6 supplementation to reduce homocysteine levels in AD patients |
| Teunissen et al. [181] | 2003 | Clinical trial/one-point/neurological patients (AD: n = 34; Parkinson’s disease: n = 46; other cognitive disorders: n = 47) and healthy controls (n = 61) | Compared to healthy individuals the median vitamin B12 concentration was decreased in all neurological patients |
4. Is There an Association of Diseases and Medications, Known to Be Linked to Vitamin B12 Deficiency, with AD?
| Link to Vitamin B12 | Link to Alzheimer’s Disease | ||
|---|---|---|---|
| Author | Main Finding | Author | Main Finding |
| Inflammatory Bowel Disease (IBD) | |||
| Weisshof et al. (2015) [198] | Micronutrient deficiencies are common (>50%) in patients with IBD with vitamin B12 deficiency belonging to the most common ones. | Zingel et al. (2021) [203] | This study analyzing 3850 patients with an initial diagnosis of inflammatory bowel diseases (IBD; Crohn’s Disease, ulcerative colitis) and 3850 patients without IBD reported that IBD is associated with a 1.22-fold increase in the risk of developing dementia. |
| Yakut et al. (2010) [199] | Patients with Crohn’s disease common have a serum vitamin B12 deficiency. | Zhan et al. (2021) [204] | An increase in the risk of developing AD was reported in IBD patients in a 16-year longitudinal study including 1742 patients with IBD. |
| Bermejo et al. (2013) [200] | 15.6% (95% CI 9.7–20%) of patients with Crohn’s disease suffer from vitamin B12 deficiency. | ||
| Park et al. (2021) [201] | Crohn’s disease patients are more often deficient in micronutrients like vitamin B12. | ||
| Ward et al. (2015) [202] | The prevalence of vitamin B12 deficiency is common in patients with Crohn’s disease. | ||
| Gastritis | |||
| Porter et al. (2021) [207] | Atrophic gastritis was associated with significantly lower serum total vitamin B12 levels and higher prevalence of vitamin B12 deficiency. | Li et al. (2018) [210] | The risk of dementia and AD is increased in patients with many types of autoimmune disorders, like pernicious anemia. |
| Green (2017) [205] | Pernicious anemia (autoimmune gastritis) is a cause of vitamin B12 deficiency. | Metzler et al. (1991) [212] | Specific clinical entities of a vitamin B12 deficiency include, amongst others, dementia. |
| Sipponen et al. (2003) [208] | Association of low vitamin B12 serum levels and atrophic gastritis in an elderly male cohort. | Kountouras et al. (2006) [213] | There is a link between an infection with Helicobacter pylori and Alzheimer’s disease. |
5. Veganism/Vegetarianism, Vitamin B12 Levels and AD
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Plassman, B.L.; Langa, K.M.; Fisher, G.G.; Heeringa, S.G.; Weir, D.R.; Ofstedal, M.B.; Burke, J.R.; Hurd, M.D.; Potter, G.G.; Rodgers, W.L.; et al. Prevalence of dementia in the united states: The aging, demographics, and memory study. Neuroepidemiology 2007, 29, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 2012, 425, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef]
- Dyrks, T.; Weidemann, A.; Multhaup, G.; Salbaum, J.M.; Lemaire, H.G.; Kang, J.; Muller-Hill, B.; Masters, C.L.; Beyreuther, K. Identification, transmembrane orientation and biogenesis of the amyloid a4 precursor of Alzheimer’s disease. EMBO J. 1988, 7, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Anderson, J.P.; Barbour, R.; Basi, G.S.; Caccavello, R.; Davis, D.; Doan, M.; Dovey, H.F.; Frigon, N.; Hong, J.; et al. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 1999, 402, 537–540. [Google Scholar] [CrossRef]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease bace. Science 1999, 286, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Bienkowski, M.J.; Shuck, M.E.; Miao, H.; Tory, M.C.; Pauley, A.M.; Brashier, J.R.; Stratman, N.C.; Mathews, W.R.; Buhl, A.E.; et al. Membrane-anchored aspartyl protease with Alzheimer’s disease beta-secretase activity. Nature 1999, 402, 533–537. [Google Scholar] [CrossRef]
- Kimberly, W.T.; LaVoie, M.J.; Ostaszewski, B.L.; Ye, W.; Wolfe, M.S.; Selkoe, D.J. Gamma-secretase is a membrane protein complex comprised of presenilin, nicastrin, aph-1, and pen-2. Proc. Natl. Acad. Sci. USA 2003, 100, 6382–6387. [Google Scholar] [CrossRef]
- Haass, C. Take five-bace and the gamma-secretase quartet conduct Alzheimer’s amyloid beta-peptide generation. EMBO J. 2004, 23, 483–488. [Google Scholar] [CrossRef]
- Grimm, M.O.; Tomic, I.; Hartmann, T. Potential external source of a beta in biological samples. Nat. Cell Biol. 2002, 4, E164–E165, author reply E165–E166. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Mett, J.; Stahlmann, C.P.; Haupenthal, V.J.; Blumel, T.; Stotzel, H.; Grimm, H.S.; Hartmann, T. Eicosapentaenoic acid and docosahexaenoic acid increase the degradation of amyloid-beta by affecting insulin-degrading enzyme. Biochem. Cell Biol. 2016, 94, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Kuchenbecker, J.; Grosgen, S.; Burg, V.K.; Hundsdorfer, B.; Rothhaar, T.L.; Friess, P.; de Wilde, M.C.; Broersen, L.M.; Penke, B.; et al. Docosahexaenoic acid reduces amyloid beta production via multiple pleiotropic mechanisms. J. Biol. Chem. 2011, 286, 14028–14039. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Haupenthal, V.J.; Rothhaar, T.L.; Zimmer, V.C.; Grosgen, S.; Hundsdorfer, B.; Lehmann, J.; Grimm, H.S.; Hartmann, T. Effect of different phospholipids on alpha-secretase activity in the non-amyloidogenic pathway of Alzheimer’s disease. Int. J. Mol. Sci. 2013, 14, 5879–5898. [Google Scholar] [CrossRef] [PubMed]
- Rothhaar, T.L.; Grosgen, S.; Haupenthal, V.J.; Burg, V.K.; Hundsdorfer, B.; Mett, J.; Riemenschneider, M.; Grimm, H.S.; Hartmann, T.; Grimm, M.O. Plasmalogens inhibit app processing by directly affecting gamma-secretase activity in Alzheimer’s disease. Sci. World J. 2012, 2012, 141240. [Google Scholar] [CrossRef]
- Grimm, M.O.; Rothhaar, T.L.; Grosgen, S.; Burg, V.K.; Hundsdorfer, B.; Haupenthal, V.J.; Friess, P.; Kins, S.; Grimm, H.S.; Hartmann, T. Trans fatty acids enhance amyloidogenic processing of the alzheimer amyloid precursor protein (app). J. Nutr. Biochem. 2012, 23, 1214–1223. [Google Scholar] [CrossRef] [PubMed]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Shirotani, K.; Lu, B.; Gerard, N.P.; Gerard, C.; Hama, E.; Lee, H.J.; Saido, T.C. Metabolic regulation of brain abeta by neprilysin. Science 2001, 292, 1550–1552. [Google Scholar] [CrossRef]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guenette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef]
- Buxbaum, J.D.; Liu, K.N.; Luo, Y.; Slack, J.L.; Stocking, K.L.; Peschon, J.J.; Johnson, R.S.; Castner, B.J.; Cerretti, D.P.; Black, R.A. Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the alzheimer amyloid protein precursor. J. Biol. Chem. 1998, 273, 27765–27767. [Google Scholar] [CrossRef] [PubMed]
- Lammich, S.; Kojro, E.; Postina, R.; Gilbert, S.; Pfeiffer, R.; Jasionowski, M.; Haass, C.; Fahrenholz, F. Constitutive and regulated alpha-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. USA 1999, 96, 3922–3927. [Google Scholar] [CrossRef]
- Kuhn, P.H.; Wang, H.; Dislich, B.; Colombo, A.; Zeitschel, U.; Ellwart, J.W.; Kremmer, E.; Rossner, S.; Lichtenthaler, S.F. Adam10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010, 29, 3020–3032. [Google Scholar] [CrossRef]
- Grundke-Iqbal, I.; Iqbal, K.; Quinlan, M.; Tung, Y.C.; Zaidi, M.S.; Wisniewski, H.M. Microtubule-associated protein tau. A component of alzheimer paired helical filaments. J. Biol. Chem. 1986, 261, 6084–6089. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Cairns, N.J.; Crowther, R.A. Tau proteins of alzheimer paired helical filaments: Abnormal phosphorylation of all six brain isoforms. Neuron 1992, 8, 159–168. [Google Scholar] [CrossRef]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef]
- Martin, L.; Latypova, X.; Wilson, C.M.; Magnaudeix, A.; Perrin, M.L.; Yardin, C.; Terro, F. Tau protein kinases: Involvement in Alzheimer’s disease. Ageing Res. Rev. 2013, 12, 289–309. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.; Latypova, X.; Wilson, C.M.; Magnaudeix, A.; Perrin, M.L.; Terro, F. Tau protein phosphatases in Alzheimer’s disease: The leading role of pp2a. Ageing Res. Rev. 2013, 12, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.E.; Yaffe, K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011, 10, 819–828. [Google Scholar] [CrossRef]
- Polidori, M.C.; Pientka, L.; Mecocci, P. A review of the major vascular risk factors related to Alzheimer’s disease. J. Alzheimers Dis. 2012, 32, 521–530. [Google Scholar] [CrossRef]
- Yusufov, M.; Weyandt, L.L.; Piryatinsky, I. Alzheimer’s disease and diet: A systematic review. Int. J. Neurosci. 2017, 127, 161–175. [Google Scholar] [CrossRef]
- Luchsinger, J.A.; Noble, J.M.; Scarmeas, N. Diet and Alzheimer’s disease. Curr. Neurol. Neurosci. Rep. 2007, 7, 366–372. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein e type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein e: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef]
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human apoe isoforms differentially regulate brain amyloid-beta peptide clearance. Sci. Transl. Med. 2011, 3, 89ra57. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Yee, A.; Brewer, H.B., Jr.; Das, S.; Potter, H. Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein e promote assembly of alzheimer beta-protein into filaments. Nature 1994, 372, 92–94. [Google Scholar] [CrossRef]
- Deane, R.; Sagare, A.; Hamm, K.; Parisi, M.; Lane, S.; Finn, M.B.; Holtzman, D.M.; Zlokovic, B.V. Apoe isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J. Clin. Investig. 2008, 118, 4002–4013. [Google Scholar] [CrossRef]
- Pappolla, M.A.; Bryant-Thomas, T.K.; Herbert, D.; Pacheco, J.; Fabra Garcia, M.; Manjon, M.; Girones, X.; Henry, T.L.; Matsubara, E.; Zambon, D.; et al. Mild hypercholesterolemia is an early risk factor for the development of alzheimer amyloid pathology. Neurology 2003, 61, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, T.; Sasaki, K.; Hata, J.; Hirakawa, Y.; Fujimi, K.; Ninomiya, T.; Suzuki, S.O.; Kanba, S.; Kiyohara, Y.; Iwaki, T. Association of alzheimer disease pathology with abnormal lipid metabolism: The hisayama study. Neurology 2011, 77, 1068–1075. [Google Scholar] [CrossRef]
- Meng, X.F.; Yu, J.T.; Wang, H.F.; Tan, M.S.; Wang, C.; Tan, C.C.; Tan, L. Midlife vascular risk factors and the risk of Alzheimer’s disease: A systematic review and meta-analysis. J. Alzheimers Dis. 2014, 42, 1295–1310. [Google Scholar] [CrossRef] [PubMed]
- Solomon, A.; Kivipelto, M.; Wolozin, B.; Zhou, J.; Whitmer, R.A. Midlife serum cholesterol and increased risk of Alzheimer’s and vascular dementia three decades later. Dement. Geriatr. Cogn. Disord. 2009, 28, 75–80. [Google Scholar] [CrossRef]
- Cutler, R.G.; Kelly, J.; Storie, K.; Pedersen, W.A.; Tammara, A.; Hatanpaa, K.; Troncoso, J.C.; Mattson, M.P. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 2070–2075. [Google Scholar] [CrossRef]
- Panchal, M.; Loeper, J.; Cossec, J.C.; Perruchini, C.; Lazar, A.; Pompon, D.; Duyckaerts, C. Enrichment of cholesterol in microdissected Alzheimer’s disease senile plaques as assessed by mass spectrometry. J. Lipid Res. 2010, 51, 598–605. [Google Scholar] [CrossRef]
- Xiong, H.; Callaghan, D.; Jones, A.; Walker, D.G.; Lue, L.F.; Beach, T.G.; Sue, L.I.; Woulfe, J.; Xu, H.; Stanimirovic, D.B.; et al. Cholesterol retention in Alzheimer’s brain is responsible for high beta- and gamma-secretase activities and abeta production. Neurobiol. Dis. 2008, 29, 422–437. [Google Scholar] [CrossRef]
- Simons, M.; Keller, P.; De Strooper, B.; Beyreuther, K.; Dotti, C.G.; Simons, K. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc. Natl. Acad. Sci. USA 1998, 95, 6460–6464. [Google Scholar] [CrossRef] [PubMed]
- Fassbender, K.; Simons, M.; Bergmann, C.; Stroick, M.; Lutjohann, D.; Keller, P.; Runz, H.; Kuhl, S.; Bertsch, T.; von Bergmann, K.; et al. Simvastatin strongly reduces levels of Alzheimer’s disease beta -amyloid peptides abeta 42 and abeta 40 in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 5856–5861. [Google Scholar] [CrossRef]
- Grimm, M.O.; Grimm, H.S.; Tomic, I.; Beyreuther, K.; Hartmann, T.; Bergmann, C. Independent inhibition of Alzheimer disease beta- and gamma-secretase cleavage by lowered cholesterol levels. J. Biol. Chem. 2008, 283, 11302–11311. [Google Scholar] [CrossRef] [PubMed]
- Osenkowski, P.; Ye, W.; Wang, R.; Wolfe, M.S.; Selkoe, D.J. Direct and potent regulation of gamma-secretase by its lipid microenvironment. J. Biol. Chem. 2008, 283, 22529–22540. [Google Scholar] [CrossRef]
- Kalvodova, L.; Kahya, N.; Schwille, P.; Ehehalt, R.; Verkade, P.; Drechsel, D.; Simons, K. Lipids as modulators of proteolytic activity of bace: Involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. J. Biol. Chem. 2005, 280, 36815–36823. [Google Scholar] [CrossRef] [PubMed]
- Cossec, J.C.; Simon, A.; Marquer, C.; Moldrich, R.X.; Leterrier, C.; Rossier, J.; Duyckaerts, C.; Lenkei, Z.; Potier, M.C. Clathrin-dependent app endocytosis and abeta secretion are highly sensitive to the level of plasma membrane cholesterol. Biochim. Biophys. Acta 2010, 1801, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Kojro, E.; Gimpl, G.; Lammich, S.; Marz, W.; Fahrenholz, F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha-secretase ADAM 10. Proc. Natl. Acad. Sci. USA 2001, 98, 5815–5820. [Google Scholar] [CrossRef]
- Hao, M.; Mukherjee, S.; Maxfield, F.R. Cholesterol depletion induces large scale domain segregation in living cell membranes. Proc. Natl. Acad. Sci. USA 2001, 98, 13072–13077. [Google Scholar] [CrossRef]
- Hicks, D.A.; Nalivaeva, N.N.; Turner, A.J. Lipid rafts and Alzheimer’s disease: Protein-lipid interactions and perturbation of signaling. Front. Physiol. 2012, 3, 189. [Google Scholar] [CrossRef]
- Schneider, A.; Schulz-Schaeffer, W.; Hartmann, T.; Schulz, J.B.; Simons, M. Cholesterol depletion reduces aggregation of amyloid-beta peptide in hippocampal neurons. Neurobiol. Dis. 2006, 23, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Ferrera, P.; Mercado-Gomez, O.; Silva-Aguilar, M.; Valverde, M.; Arias, C. Cholesterol potentiates beta-amyloid-induced toxicity in human neuroblastoma cells: Involvement of oxidative stress. Neurochem. Res. 2008, 33, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Ionov, M.; Pavlov, E.; Duchen, M.R. Membrane cholesterol content plays a key role in the neurotoxicity of beta-amyloid: Implications for Alzheimer’s disease. Aging Cell 2011, 10, 595–603. [Google Scholar] [CrossRef]
- Sparks, D.L.; Scheff, S.W.; Hunsaker, J.C., 3rd; Liu, H.; Landers, T.; Gross, D.R. Induction of alzheimer-like beta-amyloid immunoreactivity in the brains of rabbits with dietary cholesterol. Exp. Neurol. 1994, 126, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Refolo, L.M.; Malester, B.; LaFrancois, J.; Bryant-Thomas, T.; Wang, R.; Tint, G.S.; Sambamurti, K.; Duff, K.; Pappolla, M.A. Hypercholesterolemia accelerates the Alzheimer’s amyloid pathology in a transgenic mouse model. Neurobiol. Dis. 2000, 7, 321–331. [Google Scholar] [CrossRef]
- Refolo, L.M.; Pappolla, M.A.; LaFrancois, J.; Malester, B.; Schmidt, S.D.; Thomas-Bryant, T.; Tint, G.S.; Wang, R.; Mercken, M.; Petanceska, S.S.; et al. A cholesterol-lowering drug reduces beta-amyloid pathology in a transgenic mouse model of Alzheimer’s disease. Neurobiol. Dis. 2001, 8, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Kurata, T.; Kawai, H.; Miyazaki, K.; Kozuki, M.; Morimoto, N.; Ohta, Y.; Ikeda, Y.; Abe, K. Statins have therapeutic potential for the treatment of Alzheimer’s disease, likely via protection of the neurovascular unit in the ad brain. J. Neurol. Sci. 2012, 322, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, S.; Beiser, A.; Selhub, J.; Jacques, P.F.; Rosenberg, I.H.; D’Agostino, R.B.; Wilson, P.W.; Wolf, P.A. Plasma homocysteine as a risk factor for dementia and alz.zheimer’s disease. N. Engl. J. Med. 2002, 346, 476–483. [Google Scholar] [CrossRef]
- Luchsinger, J.A.; Tang, M.X.; Shea, S.; Miller, J.; Green, R.; Mayeux, R. Plasma homocysteine levels and risk of alzheimer disease. Neurology 2004, 62, 1972–1976. [Google Scholar] [CrossRef]
- Van Dam, F.; Van Gool, W.A. Hyperhomocysteinemia and Alzheimer’s disease: A systematic review. Arch. Gerontol. Geriatr. 2009, 48, 425–430. [Google Scholar] [CrossRef]
- Isobe, C.; Murata, T.; Sato, C.; Terayama, Y. Increase of total homocysteine concentration in cerebrospinal fluid in patients with Alzheimer’s disease and parkinson’s disease. Life Sci. 2005, 77, 1836–1843. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.S.P.; Liu, C.S.; Rau, A.; Lanctot, K.L.; Kohler, C.A.; Pakosh, M.; Carvalho, A.F.; Herrmann, N. Peripheral inflammatory markers in Alzheimer’s disease: A systematic review and meta-analysis of 175 studies. J. Neurol. Neurosurg. Psychiatry 2017, 88, 876–882. [Google Scholar] [CrossRef] [PubMed]
- McCaddon, A.; Hudson, P.; Davies, G.; Hughes, A.; Williams, J.H.; Wilkinson, C. Homocysteine and cognitive decline in healthy elderly. Dement. Geriatr. Cogn. Disord. 2001, 12, 309–313. [Google Scholar] [CrossRef]
- Zylberstein, D.E.; Lissner, L.; Bjorkelund, C.; Mehlig, K.; Thelle, D.S.; Gustafson, D.; Ostling, S.; Waern, M.; Guo, X.; Skoog, I. Midlife homocysteine and late-life dementia in women. A prospective population study. Neurobiol. Aging 2011, 32, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Kamath, A.F.; Chauhan, A.K.; Kisucka, J.; Dole, V.S.; Loscalzo, J.; Handy, D.E.; Wagner, D.D. Elevated levels of homocysteine compromise blood-brain barrier integrity in mice. Blood 2006, 107, 591–593. [Google Scholar] [CrossRef]
- Kruman, I.I.; Kumaravel, T.S.; Lohani, A.; Pedersen, W.A.; Cutler, R.G.; Kruman, Y.; Haughey, N.; Lee, J.; Evans, M.; Mattson, M.P. Folic acid deficiency and homocysteine impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental models of Alzheimer’s disease. J. Neurosci. 2002, 22, 1752–1762. [Google Scholar] [CrossRef] [PubMed]
- Maler, J.M.; Seifert, W.; Huther, G.; Wiltfang, J.; Ruther, E.; Kornhuber, J.; Bleich, S. Homocysteine induces cell death of rat astrocytes in vitro. Neurosci. Lett. 2003, 347, 85–88. [Google Scholar] [CrossRef]
- Fighera, M.R.; Royes, L.F.; Furian, A.F.; Oliveira, M.S.; Fiorenza, N.G.; Frussa-Filho, R.; Petry, J.C.; Coelho, R.C.; Mello, C.F. Gm1 ganglioside prevents seizures, Na+, K+-atpase activity inhibition and oxidative stress induced by glutaric acid and pentylenetetrazole. Neurobiol. Dis. 2006, 22, 611–623. [Google Scholar] [CrossRef]
- Zhang, C.E.; Wei, W.; Liu, Y.H.; Peng, J.H.; Tian, Q.; Liu, G.P.; Zhang, Y.; Wang, J.Z. Hyperhomocysteinemia increases beta-amyloid by enhancing expression of gamma-secretase and phosphorylation of amyloid precursor protein in rat brain. Am. J. Pathol. 2009, 174, 1481–1491. [Google Scholar] [CrossRef]
- White, A.R.; Huang, X.; Jobling, M.F.; Barrow, C.J.; Beyreuther, K.; Masters, C.L.; Bush, A.I.; Cappai, R. Homocysteine potentiates copper- and amyloid beta peptide-mediated toxicity in primary neuronal cultures: Possible risk factors in the Alzheimer’s-type neurodegenerative pathways. J. Neurochem. 2001, 76, 1509–1520. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Oxidative stress in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef]
- Pocernich, C.B.; Butterfield, D.A. Elevation of glutathione as a therapeutic strategy in Alzheimer disease. Biochim. Biophys. Acta 2012, 1822, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Nunomura, A.; Castellani, R.J.; Zhu, X.; Moreira, P.I.; Perry, G.; Smith, M.A. Involvement of oxidative stress in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2006, 65, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Haupenthal, V.J.; Mett, J.; Stahlmann, C.P.; Blumel, T.; Mylonas, N.T.; Endres, K.; Grimm, H.S.; Hartmann, T. Oxidized docosahexaenoic acid species and lipid peroxidation products increase amyloidogenic amyloid precursor protein processing. Neurodegener. Dis. 2016, 16, 44–54. [Google Scholar] [CrossRef]
- Keller, J.N.; Schmitt, F.A.; Scheff, S.W.; Ding, Q.; Chen, Q.; Butterfield, D.A.; Markesbery, W.R. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology 2005, 64, 1152–1156. [Google Scholar] [CrossRef]
- Mecocci, P.; MacGarvey, U.; Beal, M.F. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann. Neurol. 1994, 36, 747–751. [Google Scholar] [CrossRef]
- Nunomura, A.; Perry, G.; Pappolla, M.A.; Wade, R.; Hirai, K.; Chiba, S.; Smith, M.A. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J. Neurosci. 1999, 19, 1959–1964. [Google Scholar] [CrossRef]
- Mullaart, E.; Boerrigter, M.E.; Ravid, R.; Swaab, D.F.; Vijg, J. Increased levels of DNA breaks in cerebral cortex of Alzheimer’s disease patients. Neurobiol. Aging 1990, 11, 169–173. [Google Scholar] [CrossRef]
- Apelt, J.; Bigl, M.; Wunderlich, P.; Schliebs, R. Aging-related increase in oxidative stress correlates with developmental pattern of beta-secretase activity and beta-amyloid plaque formation in transgenic tg2576 mice with alzheimer-like pathology. Int. J. Dev. Neurosci. 2004, 22, 475–484. [Google Scholar] [CrossRef]
- Simpson, J.E.; Ince, P.G.; Haynes, L.J.; Theaker, R.; Gelsthorpe, C.; Baxter, L.; Forster, G.; Lace, G.L.; Shaw, P.J.; Matthews, F.E.; et al. Population variation in oxidative stress and astrocyte DNA damage in relation to alzheimer-type pathology in the ageing brain. Neuropathol. Appl. Neurobiol. 2010, 36, 25–40. [Google Scholar] [CrossRef]
- Tamagno, E.; Guglielmotto, M.; Aragno, M.; Borghi, R.; Autelli, R.; Giliberto, L.; Muraca, G.; Danni, O.; Zhu, X.; Smith, M.A.; et al. Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J. Neurochem. 2008, 104, 683–695. [Google Scholar] [PubMed]
- Tong, Y.; Zhou, W.; Fung, V.; Christensen, M.A.; Qing, H.; Sun, X.; Song, W. Oxidative stress potentiates bace1 gene expression and abeta generation. J. Neural Transm. 2005, 112, 455–469. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid. Med. Cell. Longev. 2013, 2013, 316523. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial defects and oxidative stress in Alzheimer disease and parkinson disease. Free Radic. Biol. Med. 2013, 62, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Mao, P.; Calkins, M.J.; Cornea, A.; Reddy, A.P.; Murphy, M.P.; Szeto, H.H.; Park, B.; Reddy, P.H. Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer’s disease neurons. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S609–S631. [Google Scholar] [CrossRef]
- Dias-Santagata, D.; Fulga, T.A.; Duttaroy, A.; Feany, M.B. Oxidative stress mediates tau-induced neurodegeneration in drosophila. J. Clin. Investig. 2007, 117, 236–245. [Google Scholar] [CrossRef]
- Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Tau blocks traffic of organelles, neurofilaments, and app vesicles in neurons and enhances oxidative stress. J. Cell Biol. 2002, 156, 1051–1063. [Google Scholar] [CrossRef]
- Candore, G.; Bulati, M.; Caruso, C.; Castiglia, L.; Colonna-Romano, G.; Di Bona, D.; Duro, G.; Lio, D.; Matranga, D.; Pellicano, M.; et al. Inflammation, cytokines, immune response, apolipoprotein e, cholesterol, and oxidative stress in Alzheimer disease: Therapeutic implications. Rejuvenation Res. 2010, 13, 301–313. [Google Scholar] [CrossRef]
- Lee, Y.J.; Han, S.B.; Nam, S.Y.; Oh, K.W.; Hong, J.T. Inflammation and Alzheimer’s disease. Arch. Pharm. Res. 2010, 33, 1539–1556. [Google Scholar] [CrossRef]
- Ayton, S.; Lei, P.; Bush, A.I. Metallostasis in Alzheimer’s disease. Free Radic. Biol. Med. 2013, 62, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Greenough, M.A.; Camakaris, J.; Bush, A.I. Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem. Int. 2013, 62, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Cardaioli, E.; Da Pozzo, P.; Formichi, P.; Gallus, G.N.; Radi, E. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 2012, 322, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of a beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef]
- Wadsworth, T.L.; Bishop, J.A.; Pappu, A.S.; Woltjer, R.L.; Quinn, J.F. Evaluation of coenzyme q as an antioxidant strategy for Alzheimer’s disease. J. Alzheimers Dis. 2008, 14, 225–234. [Google Scholar] [CrossRef]
- Yang, X.; Dai, G.; Li, G.; Yang, E.S. Coenzyme q10 reduces beta-amyloid plaque in an app/ps1 transgenic mouse model of Alzheimer’s disease. J. Mol. Neurosci. 2010, 41, 110–113. [Google Scholar] [CrossRef]
- Senin, U.; Parnetti, L.; Barbagallo-Sangiorgi, G.; Bartorelli, L.; Bocola, V.; Capurso, A.; Cuzzupoli, M.; Denaro, M.; Marigliano, V.; Tammaro, A.E.; et al. Idebenone in senile dementia of alzheimer type: A multicentre study. Arch. Gerontol. Geriatr. 1992, 15, 249–260. [Google Scholar] [CrossRef]
- Smith, R.A.; Murphy, M.P. Animal and human studies with the mitochondria-targeted antioxidant mitoq. Ann. N. Y. Acad. Sci. 2010, 1201, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Gavrilova, S.I.; Sano, M.; Thomas, R.G.; Aisen, P.S.; Bachurin, S.O.; Seely, L.; Hung, D.; Dimebon, I. Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer’s disease: A randomised, double-blind, placebo-controlled study. Lancet 2008, 372, 207–215. [Google Scholar] [CrossRef]
- Grimm, M.O.; Mett, J.; Hartmann, T. The impact of vitamin e and other fat-soluble vitamins on Alzheimer’s disease. Int. J. Mol. Sci. 2016, 17, 1785. [Google Scholar] [CrossRef]
- Grimm, M.O.W.; Thiel, A.; Lauer, A.A.; Winkler, J.; Lehmann, J.; Regner, L.; Nelke, C.; Janitschke, D.; Benoist, C.; Streidenberger, O.; et al. Vitamin d and its analogues decrease amyloid-beta (abeta) formation and increase abeta-degradation. Int. J. Mol. Sci. 2017, 18, 2764. [Google Scholar] [CrossRef]
- Grimm, M.O.; Lehmann, J.; Mett, J.; Zimmer, V.C.; Grosgen, S.; Stahlmann, C.P.; Hundsdorfer, B.; Haupenthal, V.J.; Rothhaar, T.L.; Herr, C.; et al. Impact of vitamin d on amyloid precursor protein processing and amyloid-beta peptide degradation in Alzheimer’s disease. Neurodegener. Dis. 2014, 13, 75–81. [Google Scholar] [CrossRef]
- Grimm, M.O.; Regner, L.; Mett, J.; Stahlmann, C.P.; Schorr, P.; Nelke, C.; Streidenberger, O.; Stoetzel, H.; Winkler, J.; Zaidan, S.R.; et al. Tocotrienol affects oxidative stress, cholesterol homeostasis and the amyloidogenic pathway in neuroblastoma cells: Consequences for Alzheimer’s disease. Int. J. Mol. Sci. 2016, 17, 1809. [Google Scholar] [CrossRef] [PubMed]
- Green, R.; Allen, L.H.; Bjorke-Monsen, A.L.; Brito, A.; Gueant, J.L.; Miller, J.W.; Molloy, A.M.; Nexo, E.; Stabler, S.; Toh, B.H.; et al. Vitamin b12 deficiency. Nat. Rev. Dis. Primers 2017, 3, 17040. [Google Scholar] [CrossRef]
- McCaddon, A.; Regland, B.; Hudson, P.; Davies, G. Functional vitamin b(12) deficiency and Alzheimer disease. Neurology 2002, 58, 1395–1399. [Google Scholar] [CrossRef] [PubMed]
- McCaddon, A. Vitamin b12 in neurology and ageing; clinical and genetic aspects. Biochimie 2013, 95, 1066–1076. [Google Scholar] [CrossRef]
- Moreira, E.S.; Brasch, N.E.; Yun, J. Vitamin b12 protects against superoxide-induced cell injury in human aortic endothelial cells. Free Radic. Biol. Med. 2011, 51, 876–883. [Google Scholar] [CrossRef]
- Chan, W.; Almasieh, M.; Catrinescu, M.M.; Levin, L.A. Cobalamin-associated superoxide scavenging in neuronal cells is a potential mechanism for vitamin b12-deprivation optic neuropathy. Am. J. Pathol. 2018, 188, 160–172. [Google Scholar] [CrossRef]
- Manzanares, W.; Hardy, G. Vitamin b12: The forgotten micronutrient for critical care. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Karamshetty, V.; Acharya, J.D.; Ghaskadbi, S.; Goel, P. Mathematical modeling of glutathione status in type 2 diabetics with vitamin b12 deficiency. Front. Cell Dev. Biol. 2016, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Politis, A.; Olgiati, P.; Malitas, P.; Albani, D.; Signorini, A.; Polito, L.; De Mauro, S.; Zisaki, A.; Piperi, C.; Stamouli, E.; et al. Vitamin b12 levels in Alzheimer’s disease: Association with clinical features and cytokine production. J. Alzheimers Dis. 2010, 19, 481–488. [Google Scholar] [CrossRef]
- Birch, C.S.; Brasch, N.E.; McCaddon, A.; Williams, J.H. A novel role for vitamin b(12): Cobalamins are intracellular antioxidants in vitro. Free Radic. Biol. Med. 2009, 47, 184–188. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Orellana, D.I.; Gonzalez-Billault, C.; Maccioni, R.B. Interleukin-6 induces alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp. Cell Res. 2004, 295, 245–257. [Google Scholar] [CrossRef]
- Weiss, N. Mechanisms of increased vascular oxidant stress in hyperhomocys-teinemia and its impact on endothelial function. Curr. Drug Metab. 2005, 6, 27–36. [Google Scholar] [CrossRef]
- Kennedy, D.O. B vitamins and the brain: Mechanisms, dose and efficacy—A review. Nutrients 2016, 8, 68. [Google Scholar] [CrossRef]
- Evans, J.R.; Lawrenson, J.G. Antioxidant vitamin and mineral supplements for preventing age-related macular degeneration. Cochrane Database Syst. Rev. 2017, 7, CD000253. [Google Scholar] [CrossRef]
- Calderon-Ospina, C.A.; Nava-Mesa, M.O. B vitamins in the nervous system: Current knowledge of the biochemical modes of action and synergies of thiamine, pyridoxine, and cobalamin. CNS Neurosci. Ther. 2020, 26, 5–13. [Google Scholar] [CrossRef]
- Miller, A.L. The methionine-homocysteine cycle and its effects on cognitive diseases. Altern. Med. Rev. 2003, 8, 7–19. [Google Scholar]
- Selhub, J. Homocysteine metabolism. Annu. Rev. Nutr. 1999, 19, 217–246. [Google Scholar] [CrossRef] [PubMed]
- Hama, Y.; Hamano, T.; Shirafuji, N.; Hayashi, K.; Ueno, A.; Enomoto, S.; Nagata, M.; Kimura, H.; Matsunaga, A.; Ikawa, M.; et al. Influences of folate supplementation on homocysteine and cognition in patients with folate deficiency and cognitive impairment. Nutrients 2020, 12, 3138. [Google Scholar] [CrossRef] [PubMed]
- Surtees, R.; Leonard, J.; Austin, S. Association of demyelination with deficiency of cerebrospinal-fluid s-adenosylmethionine in inborn errors of methyl-transfer pathway. Lancet 1991, 338, 1550–1554. [Google Scholar] [CrossRef]
- Walker, A.K.; Jacobs, R.L.; Watts, J.L.; Rottiers, V.; Jiang, K.; Finnegan, D.M.; Shioda, T.; Hansen, M.; Yang, F.; Niebergall, L.J.; et al. A conserved srebp-1/phosphatidylcholine feedback circuit regulates lipogenesis in metazoans. Cell 2011, 147, 840–852. [Google Scholar] [CrossRef] [PubMed]
- van Deijk, A.F.; Broersen, L.M.; Verkuyl, J.M.; Smit, A.B.; Verheijen, M.H.G. High content analysis of hippocampal neuron-astrocyte co-cultures shows a positive effect of fortasyn connect on neuronal survival and postsynaptic maturation. Front. Neurosci. 2017, 11, 440. [Google Scholar] [CrossRef]
- Wurtman, R.J. A nutrient combination that can affect synapse formation. Nutrients 2014, 6, 1701–1710. [Google Scholar] [CrossRef]
- Soininen, H.; Solomon, A.; Visser, P.J.; Hendrix, S.B.; Blennow, K.; Kivipelto, M.; Hartmann, T. LipiDiDiet clinical study, g: 24-month intervention with a specific multinutrient in people with prodromal Alzheimer’s disease (LipiDiDiet): A randomised, double-blind, controlled trial. Lancet Neurol. 2017, 16, 965–975. [Google Scholar] [CrossRef]
- Herrmann, W.; Obeid, R. Causes and early diagnosis of vitamin b12 deficiency. Dtsch. Arztebl. Int. 2008, 105, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Briani, C.; Dalla Torre, C.; Citton, V.; Manara, R.; Pompanin, S.; Binotto, G.; Adami, F. Cobalamin deficiency: Clinical picture and radiological findings. Nutrients 2013, 5, 4521–4539. [Google Scholar] [CrossRef]
- Adamo, A.M. Nutritional factors and aging in demyelinating diseases. Genes Nutr. 2014, 9, 360. [Google Scholar] [CrossRef] [PubMed]
- Papuc, E.; Rejdak, K. The role of myelin damage in Alzheimer’s disease pathology. Arch. Med. Sci. 2020, 16, 345–351. [Google Scholar] [CrossRef]
- Zhuo, J.M.; Pratico, D. Acceleration of brain amyloidosis in an Alzheimer’s disease mouse model by a folate, vitamin b6 and b12-deficient diet. Exp. Gerontol. 2010, 45, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Riddell, D.R.; Christie, G.; Hussain, I.; Dingwall, C. Compartmentalization of beta-secretase (asp2) into low-buoyant density, noncaveolar lipid rafts. Curr. Biol. 2001, 11, 1288–1293. [Google Scholar] [CrossRef]
- Vetrivel, K.S.; Cheng, H.; Lin, W.; Sakurai, T.; Li, T.; Nukina, N.; Wong, P.C.; Xu, H.; Thinakaran, G. Association of gamma-secretase with lipid rafts in post-golgi and endosome membranes. J. Biol. Chem. 2004, 279, 44945–44954. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, J.M.; Pratico, D. Severe in vivo hyper-homocysteinemia is not associatedwith elevation of amyloid-beta peptides in the tg2576 mice. J. Alzheimers Dis. 2010, 21, 133–140. [Google Scholar] [CrossRef]
- Fuso, A.; Nicolia, V.; Cavallaro, R.A.; Ricceri, L.; D’Anselmi, F.; Coluccia, P.; Calamandrei, G.; Scarpa, S. B-vitamin deprivation induces hyperhomocysteinemia and brain s-adenosylhomocysteine, depletes brain s-adenosylmethionine, and enhances ps1 and bace expression and amyloid-beta deposition in mice. Mol. Cell Neurosci. 2008, 37, 731–746. [Google Scholar] [CrossRef]
- Fuso, A.; Nicolia, V.; Pasqualato, A.; Fiorenza, M.T.; Cavallaro, R.A.; Scarpa, S. Changes in presenilin 1 gene methylation pattern in diet-induced b vitamin deficiency. Neurobiol. Aging 2011, 32, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Honarpisheh, P.; Reynolds, C.R.; Blasco Conesa, M.P.; Moruno Manchon, J.F.; Putluri, N.; Bhattacharjee, M.B.; Urayama, A.; McCullough, L.D.; Ganesh, B.P. Dysregulated gut homeostasis observed prior to the accumulation of the brain amyloid-beta in tg2576 mice. Int. J. Mol. Sci. 2020, 21, 1711. [Google Scholar] [CrossRef]
- Christensen, E.I.; Nielsen, R.; Birn, H. From bowel to kidneys: The role of cubilin in physiology and disease. Nephrol. Dial. Transplant. 2013, 28, 274–281. [Google Scholar] [CrossRef]
- Vingtdeux, V.; Hamdane, M.; Gompel, M.; Begard, S.; Drobecq, H.; Ghestem, A.; Grosjean, M.E.; Kostanjevecki, V.; Grognet, P.; Vanmechelen, E.; et al. Phosphorylation of amyloid precursor carboxy-terminal fragments enhances their processing by a gamma-secretase-dependent mechanism. Neurobiol. Dis. 2005, 20, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.E.; Tian, Q.; Wei, W.; Peng, J.H.; Liu, G.P.; Zhou, X.W.; Wang, Q.; Wang, D.W.; Wang, J.Z. Homocysteine induces tau phosphorylation by inactivating protein phosphatase 2a in rat hippocampus. Neurobiol. Aging 2008, 29, 1654–1665. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Liu, Y.H.; Zhang, C.E.; Wang, Q.; Wei, Z.; Mousseau, D.D.; Wang, J.Z.; Tian, Q.; Liu, G.P. Folate/vitamin-b12 prevents chronic hyperhomocysteinemia-induced tau hyperphosphorylation and memory deficits in aged rats. J. Alzheimers Dis. 2011, 27, 639–650. [Google Scholar] [CrossRef]
- Koronyo-Hamaoui, M.; Koronyo, Y.; Ljubimov, A.V.; Miller, C.A.; Ko, M.K.; Black, K.L.; Schwartz, M.; Farkas, D.L. Identification of amyloid plaques in retinas from Alzheimer’s patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. Neuroimage 2011, 54 (Suppl. 1), S204–S217. [Google Scholar] [CrossRef]
- La Morgia, C.; Ross-Cisneros, F.N.; Koronyo, Y.; Hannibal, J.; Gallassi, R.; Cantalupo, G.; Sambati, L.; Pan, B.X.; Tozer, K.R.; Barboni, P.; et al. Melanopsin retinal ganglion cell loss in Alzheimer disease. Ann. Neurol. 2016, 79, 90–109. [Google Scholar] [CrossRef]
- Guo, J.; Ni, S.; Li, Q.; Wang, J.Z.; Yang, Y. Folate/vitamin b alleviates hyperhomocysteinemia-induced alzheimer-like pathologies in rat retina. Neurosci. Bull. 2019, 35, 325–335. [Google Scholar] [CrossRef]
- Andra, A.; Tanigawa, S.; Bito, T.; Ishihara, A.; Watanabe, F.; Yabuta, Y. Effects of vitamin b12 deficiency on amyloid-beta toxicity in caenorhabditis elegans. Antioxidants 2021, 10, 962. [Google Scholar] [CrossRef]
- Lam, A.B.; Kervin, K.; Tanis, J.E. Vitamin b12 impacts amyloid beta-induced proteotoxicity by regulating the methionine/s-adenosylmethionine cycle. Cell Rep. 2021, 36, 109753. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.; Link, C.D.; Butterfield, D.A. Oxidative stress precedes fibrillar deposition of Alzheimer’s disease amyloid beta-peptide (1-42) in a transgenic caenorhabditis elegans model. Neurobiol. Aging 2003, 24, 415–420. [Google Scholar] [CrossRef]
- Fong, S.; Teo, E.; Ng, L.F.; Chen, C.B.; Lakshmanan, L.N.; Tsoi, S.Y.; Moore, P.K.; Inoue, T.; Halliwell, B.; Gruber, J. Energy crisis precedes global metabolic failure in a novel caenorhabditis elegans Alzheimer disease model. Sci. Rep. 2016, 6, 33781. [Google Scholar] [CrossRef]
- McColl, G.; Roberts, B.R.; Pukala, T.L.; Kenche, V.B.; Roberts, C.M.; Link, C.D.; Ryan, T.M.; Masters, C.L.; Barnham, K.J.; Bush, A.I.; et al. Utility of an improved model of amyloid-beta (αβ1−42) toxicity in Caenorhabditis elegans for drug screening for Alzheimer’s disease. Mol. Neurodegener. 2012, 7, 57. [Google Scholar] [CrossRef]
- Alam, P.; Siddiqi, M.K.; Chaturvedi, S.K.; Zaman, M.; Khan, R.H. Vitamin b12 offers neuronal cell protection by inhibiting abeta-42 amyloid fibrillation. Int. J. Biol. Macromol. 2017, 99, 477–482. [Google Scholar] [CrossRef]
- Andrade, S.; Loureiro, J.A.; Pereira, M.C. Vitamin b12 inhibits abeta fibrillation and disaggregates preformed fibrils in the presence of synthetic neuronal membranes. ACS Chem. Neurosci. 2021, 12, 2491–2502. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xu, T. Methyl b12 protects pc12 cells against cytotoxicity induced by abeta(25–35). J. Cell Biochem. 2019; in press. [Google Scholar]
- Kaminsky, Y.G.; Marlatt, M.W.; Smith, M.A.; Kosenko, E.A. Subcellular and metabolic examination of amyloid-beta peptides in Alzheimer disease pathogenesis: Evidence for abeta(25–35). Exp. Neurol. 2010, 221, 26–37. [Google Scholar] [CrossRef]
- Malyshev, I.Y.; Wiegant, F.A.; Mashina, S.Y.; Torshin, V.I.; Goryacheva, A.V.; Khomenko, I.P.; Kruglov, S.V.; Pokidyshev, D.A.; Popkova, E.V.; Pshennikova, M.G.; et al. Possible use of adaptation to hypoxia in Alzheimer’s disease: A hypothesis. Med. Sci. Monit. 2005, 11, HY31–HY38. [Google Scholar]
- Zeng, H.; Chen, Q.; Zhao, B. Genistein ameliorates beta-amyloid peptide (25–35)-induced hippocampal neuronal apoptosis. Free Radic. Biol. Med. 2004, 36, 180–188. [Google Scholar] [CrossRef]
- Zhong, L.; Zhou, J.; Chen, X.; Lou, Y.; Liu, D.; Zou, X.; Yang, B.; Yin, Y.; Pan, Y. Quantitative proteomics study of the neuroprotective effects of b12 on hydrogen peroxide-induced apoptosis in sh-sy5y cells. Sci. Rep. 2016, 6, 22635. [Google Scholar] [CrossRef] [PubMed]
- Fuso, A.; Seminara, L.; Cavallaro, R.A.; D’Anselmi, F.; Scarpa, S. S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of ps1 and bace and beta-amyloid production. Mol. Cell Neurosci. 2005, 28, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, S.; Fuso, A.; D’Anselmi, F.; Cavallaro, R.A. Presenilin 1 gene silencing by s-adenosylmethionine: A treatment for Alzheimer disease? FEBS Lett. 2003, 541, 145–148. [Google Scholar] [CrossRef]
- Grimm, M.O.; Zimmer, V.C.; Lehmann, J.; Grimm, H.S.; Hartmann, T. The impact of cholesterol, dha, and sphingolipids on Alzheimer’s disease. Biomed. Res. Int. 2013, 2013, 814390. [Google Scholar] [CrossRef] [PubMed]
- Feringa, F.M.; van der Kant, R. Cholesterol and Alzheimer’s disease; from risk genes to pathological effects. Front. Aging Neurosci. 2021, 13, 690372. [Google Scholar] [CrossRef] [PubMed]
- Adaikalakoteswari, A.; Finer, S.; Voyias, P.D.; McCarthy, C.M.; Vatish, M.; Moore, J.; Smart-Halajko, M.; Bawazeer, N.; Al-Daghri, N.M.; McTernan, P.G.; et al. Vitamin b12 insufficiency induces cholesterol biosynthesis by limiting s-adenosylmethionine and modulating the methylation of srebf1 and ldlr genes. Clin. Epigenet. 2015, 7, 14. [Google Scholar] [CrossRef]
- Perla-Kajan, J.; Wloczkowska, O.; Ziola-Frankowska, A.; Frankowski, M.; Smith, A.D.; de Jager, C.A.; Refsum, H.; Jakubowski, H. Paraoxonase 1, b vitamins supplementation, and mild cognitive impairment. J. Alzheimers Dis. 2021, 81, 1211–1229. [Google Scholar] [CrossRef] [PubMed]
- Ford, A.H.; Flicker, L.; Alfonso, H.; Thomas, J.; Clarnette, R.; Martins, R.; Almeida, O.P. Vitamins b(12), b(6), and folic acid for cognition in older men. Neurology 2010, 75, 1540–1547. [Google Scholar] [CrossRef]
- Zhang, C.; Luo, J.; Yuan, C.; Ding, D. Vitamin b12, b6, or folate and cognitive function in community-dwelling older adults: A systematic review and meta-analysis. J. Alzheimers Dis. 2020, 77, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Guo, Y.; Men, J.; Fu, H.; Xu, T. The preventive efficacy of vitamin b supplements on the cognitive decline of elderly adults: A systematic review and meta-analysis. BMC Geriatr. 2021, 21, 367. [Google Scholar] [CrossRef]
- Ma, F.; Zhou, X.; Li, Q.; Zhao, J.; Song, A.; An, P.; Du, Y.; Xu, W.; Huang, G. Effects of folic acid and vitamin b12, alone and in combination on cognitive function and inflammatory factors in the elderly with mild cognitive impairment: A single-blind experimental design. Curr. Alzheimer Res. 2019, 16, 622–632. [Google Scholar] [CrossRef]
- Smith, A.D.; Smith, S.M.; de Jager, C.A.; Whitbread, P.; Johnston, C.; Agacinski, G.; Oulhaj, A.; Bradley, K.M.; Jacoby, R.; Refsum, H. Homocysteine-lowering by b vitamins slows the rate of accelerated brain atrophy in mild cognitive impairment: A randomized controlled trial. PLoS ONE 2010, 5, e12244. [Google Scholar] [CrossRef]
- de Jager, C.A.; Oulhaj, A.; Jacoby, R.; Refsum, H.; Smith, A.D. Cognitive and clinical outcomes of homocysteine-lowering b-vitamin treatment in mild cognitive impairment: A randomized controlled trial. Int. J. Geriatr. Psychiatry 2012, 27, 592–600. [Google Scholar] [CrossRef]
- Douaud, G.; Refsum, H.; de Jager, C.A.; Jacoby, R.; Nichols, T.E.; Smith, S.M.; Smith, A.D. Preventing Alzheimer’s disease-related gray matter atrophy by b-vitamin treatment. Proc. Natl. Acad. Sci. USA 2013, 110, 9523–9528. [Google Scholar] [CrossRef]
- Jerneren, F.; Elshorbagy, A.K.; Oulhaj, A.; Smith, S.M.; Refsum, H.; Smith, A.D. Brain atrophy in cognitively impaired elderly: The importance of long-chain omega-3 fatty acids and b vitamin status in a randomized controlled trial. Am. J. Clin. Nutr. 2015, 102, 215–221. [Google Scholar] [CrossRef]
- Oulhaj, A.; Jerneren, F.; Refsum, H.; Smith, A.D.; de Jager, C.A. Omega-3 fatty acid status enhances the prevention of cognitive decline by b vitamins in mild cognitive impairment. J. Alzheimers Dis. 2016, 50, 547–557. [Google Scholar] [CrossRef]
- Remington, R.; Lortie, J.J.; Hoffmann, H.; Page, R.; Morrell, C.; Shea, T.B. A nutritional formulation for cognitive performance in mild cognitive impairment: A placebo-controlled trial with an open-label extension. J. Alzheimers Dis. 2015, 48, 591–595. [Google Scholar] [CrossRef]
- Flicker, L.; Martins, R.N.; Thomas, J.; Acres, J.; Taddei, K.; Vasikaran, S.D.; Norman, P.; Jamrozik, K.; Almeida, O.P. B-vitamins reduce plasma levels of beta amyloid. Neurobiol. Aging 2008, 29, 303–305. [Google Scholar] [CrossRef]
- Frick, B.; Gruber, B.; Schroecksnadel, K.; Leblhuber, F.; Fuchs, D. Homocysteine but not neopterin declines in demented patients on b vitamins. J. Neural Transm. 2006, 113, 1815–1819. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Feng, L.; Zhang, X.; Wang, Y.; Wang, Y.; Tao, L.; Qin, Z.; Xiao, R. Dietary intakes and biomarker patterns of folate, vitamin b6, and vitamin b12 can be associated with cognitive impairment by hypermethylation of redox-related genes nudt15 and txnrd1. Clin. Epigenet. 2019, 11, 139. [Google Scholar] [CrossRef] [PubMed]
- van Dyck, C.H.; Lyness, J.M.; Rohrbaugh, R.M.; Siegal, A.P. Cognitive and psychiatric effects of vitamin b12 replacement in dementia with low serum b12 levels: A nursing home study. Int. Psychogeriatr. 2009, 21, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Ji, H.F. Associations between homocysteine, folic acid, vitamin b12 and Alzheimer’s disease: Insights from meta-analyses. J. Alzheimers Dis. 2015, 46, 777–790. [Google Scholar] [CrossRef]
- Lopes da Silva, S.; Vellas, B.; Elemans, S.; Luchsinger, J.; Kamphuis, P.; Yaffe, K.; Sijben, J.; Groenendijk, M.; Stijnen, T. Plasma nutrient status of patients with Alzheimer’s disease: Systematic review and meta-analysis. Alzheimers Dement. 2014, 10, 485–502. [Google Scholar] [CrossRef]
- Teunissen, C.E.; Lutjohann, D.; von Bergmann, K.; Verhey, F.; Vreeling, F.; Wauters, A.; Bosmans, E.; Bosma, H.; van Boxtel, M.P.; Maes, M.; et al. Combination of serum markers related to several mechanisms in Alzheimer’s disease. Neurobiol. Aging 2003, 24, 893–902. [Google Scholar] [CrossRef]
- Aisen, P.S.; Egelko, S.; Andrews, H.; Diaz-Arrastia, R.; Weiner, M.; DeCarli, C.; Jagust, W.; Miller, J.W.; Green, R.; Bell, K.; et al. A pilot study of vitamins to lower plasma homocysteine levels in Alzheimer disease. Am. J. Geriatr. Psychiatry 2003, 11, 246–249. [Google Scholar] [CrossRef]
- Sun, Y.; Lu, C.J.; Chien, K.L.; Chen, S.T.; Chen, R.C. Efficacy of multivitamin supplementation containing vitamins b6 and b12 and folic acid as adjunctive treatment with a cholinesterase inhibitor in Alzheimer’s disease: A 26-week, randomized, double-blind, placebo-controlled study in taiwanese patients. Clin. Ther. 2007, 29, 2204–2214. [Google Scholar] [CrossRef]
- Aisen, P.S.; Schneider, L.S.; Sano, M.; Diaz-Arrastia, R.; van Dyck, C.H.; Weiner, M.F.; Bottiglieri, T.; Jin, S.; Stokes, K.T.; Thomas, R.G.; et al. High-dose b vitamin supplementation and cognitive decline in Alzheimer disease: A randomized controlled trial. JAMA 2008, 300, 1774–1783. [Google Scholar] [CrossRef]
- Zhang, D.M.; Ye, J.X.; Mu, J.S.; Cui, X.P. Efficacy of vitamin b supplementation on cognition in elderly patients with cognitive-related diseases. J. Geriatr. Psychiatry Neurol. 2017, 30, 50–59. [Google Scholar] [CrossRef]
- Chan, A.; Paskavitz, J.; Remington, R.; Rasmussen, S.; Shea, T.B. Efficacy of a vitamin/nutriceutical formulation for early-stage Alzheimer’s disease: A 1-year, open-label pilot study with an 16-month caregiver extension. Am. J. Alzheimers Dis. Other Dement. 2008, 23, 571–585. [Google Scholar] [CrossRef]
- Remington, R.; Chan, A.; Paskavitz, J.; Shea, T.B. Efficacy of a vitamin/nutriceutical formulation for moderate-stage to later-stage Alzheimer’s disease: A placebo-controlled pilot study. Am. J. Alzheimers Dis. Other Dement. 2009, 24, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Remington, R.; Bechtel, C.; Larsen, D.; Samar, A.; Doshanjh, L.; Fishman, P.; Luo, Y.; Smyers, K.; Page, R.; Morrell, C.; et al. A phase ii randomized clinical trial of a nutritional formulation for cognition and mood in Alzheimer’s disease. J. Alzheimers Dis. 2015, 45, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Remington, R.; Bechtel, C.; Larsen, D.; Samar, A.; Page, R.; Morrell, C.; Shea, T.B. Maintenance of cognitive performance and mood for individuals with Alzheimer’s disease following consumption of a nutraceutical formulation: A one-year, open-label study. J. Alzheimers Dis. 2016, 51, 991–995. [Google Scholar] [CrossRef] [PubMed]
- Cornelli, U. Treatment of Alzheimer’s disease with a cholinesterase inhibitor combined with antioxidants. Neurodegener. Dis. 2010, 7, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Vakilian, A.; Razavi-Nasab, S.M.; Ravari, A.; Mirzaei, T.; Moghadam-Ahmadi, A.; Jalali, N.; Bahramabadi, R.; Rezayati, M.; Yazdanpanah-Ravari, A.; Bahmaniar, F.; et al. Vitamin b12 in association with antipsychotic drugs can modulate the expression of pro-/anti-inflammatory cytokines in Alzheimer disease patients. Neuroimmunomodulation 2017, 24, 310–319. [Google Scholar] [CrossRef]
- Rommer, P.S.; Fuchs, D.; Leblhuber, F.; Schroth, R.; Greilberger, M.; Tafeit, E.; Greilberger, J. Lowered levels of carbonyl proteins after vitamin b supplementation in patients with mild cognitive impairment and Alzheimer’s disease. Neurodegener. Dis. 2016, 16, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, J. The lipididiet trial: What does it add to the current evidence for fortasyn connect in early Alzheimer’s disease? Clin. Interv. Aging 2019, 14, 1481–1492. [Google Scholar] [CrossRef]
- Rijpma, A.; Meulenbroek, O.; van Hees, A.M.; Sijben, J.W.; Vellas, B.; Shah, R.C.; Bennett, D.A.; Scheltens, P.; Olde Rikkert, M.G. Effects of souvenaid on plasma micronutrient levels and fatty acid profiles in mild and mild-to-moderate Alzheimer’s disease. Alzheimers Res. Ther. 2015, 7, 51. [Google Scholar] [CrossRef]
- Scheltens, P.; Kamphuis, P.J.; Verhey, F.R.; Olde Rikkert, M.G.; Wurtman, R.J.; Wilkinson, D.; Twisk, J.W.; Kurz, A. Efficacy of a medical food in mild Alzheimer’s disease: A randomized, controlled trial. Alzheimers Dement. 2010, 6, 1–10.e11. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Martinez, L.; Farias, G.A.; Tapia, J.P.; Sanchez, M.P.; Fuentes, P.; Gloger, S.; Maccioni, R.B. Interventional study to evaluate the clinical effects and safety of the nutraceutical compound brainup-10(r) in a cohort of patients with Alzheimer’s disease: A multicenter, randomized, double-blind, and placebo-controlled trial. J. Alzheimers Dis. 2021, 81, 1231–1241. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liu, S.; Ge, B.; Zhou, D.; Li, M.; Li, W.; Ma, F.; Liu, Z.; Ji, Y.; Huang, G. Effects of folic acid and vitamin b12 supplementation on cognitive impairment and inflammation in patients with Alzheimer’s disease: A randomized, single-blinded, placebo-controlled trial. J. Prev. Alzheimers Dis. 2021, 8, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Weisshof, R.; Chermesh, I. Micronutrient deficiencies in inflammatory bowel disease. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 576–581. [Google Scholar] [CrossRef]
- Yakut, M.; Ustun, Y.; Kabacam, G.; Soykan, I. Serum vitamin b12 and folate status in patients with inflammatory bowel diseases. Eur. J. Intern. Med. 2010, 21, 320–323. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, F.; Algaba, A.; Guerra, I.; Chaparro, M.; De-La-Poza, G.; Valer, P.; Piqueras, B.; Bermejo, A.; Garcia-Alonso, J.; Perez, M.J.; et al. Should we monitor vitamin b12 and folate levels in crohn’s disease patients? Scand. J. Gastroenterol. 2013, 48, 1272–1277. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.E.; Park, S.J.; Park, J.J.; Cheon, J.H.; Kim, T.; Kim, W.H. Incidence and risk factors of micronutrient deficiency in patients with ibd and intestinal behcet’s disease: Folate, vitamin b12, 25-oh-vitamin d, and ferritin. BMC Gastroenterol. 2021, 21, 32. [Google Scholar] [CrossRef]
- Ward, M.G.; Kariyawasam, V.C.; Mogan, S.B.; Patel, K.V.; Pantelidou, M.; Sobczynska-Malefora, A.; Porte, F.; Griffin, N.; Anderson, S.H.; Sanderson, J.D.; et al. Prevalence and risk factors for functional vitamin b12 deficiency in patients with crohn’s disease. Inflamm. Bowel Dis. 2015, 21, 2839–2847. [Google Scholar] [CrossRef] [PubMed]
- Zingel, R.; Bohlken, J.; Kostev, K. Association between inflammatory bowel disease and dementia: A retrospective cohort study. J. Alzheimers Dis. 2021, 80, 1471–1478. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, H.E.; Bai, Y.M.; Tsai, S.J.; Su, T.P.; Chen, T.J.; Wang, Y.P.; Chen, M.H. Inflammatory bowel disease is associated with higher dementia risk: A nationwide longitudinal study. Gut 2021, 70, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Green, R. Vitamin b12 deficiency from the perspective of a practicing hematologist. Blood 2017, 129, 2603–2611. [Google Scholar] [CrossRef]
- Minalyan, A.; Benhammou, J.N.; Artashesyan, A.; Lewis, M.S.; Pisegna, J.R. Autoimmune atrophic gastritis: Current perspectives. Clin. Exp. Gastroenterol. 2017, 10, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.M.; Hoey, L.; Hughes, C.F.; Ward, M.; Clements, M.; Strain, J.; Cunningham, C.; Casey, M.C.; Tracey, F.; O’Kane, M.; et al. Associations of atrophic gastritis and proton-pump inhibitor drug use with vitamin b-12 status, and the impact of fortified foods, in older adults. Am. J. Clin. Nutr. 2021, 114, 1286–1294. [Google Scholar] [CrossRef]
- Sipponen, P.; Laxen, F.; Huotari, K.; Harkonen, M. Prevalence of low vitamin b12 and high homocysteine in serum in an elderly male population: Association with atrophic gastritis and helicobacter pylori infection. Scand. J. Gastroenterol. 2003, 38, 1209–1216. [Google Scholar] [CrossRef]
- Momtaz, Y.A.; Hamid, T.A.; Ibrahim, R. Gastritis may boost odds of dementia. Am. J. Alzheimers Dis. Other Dement. 2014, 29, 452–456. [Google Scholar] [CrossRef]
- Li, X.; Sundquist, J.; Zoller, B.; Sundquist, K. Dementia and Alzheimer’s disease risks in patients with autoimmune disorders. Geriatr. Gerontol. Int. 2018, 18, 1350–1355. [Google Scholar] [CrossRef]
- Lahner, E.; Annibale, B. Pernicious anemia: New insights from a gastroenterological point of view. World J. Gastroenterol. 2009, 15, 5121–5128. [Google Scholar] [CrossRef]
- Metzler, D.; Miller, W.H.; Edwards, S.C. Psychiatric manifestation of vitamin b-12 deficiency: An update. Jefferson J. Psychiatry 1991, 9, 8. [Google Scholar] [CrossRef][Green Version]
- Kountouras, J.; Tsolaki, M.; Gavalas, E.; Boziki, M.; Zavos, C.; Karatzoglou, P.; Chatzopoulos, D.; Venizelos, I. Relationship between helicobacter pylori infection and Alzheimer disease. Neurology 2006, 66, 938–940. [Google Scholar] [CrossRef]
- Choi, Y.J.; Shin, D.W.; Jang, W.; Lee, D.H.; Jeong, S.M.; Park, S.; Han, K.D.; Park, Y.G. Risk of dementia in gastric cancer survivors who underwent gastrectomy: A nationwide study in korea. Ann. Surg. Oncol. 2019, 26, 4229–4237. [Google Scholar] [CrossRef]
- Rotman, S.R.; Bishop, T.F. Proton pump inhibitor use in the U.S. Ambulatory setting, 2002–2009. PLoS ONE 2013, 8, e56060. [Google Scholar] [CrossRef]
- Saltzman, J.R.; Kemp, J.A.; Golner, B.B.; Pedrosa, M.C.; Dallal, G.E.; Russell, R.M. Effect of hypochlorhydria due to omeprazole treatment or atrophic gastritis on protein-bound vitamin b12 absorption. J. Am. Coll. Nutr. 1994, 13, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Schenk, B.E.; Festen, H.P.; Kuipers, E.J.; Klinkenberg-Knol, E.C.; Meuwissen, S.G. Effect of short- and long-term treatment with omeprazole on the absorption and serum levels of cobalamin. Aliment. Pharmacol. Ther. 1996, 10, 541–545. [Google Scholar] [CrossRef]
- Bradford, G.S.; Taylor, C.T. Omeprazole and vitamin b12 deficiency. Ann. Pharmacother. 1999, 33, 641–643. [Google Scholar] [CrossRef] [PubMed]
- Bellou, A.; Aimone-Gastin, I.; De Korwin, J.D.; Bronowicki, J.P.; Moneret-Vautrin, A.; Nicolas, J.P.; Bigard, M.A.; Gueant, J.L. Cobalamin deficiency with megaloblastic anaemia in one patient under long-term omeprazole therapy. J. Intern. Med. 1996, 240, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.R.; Schneider, J.L.; Zhao, W.; Corley, D.A. Proton pump inhibitor and histamine 2 receptor antagonist use and vitamin b12 deficiency. JAMA 2013, 310, 2435–2442. [Google Scholar] [CrossRef] [PubMed]
- Marcuard, S.P.; Albernaz, L.; Khazanie, P.G. Omeprazole therapy causes malabsorption of cyanocobalamin (vitamin b12). Ann. Intern. Med. 1994, 120, 211–215. [Google Scholar] [CrossRef]
- Ruscin, J.M.; Page, R.L., 2nd; Valuck, R.J. Vitamin b(12) deficiency associated with histamine(2)-receptor antagonists and a proton-pump inhibitor. Ann. Pharmacother. 2002, 36, 812–816. [Google Scholar] [CrossRef] [PubMed]
- Akter, S.; Hassan, M.R.; Shahriar, M.; Akter, N.; Abbas, M.G.; Bhuiyan, M.A. Cognitive impact after short-term exposure to different proton pump inhibitors: Assessment using cantab software. Alzheimers Res. Ther. 2015, 7, 79. [Google Scholar] [CrossRef]
- Song, Y.Q.; Li, Y.; Zhang, S.L.; Gao, J.; Feng, S.Y. Proton pump inhibitor use does not increase dementia and Alzheimer’s disease risk: An updated meta-analysis of published studies involving 642305 patients. PLoS ONE 2019, 14, e0219213. [Google Scholar] [CrossRef]
- Li, M.; Luo, Z.; Yu, S.; Tang, Z. Proton pump inhibitor use and risk of dementia: Systematic review and meta-analysis. Medicine 2019, 98, e14422. [Google Scholar] [CrossRef]
- Goldstein, F.C.; Steenland, K.; Zhao, L.; Wharton, W.; Levey, A.I.; Hajjar, I. Proton pump inhibitors and risk of mild cognitive impairment and dementia. J. Am. Geriatr. Soc. 2017, 65, 1969–1974. [Google Scholar] [CrossRef]
- Gomm, W.; von Holt, K.; Thome, F.; Broich, K.; Maier, W.; Fink, A.; Doblhammer, G.; Haenisch, B. Association of proton pump inhibitors with risk of dementia: A pharmacoepidemiological claims data analysis. JAMA Neurol. 2016, 73, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Haenisch, B.; von Holt, K.; Wiese, B.; Prokein, J.; Lange, C.; Ernst, A.; Brettschneider, C.; Konig, H.H.; Werle, J.; Weyerer, S.; et al. Risk of dementia in elderly patients with the use of proton pump inhibitors. Eur. Arch. Psychiatry Clin. Neurosci. 2015, 265, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Novotny, M.; Klimova, B.; Valis, M. PPI long term use: Risk of neurological adverse events? Front. Neurol. 2018, 9, 1142. [Google Scholar] [CrossRef]
- Ortiz-Guerrero, G.; Amador-Munoz, D.; Calderon-Ospina, C.A.; Lopez-Fuentes, D.; Nava Mesa, M.O. Proton pump inhibitors and dementia: Physiopathological mechanisms and clinical consequences. Neural Plast. 2018, 2018, 5257285. [Google Scholar] [CrossRef]
- Kumar, R.; Kumar, A.; Nordberg, A.; Langstrom, B.; Darreh-Shori, T. Proton pump inhibitors act with unprecedented potencies as inhibitors of the acetylcholine biosynthesizing enzyme-a plausible missing link for their association with incidence of dementia. Alzheimers Dement. 2020, 16, 1031–1042. [Google Scholar] [CrossRef]
- Schupbach, R.; Wegmuller, R.; Berguerand, C.; Bui, M.; Herter-Aeberli, I. Micronutrient status and intake in omnivores, vegetarians and vegans in Switzerland. Eur. J. Nutr. 2017, 56, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Fallon, N.; Dillon, S.A. Low intakes of iodine and selenium amongst vegan and vegetarian women highlight a potential nutritional vulnerability. Front. Nutr. 2020, 7, 72. [Google Scholar] [CrossRef]
- Elorinne, A.L.; Alfthan, G.; Erlund, I.; Kivimaki, H.; Paju, A.; Salminen, I.; Turpeinen, U.; Voutilainen, S.; Laakso, J. Food and nutrient intake and nutritional status of finnish vegans and non-vegetarians. PLoS ONE 2016, 11, e0148235. [Google Scholar] [CrossRef]
- Gallego-Narbon, A.; Zapatera, B.; Barrios, L.; Vaquero, M.P. Vitamin b12 and folate status in spanish lacto-ovo vegetarians and vegans. J. Nutr. Sci. 2019, 8, e7. [Google Scholar] [CrossRef]
- Lee, Y.P.; Loh, C.H.; Hwang, M.J.; Lin, C.P. Vitamin b12 deficiency and anemia in 140 taiwanese female lacto-vegetarians. J. Formos. Med. Assoc. 2021, 120, 2003–2009. [Google Scholar] [CrossRef] [PubMed]
- Alles, B.; Baudry, J.; Mejean, C.; Touvier, M.; Peneau, S.; Hercberg, S.; Kesse-Guyot, E. Comparison of sociodemographic and nutritional characteristics between self-reported vegetarians, vegans, and meat-eaters from the nutrinet-sante study. Nutrients 2017, 9, 1023. [Google Scholar] [CrossRef]
- Gilsing, A.M.; Crowe, F.L.; Lloyd-Wright, Z.; Sanders, T.A.; Appleby, P.N.; Allen, N.E.; Key, T.J. Serum concentrations of vitamin b12 and folate in british male omnivores, vegetarians and vegans: Results from a cross-sectional analysis of the epic-oxford cohort study. Eur. J. Clin. Nutr. 2010, 64, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Naik, S.; Mahalle, N.; Bhide, V. Identification of vitamin b12 deficiency in vegetarian indians. Br. J. Nutr. 2018, 119, 629–635. [Google Scholar] [CrossRef]
- Bakaloudi, D.R.; Halloran, A.; Rippin, H.L.; Oikonomidou, A.C.; Dardavesis, T.I.; Williams, J.; Wickramasinghe, K.; Breda, J.; Chourdakis, M. Intake and adequacy of the vegan diet. A systematic review of the evidence. Clin. Nutr. 2021, 40, 3503–3521. [Google Scholar] [CrossRef]
- Sofi, F.; Dinu, M.; Pagliai, G.; Cesari, F.; Gori, A.M.; Sereni, A.; Becatti, M.; Fiorillo, C.; Marcucci, R.; Casini, A. Low-calorie vegetarian versus mediterranean diets for reducing body weight and improving cardiovascular risk profile: Cardiveg study (cardiovascular prevention with vegetarian diet). Circulation 2018, 137, 1103–1113. [Google Scholar] [CrossRef]
- Lederer, A.K.; Hannibal, L.; Hettich, M.; Behringer, S.; Spiekerkoetter, U.; Steinborn, C.; Grundemann, C.; Zimmermann-Klemd, A.M.; Muller, A.; Simmet, T.; et al. Vitamin b12 status upon short-term intervention with a vegan diet-a randomized controlled trial in healthy participants. Nutrients 2019, 11, 2815. [Google Scholar] [CrossRef]
- Obeid, R.; Heil, S.G.; Verhoeven, M.M.A.; van den Heuvel, E.; de Groot, L.; Eussen, S. Vitamin b12 intake from animal foods, biomarkers, and health aspects. Front. Nutr. 2019, 6, 93. [Google Scholar] [CrossRef]
- Engel, M.G.; Kern, H.J.; Brenna, J.T.; Mitmesser, S.H. Micronutrient gaps in three commercial weight-loss diet plans. Nutrients 2018, 10, 108. [Google Scholar] [CrossRef]
- Watanabe, F.; Bito, T. Vitamin b12 sources and microbial interaction. Exp. Biol. Med. 2018, 243, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Sela, I.; Yaskolka Meir, A.; Brandis, A.; Krajmalnik-Brown, R.; Zeibich, L.; Chang, D.; Dirks, B.; Tsaban, G.; Kaplan, A.; Rinott, E.; et al. Wolffia globosa-mankai plant-based protein contains bioactive vitamin b12 and is well absorbed in humans. Nutrients 2020, 12, 3067. [Google Scholar] [CrossRef] [PubMed]
- Skrovankova, S. Seaweed vitamins as nutraceuticals. Adv. Food Nutr. Res. 2011, 64, 357–369. [Google Scholar]
- Nakos, M.; Pepelanova, I.; Beutel, S.; Krings, U.; Berger, R.G.; Scheper, T. Isolation and analysis of vitamin b12 from plant samples. Food Chem. 2017, 216, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Croft, M.T.; Lawrence, A.D.; Raux-Deery, E.; Warren, M.J.; Smith, A.G. Algae acquire vitamin b12 through a symbiotic relationship with bacteria. Nature 2005, 438, 90–93. [Google Scholar] [CrossRef]
- McClements, D.J. Development of next-generation nutritionally fortified plant-based milk substitutes: Structural design principles. Foods 2020, 9, 421. [Google Scholar] [CrossRef]
- Brito, A.; Habeych, E.; Silva-Zolezzi, I.; Galaffu, N.; Allen, L.H. Methods to assess vitamin b12 bioavailability and technologies to enhance its absorption. Nutr. Rev. 2018, 76, 778–792. [Google Scholar] [CrossRef]
- Del Bo, C.; Riso, P.; Gardana, C.; Brusamolino, A.; Battezzati, A.; Ciappellano, S. Effect of two different sublingual dosages of vitamin b12 on cobalamin nutritional status in vegans and vegetarians with a marginal deficiency: A randomized controlled trial. Clin. Nutr. 2019, 38, 575–583. [Google Scholar] [CrossRef]
- Medawar, E.; Huhn, S.; Villringer, A.; Veronica Witte, A. The effects of plant-based diets on the body and the brain: A systematic review. Transl. Psychiatry 2019, 9, 226. [Google Scholar] [CrossRef] [PubMed]
- Bowman, S.A. A vegetarian-style dietary pattern is associated with lower energy, saturated fat, and sodium intakes; and higher whole grains, legumes, nuts, and soy intakes by adults: National health and nutrition examination surveys 2013–2016. Nutrients 2020, 12, 2668. [Google Scholar] [CrossRef] [PubMed]
- Pistollato, F.; Iglesias, R.C.; Ruiz, R.; Aparicio, S.; Crespo, J.; Lopez, L.D.; Manna, P.P.; Giampieri, F.; Battino, M. Nutritional patterns associated with the maintenance of neurocognitive functions and the risk of dementia and Alzheimer’s disease: A focus on human studies. Pharmacol. Res. 2018, 131, 32–43. [Google Scholar] [CrossRef]
- Jiang, Y.W.; Sheng, L.T.; Pan, X.F.; Feng, L.; Yuan, J.M.; Pan, A.; Koh, W.P. Meat consumption in midlife and risk of cognitive impairment in old age: The singapore chinese health study. Eur. J. Nutr. 2020, 59, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.J. Dietary patterns and cognitive decline in taiwanese aged 65 years and older. Int. J. Geriatr. Psychiatry 2015, 30, 523–530. [Google Scholar] [CrossRef]
- Li, D. Chemistry behind vegetarianism. J. Agric. Food Chem. 2011, 59, 777–784. [Google Scholar] [CrossRef]
- Leitzmann, C. Vegetarian diets: What are the advantages? Forum Nutr. 2005, 57, 147–156. [Google Scholar]
- Malcomson, F.C.; Mathers, J.C. Nutrition and ageing. Subcell Biochem. 2018, 90, 373–424. [Google Scholar]
- Fabek, H.; Sanchez-Hernandez, D.; Ahmed, M.; Marinangeli, C.P.F.; House, J.D.; Anderson, G.H. An examination of contributions of animal-and plant-based dietary patterns on the nutrient quality of diets of adult canadians. Appl. Physiol. Nutr. Metab. 2021, 46, 877–886. [Google Scholar] [CrossRef]
- Weikert, C.; Trefflich, I.; Menzel, J.; Obeid, R.; Longree, A.; Dierkes, J.; Meyer, K.; Herter-Aeberli, I.; Mai, K.; Stangl, G.I.; et al. Vitamin and mineral status in a vegan diet. Dtsch. Arztebl. Int. 2020, 117, 575–582. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lauer, A.A.; Grimm, H.S.; Apel, B.; Golobrodska, N.; Kruse, L.; Ratanski, E.; Schulten, N.; Schwarze, L.; Slawik, T.; Sperlich, S.; et al. Mechanistic Link between Vitamin B12 and Alzheimer’s Disease. Biomolecules 2022, 12, 129. https://doi.org/10.3390/biom12010129
Lauer AA, Grimm HS, Apel B, Golobrodska N, Kruse L, Ratanski E, Schulten N, Schwarze L, Slawik T, Sperlich S, et al. Mechanistic Link between Vitamin B12 and Alzheimer’s Disease. Biomolecules. 2022; 12(1):129. https://doi.org/10.3390/biom12010129
Chicago/Turabian StyleLauer, Anna Andrea, Heike Sabine Grimm, Birgit Apel, Nataliya Golobrodska, Lara Kruse, Elina Ratanski, Noemi Schulten, Laura Schwarze, Thomas Slawik, Saskia Sperlich, and et al. 2022. "Mechanistic Link between Vitamin B12 and Alzheimer’s Disease" Biomolecules 12, no. 1: 129. https://doi.org/10.3390/biom12010129
APA StyleLauer, A. A., Grimm, H. S., Apel, B., Golobrodska, N., Kruse, L., Ratanski, E., Schulten, N., Schwarze, L., Slawik, T., Sperlich, S., Vohla, A., & Grimm, M. O. W. (2022). Mechanistic Link between Vitamin B12 and Alzheimer’s Disease. Biomolecules, 12(1), 129. https://doi.org/10.3390/biom12010129