
The Builders of the Junction: Roles of Junctophilin1 and Junctophilin2 in the Assembly of the Sarcoplasmic Reticulum–Plasma Membrane Junctions in Striated Muscle

Abstract
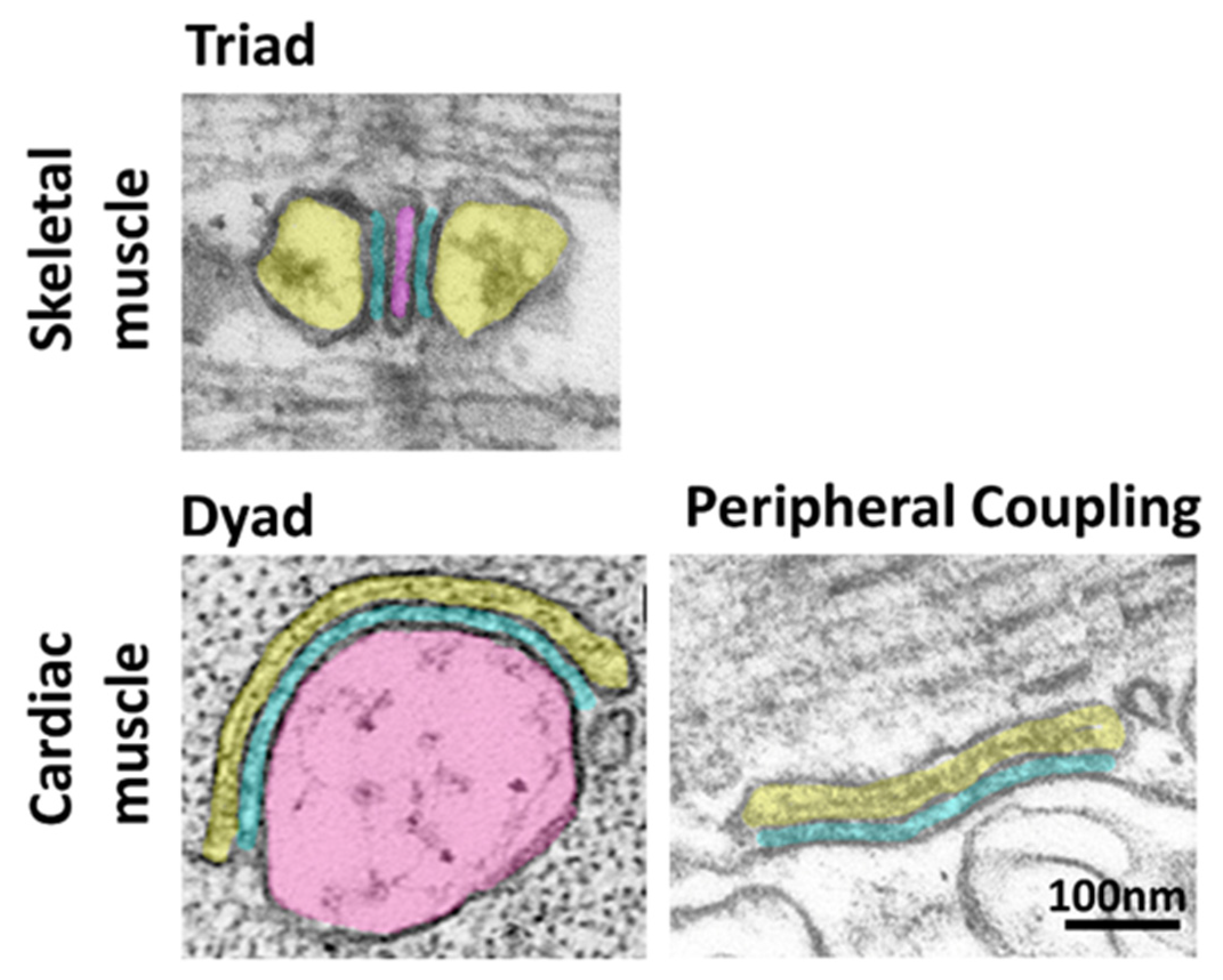
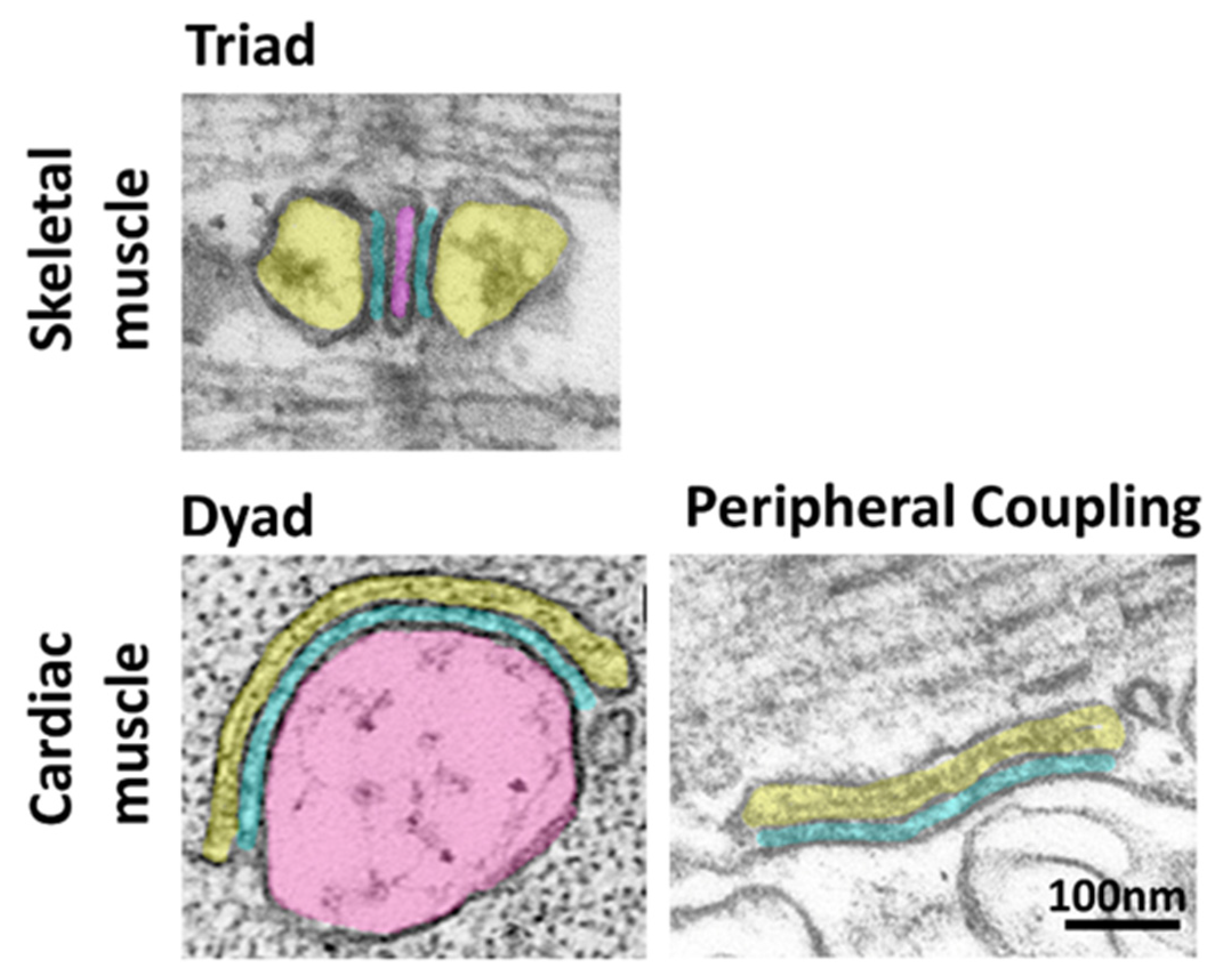
:1. Introduction
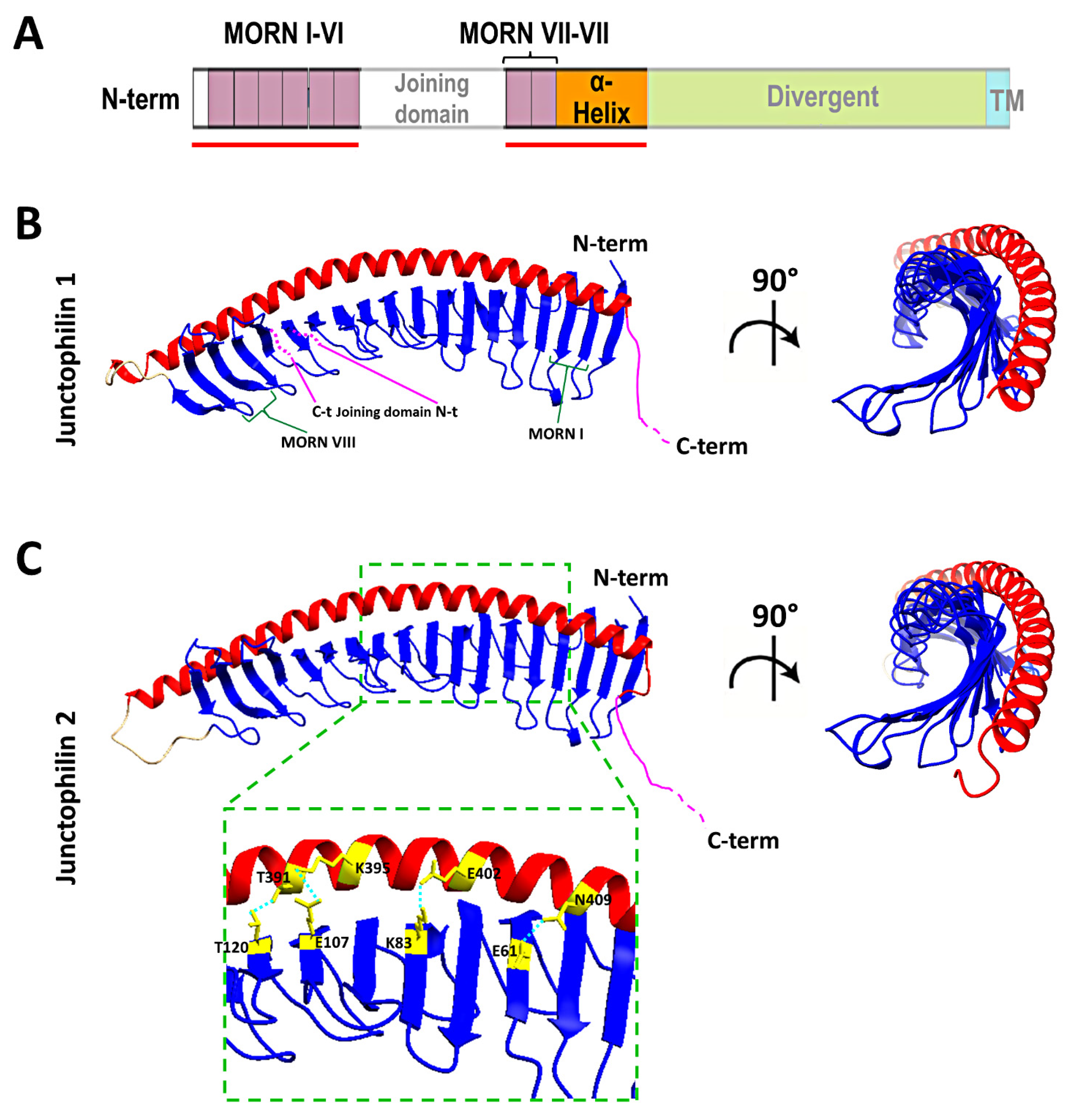
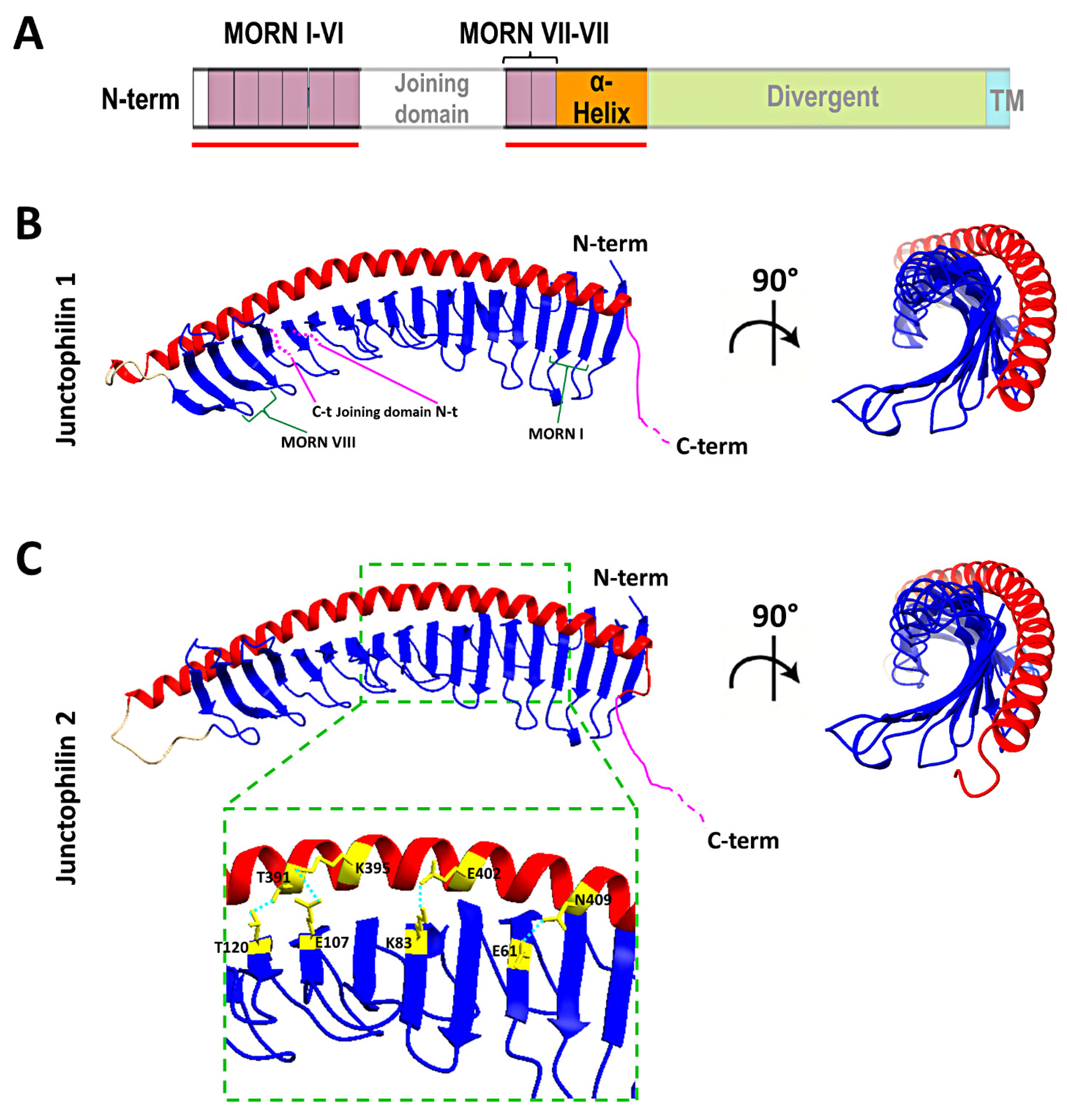
2. The Junctophilin Family
3. Recruitment of Junctional Proteins by Junctophilins1 and 2
3.1. Recruitment of Skeletal Muscle Junctional Proteins
3.2. Recruitment of Cardiac Muscle Junctional Proteins
4. Functional Studies on Junctophilins 1 and 2
4.1. Junctophilin 1
4.2. Junctophilin 2
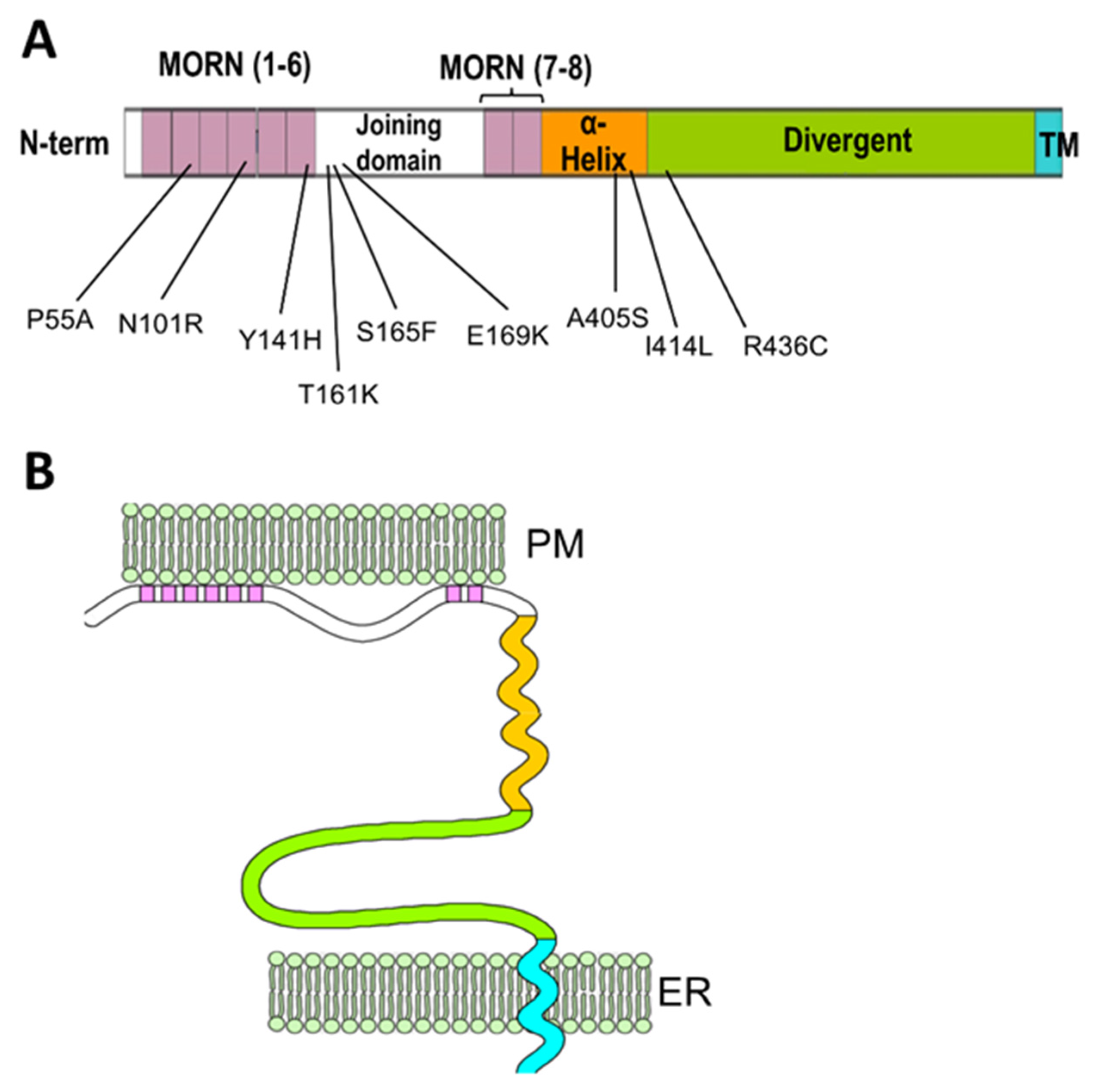
5. Post-Transcriptional Regulation of JPH1 and JPH2
6. New Insights from Deep Learning Protein Structure Prediction
7. Closing Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huxley, A.F.; Taylor, R.E. Local activation of striated muscle fibres. J. Physiol. 1958, 144, 426–441. [Google Scholar] [CrossRef]
- Huxley, A.F. Local activation of striated muscle from the frog and the crab. J. Physiol. 1957, 135, 17–18. [Google Scholar] [PubMed]
- Porter, K.R.; Palade, G.E. Studies on the endoplasmic reticulum. III. Its form and distribution in striated muscle cells. J. Biophys. Biochem. Cytol. 1957, 3, 269–300. [Google Scholar] [CrossRef]
- Perni, S.; Marsden, K.C.; Escobar, M.; Hollingworth, S.; Baylor, S.M.; Franzini-Armstrong, C. Structural and functional properties of ryanodine receptor type 3 in zebrafish tail muscle. J. Gen. Physiol. 2015, 145, 253. [Google Scholar] [CrossRef] [Green Version]
- Lavorato, M.; Huang, T.Q.; Iyer, V.R.; Perni, S.; Meissner, G.; Franzini-Armstrong, C. Dyad content is reduced in cardiac myocytes of mice with impaired calmodulin regulation of RyR2. J. Muscle Res. Cell Motil. 2015, 36, 205–214. [Google Scholar] [CrossRef]
- Perni, S.; Iyer, V.R.; Franzini-Armstrong, C. Ultrastructure of cardiac muscle in reptiles and birds: Optimizing and/or reducing the probability of transmission between calcium release units. J. Muscle Res. Cell Motil. 2012, 33, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Franzini-Armstrong, C. Fine Structure of Sarcoplasmic Reticulum and Tranverse Tubular System in Muscle Fibers. Fed. Proc. 1964, 23, 887–895. [Google Scholar] [PubMed]
- Franzini-Armstrong, C. STUDIES OF THE TRIAD: I. Structure of the Junction in Frog Twitch Fibers. J. Cell Biol. 1970, 47, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Franzini-Armstrong, C.; Protasi, F.; Ramesh, V. Shape, size, and distribution of Ca2+ release units and couplons in skeletal and cardiac muscles. Biophys. J. 1999, 77, 1528–1539. [Google Scholar] [CrossRef] [Green Version]
- Bossen, E.H.; Sommer, J.R. Comparative stereology of the lizard and frog myocardium. Tissue Cell 1984, 16, 173–178. [Google Scholar] [CrossRef]
- Bossen, E.H.; Sommer, J.R.; Waugh, R.A. Comparative stereology of the mouse and finch left ventricle. Tissue Cell 1978, 10, 773–784. [Google Scholar] [CrossRef]
- Grimley, P.M.; Edwards, G.A. The ultrastructure of cardiac desnosomes in the toad and their relationship to the intercalated disc. J. Biophys. Biochem. Cytol. 1960, 8, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Santer, R.M.; Cobb, J.L. The fine structure of the heart of the teleost, Pleuronectes platessa L. Z. Zellforsch. Mikrosk. Anat. 1972, 131, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Fabiato, A. Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J. Gen. Physiol. 1985, 85, 247–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Block, B.A.; Imagawa, T.; Campbell, K.P.; Franzini-Armstrong, C. Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J. Cell Biol. 1988, 107, 2587–2600. [Google Scholar] [CrossRef] [PubMed]
- Schredelseker, J.; Dayal, A.; Schwerte, T.; Franzini-Armstrong, C.; Grabner, M. Proper restoration of excitation-contraction coupling in the dihydropyridine receptor β1-null zebrafish relaxed is an exclusive function of the β1a subunit. J. Biol. Chem. 2009, 284, 1242–1251. [Google Scholar] [CrossRef] [Green Version]
- Weissgerber, P.; Held, B.; Bloch, W.; Kaestner, L.; Chien, K.R.; Fleischmann, B.K.; Lipp, P.; Flockerzi, V.; Freichel, M. Reduced cardiac L-type Ca2+ current in Cavβ2−/− embryos impairs cardiac development and contraction with secondary defects in vascular maturation. Circ. Res. 2006, 99, 749–757. [Google Scholar] [CrossRef] [Green Version]
- Pragnell, M.; De Waard, M.; Mori, Y.; Tanabe, T.; Snutch, T.P.; Campbell, K.P. Calcium channel β-subunit binds to a conserved motif in the I-II cytoplasmic linker of the alpha 1-subunit. Nature 1994, 368, 67–70. [Google Scholar] [CrossRef]
- Chien, A.J.; Zhao, X.; Shirokov, R.E.; Puri, T.S.; Chang, C.F.; Sun, D.; Rios, E.; Hosey, M.M. Roles of a membrane-localized β subunit in the formation and targeting of functional L-type Ca2+ channels. J. Biol. Chem. 1995, 270, 30036–30044. [Google Scholar] [CrossRef] [Green Version]
- Nelson, B.R.; Wu, F.; Liu, Y.; Anderson, D.M.; McAnally, J.; Lin, W.; Cannon, S.C.; Bassel-Duby, R.; Olson, E.N. Skeletal muscle-specific T-tubule protein STAC3 mediates voltage-induced Ca2+ release and contractility. Proc. Natl. Acad. Sci. USA 2013, 110, 11881–11886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horstick, E.J.; Linsley, J.W.; Dowling, J.J.; Hauser, M.A.; McDonald, K.K.; Ashley-Koch, A.; Saint-Amant, L.; Satish, A.; Cui, W.W.; Zhou, W.; et al. Stac3 is a component of the excitation-contraction coupling machinery and mutated in Native American myopathy. Nat. Commun. 2013, 4, 1952. [Google Scholar] [CrossRef] [Green Version]
- Polster, A.; Perni, S.; Bichraoui, H.; Beam, K.G. Stac adaptor proteins regulate trafficking and function of muscle and neuronal L-type Ca2+ channels. Proc. Natl. Acad. Sci. USA 2015, 112, 602–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polster, A.; Nelson, B.R.; Olson, E.N.; Beam, K.G. Stac3 has a direct role in skeletal muscle-type excitation-contraction coupling that is disrupted by a myopathy-causing mutation. Proc. Natl. Acad. Sci. USA 2016, 113, 10986–10991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polster, A.; Nelson, B.R.; Papadopoulos, S.; Olson, E.N.; Beam, K.G. Stac proteins associate with the critical domain for excitation-contraction coupling in the II-III loop of CaV1.1. J. Gen. Physiol. 2018, 150, 613–624. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, T.; Beam, K.G.; Adams, B.A.; Niidome, T.; Numa, S. Regions of the skeletal muscle dihydropyridine receptor critical for excitation-contraction coupling. Nature 1990, 346, 567–569. [Google Scholar] [CrossRef] [PubMed]
- Nishi, M.; Sakagami, H.; Komazaki, S.; Kondo, H.; Takeshima, H. Coexpression of junctophilin type 3 and type 4 in brain. Brain Res. Mol. Brain Res. 2003, 118, 102–110. [Google Scholar] [CrossRef]
- Takeshima, H.; Komazaki, S.; Nishi, M.; Iino, M.; Kangawa, K. Junctophilins: A novel family of junctional membrane complex proteins. Mol. Cell 2000, 6, 11–22. [Google Scholar] [CrossRef]
- Garbino, A.; van Oort, R.J.; Dixit, S.S.; Landstrom, A.P.; Ackerman, M.J.; Wehrens, X.H. Molecular evolution of the junctophilin gene family. Physiol. Genom. 2009, 37, 175–186. [Google Scholar] [CrossRef] [Green Version]
- Bennett, H.J.; Davenport, J.B.; Collins, R.F.; Trafford, A.W.; Pinali, C.; Kitmitto, A. Human junctophilin-2 undergoes a structural rearrangement upon binding PtdIns(3,4,5)P3 and the S101R mutation identified in hypertrophic cardiomyopathy obviates this response. Biochem. J. 2013, 456, 205–217. [Google Scholar] [CrossRef] [Green Version]
- Kakizawa, S.; Moriguchi, S.; Ikeda, A.; Iino, M.; Takeshima, H. Functional cross-talk between cell-surface and intracellular channels mediated by junctophilins essential for neuronal functions. Cerebellum 2008, 7, 385–391. [Google Scholar] [CrossRef]
- Rossi, D.; Scarcella, A.M.; Liguori, E.; Lorenzini, S.; Pierantozzi, E.; Kutchukian, C.; Jacquemond, V.; Messa, M.; De Camilli, P.; Sorrentino, V. Molecular determinants of homo- and heteromeric interactions of Junctophilin-1 at triads in adult skeletal muscle fibers. Proc. Natl. Acad. Sci. USA 2019, 116, 15716–15724. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liu, H.; Raval, M.H.; Wan, J.; Yengo, C.M.; Liu, W.; Zhang, M. Structure of the MORN4/Myo3a Tail Complex Reveals MORN Repeats as Protein Binding Modules. Structure 2019, 27, 1366–1374.e1363. [Google Scholar] [CrossRef]
- Sajko, S.; Grishkovskaya, I.; Kostan, J.; Graewert, M.; Setiawan, K.; Trubestein, L.; Niedermuller, K.; Gehin, C.; Sponga, A.; Puchinger, M.; et al. Structures of three MORN repeat proteins and a re-evaluation of the proposed lipid-binding properties of MORN repeats. PLoS ONE 2020, 15, e0242677. [Google Scholar] [CrossRef]
- Mikami, K.; Saavedra, L.; Hiwatashi, Y.; Uji, T.; Hasebe, M.; Sommarin, M. A dibasic amino acid pair conserved in the activation loop directs plasma membrane localization and is necessary for activity of plant type I/II phosphatidylinositol phosphate kinase. Plant Physiol. 2010, 153, 1004–1015. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Hu, J.; White, F.K.H.; Williamson, J.; Klymchenko, A.S.; Murthy, A.; Workman, S.W.; Tseng, G.N. S-Palmitoylation of junctophilin-2 is critical for its role in tethering the sarcoplasmic reticulum to the plasma membrane. J. Biol. Chem. 2019, 294, 13487–13501. [Google Scholar] [CrossRef] [PubMed]
- Hogea, A.; Shah, S.; Jones, F.; Carver, C.M.; Hao, H.; Liang, C.; Huang, D.; Du, X.; Gamper, N. Junctophilin-4 facilitates inflammatory signalling at plasma membrane-endoplasmic reticulum junctions in sensory neurons. J. Physiol. 2021, 599, 2103–2123. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Pan, Z.F.; Huang, X.; Wu, B.W.; Li, T.; Kang, M.X.; Ge, R.S.; Hu, X.Y.; Zhang, Y.H.; Ge, L.J.; et al. Junctophilin 3 expresses in pancreatic beta cells and is required for glucose-stimulated insulin secretion. Cell Death Dis. 2016, 7, e2275. [Google Scholar] [CrossRef] [Green Version]
- Woo, J.S.; Srikanth, S.; Nishi, M.; Ping, P.; Takeshima, H.; Gwack, Y. Junctophilin-4, a component of the endoplasmic reticulum-plasma membrane junctions, regulates Ca2+ dynamics in T cells. Proc. Natl. Acad. Sci. USA 2016, 113, 2762–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pla-Martin, D.; Calpena, E.; Lupo, V.; Marquez, C.; Rivas, E.; Sivera, R.; Sevilla, T.; Palau, F.; Espinos, C. Junctophilin-1 is a modifier gene of GDAP1-related Charcot-Marie-Tooth disease. Hum. Mol. Genet. 2015, 24, 213–229. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, H.A.T.; Griffin, C.S.; Yamasaki, E.; Thakore, P.; Lane, C.; Greenstein, A.S.; Earley, S. Nanoscale coupling of junctophilin-2 and ryanodine receptors regulates vascular smooth muscle cell contractility. Proc. Natl. Acad. Sci. USA 2019, 116, 21874–21881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.J.; Quintanilla, C.G.; Liou, J. Recent insights into mammalian ER-PM junctions. Curr. Opin. Cell Biol. 2019, 57, 99–105. [Google Scholar] [CrossRef]
- Close, M.; Perni, S.; Franzini-Armstrong, C.; Cundall, D. Highly extensible skeletal muscle in snakes. J. Exp. Biol. 2014, 217, 2445–2448. [Google Scholar] [CrossRef] [Green Version]
- Rome, L.C.; Syme, D.A.; Hollingworth, S.; Lindstedt, S.L.; Baylor, S.M. The whistle and the rattle: The design of sound producing muscles. Proc. Natl. Acad. Sci. USA 1996, 93, 8095–8100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golini, L.; Chouabe, C.; Berthier, C.; Cusimano, V.; Fornaro, M.; Bonvallet, R.; Formoso, L.; Giacomello, E.; Jacquemond, V.; Sorrentino, V. Junctophilin 1 and 2 proteins interact with the L-type Ca2+ channel dihydropyridine receptors (DHPRs) in skeletal muscle. J. Biol. Chem. 2011, 286, 43717–43725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakada, T.; Kashihara, T.; Komatsu, M.; Kojima, K.; Takeshita, T.; Yamada, M. Physical interaction of junctophilin and the CaV1.1 C terminus is crucial for skeletal muscle contraction. Proc. Natl. Acad. Sci. USA 2018, 115, 4507–4512. [Google Scholar] [CrossRef] [Green Version]
- Perni, S.; Lavorato, M.; Beam, K.G. De novo reconstitution reveals the proteins required for skeletal muscle voltage-induced Ca2+ release. Proc. Natl. Acad. Sci. USA 2017, 114, 13822–13827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phimister, A.J.; Lango, J.; Lee, E.H.; Ernst-Russell, M.A.; Takeshima, H.; Ma, J.; Allen, P.D.; Pessah, I.N. Conformation-dependent stability of junctophilin 1 (JP1) and ryanodine receptor type 1 (RyR1) channel complex is mediated by their hyper-reactive thiols. J. Biol. Chem. 2007, 282, 8667–8677. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Komazaki, S.; Sasamoto, K.; Yoshida, M.; Nishi, M.; Kitamura, K.; Takeshima, H. Deficiency of triad junction and contraction in mutant skeletal muscle lacking junctophilin type 1. J. Cell Biol. 2001, 154, 1059–1067. [Google Scholar] [CrossRef] [Green Version]
- Perni, S.; Beam, K. Neuronal junctophilins recruit specific CaV and RyR isoforms to ER-PM junctions and functionally alter CaV2.1 and CaV2.2. eLife 2021, 10, e64249. [Google Scholar] [CrossRef]
- Jiang, M.; Zhang, M.; Howren, M.; Wang, Y.; Tan, A.; Balijepalli, R.C.; Huizar, J.F.; Tseng, G.N. JPH-2 interacts with Cai-handling proteins and ion channels in dyads: Contribution to premature ventricular contraction-induced cardiomyopathy. Heart Rhythm 2016, 13, 743–752. [Google Scholar] [CrossRef] [Green Version]
- Guo, A.; Hall, D.; Zhang, C.; Peng, T.; Miller, J.D.; Kutschke, W.; Grueter, C.E.; Johnson, F.L.; Lin, R.Z.; Song, L.S. Molecular Determinants of Calpain-dependent Cleavage of Junctophilin-2 Protein in Cardiomyocytes. J. Biol. Chem. 2015, 290, 17946–17955. [Google Scholar] [CrossRef] [Green Version]
- Beavers, D.L.; Wang, W.; Ather, S.; Voigt, N.; Garbino, A.; Dixit, S.S.; Landstrom, A.P.; Li, N.; Wang, Q.; Olivotto, I.; et al. Mutation E169K in junctophilin-2 causes atrial fibrillation due to impaired RyR2 stabilization. J. Am. Coll. Cardiol. 2013, 62, 2010–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, L.; Zahradnikova, A., Jr.; Rizzetto, R.; Boncompagni, S.; Rabesahala de Meritens, C.; Zhang, Y.; Joanne, P.; Marques-Sule, E.; Aguilar-Sanchez, Y.; Fernandez-Tenorio, M.; et al. Impaired Binding to Junctophilin-2 and Nanostructural Alteration in CPVT Mutation. Circ. Res. 2021, 129, e35–e52. [Google Scholar] [CrossRef] [PubMed]
- Komazaki, S.; Ito, K.; Takeshima, H.; Nakamura, H. Deficiency of triad formation in developing skeletal muscle cells lacking junctophilin type 1. FEBS Lett. 2002, 524, 225–229. [Google Scholar] [CrossRef] [Green Version]
- Hirata, Y.; Brotto, M.; Weisleder, N.; Chu, Y.; Lin, P.; Zhao, X.; Thornton, A.; Komazaki, S.; Takeshima, H.; Ma, J.; et al. Uncoupling store-operated Ca2+ entry and altered Ca2+ release from sarcoplasmic reticulum through silencing of junctophilin genes. Biophys. J. 2006, 90, 4418–4427. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Ding, X.; Lopez, J.R.; Takeshima, H.; Ma, J.; Allen, P.D.; Eltit, J.M. Impaired Orai1-mediated resting Ca2+ entry reduces the cytosolic [Ca2+] and sarcoplasmic reticulum Ca2+ loading in quiescent junctophilin 1 knock-out myotubes. J. Biol. Chem. 2010, 285, 39171–39179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Oort, R.J.; Garbino, A.; Wang, W.; Dixit, S.S.; Landstrom, A.P.; Gaur, N.; De Almeida, A.C.; Skapura, D.G.; Rudy, Y.; Burns, A.R.; et al. Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation 2011, 123, 979–988. [Google Scholar] [CrossRef] [Green Version]
- De Bruijn, S.; Galloo, X.; De Keulenaer, G.; Prihadi, E.A.; Brands, C.; Helbert, M. A special case of hypertrophic cardiomyopathy with a differential diagnosis of isolated cardiac amyloidosis or junctophilin type 2 associated cardiomyopathy. Acta Clin. Belg. 2021, 76, 136–143. [Google Scholar] [CrossRef]
- Landstrom, A.P.; Weisleder, N.; Batalden, K.B.; Bos, J.M.; Tester, D.J.; Ommen, S.R.; Wehrens, X.H.; Claycomb, W.C.; Ko, J.K.; Hwang, M.; et al. Mutations in JPH2-encoded junctophilin-2 associated with hypertrophic cardiomyopathy in humans. J. Mol. Cell. Cardiol. 2007, 42, 1026–1035. [Google Scholar] [CrossRef] [Green Version]
- Woo, J.S.; Cho, C.H.; Lee, K.J.; Kim, D.H.; Ma, J.; Lee, E.H. Hypertrophy in skeletal myotubes induced by junctophilin-2 mutant, Y141H, involves an increase in store-operated Ca2+ entry via Orai1. J. Biol. Chem. 2012, 287, 14336–14348. [Google Scholar] [CrossRef] [Green Version]
- Vanninen, S.U.M.; Leivo, K.; Seppala, E.H.; Aalto-Setala, K.; Pitkanen, O.; Suursalmi, P.; Annala, A.P.; Anttila, I.; Alastalo, T.P.; Myllykangas, S.; et al. Heterozygous junctophilin-2 (JPH2) p.(Thr161Lys) is a monogenic cause for HCM with heart failure. PLoS ONE 2018, 13, e0203422. [Google Scholar] [CrossRef]
- Woo, J.S.; Hwang, J.H.; Ko, J.K.; Weisleder, N.; Kim, D.H.; Ma, J.; Lee, E.H. S165F mutation of junctophilin 2 affects Ca2+ signalling in skeletal muscle. Biochem. J. 2010, 427, 125–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quick, A.P.; Landstrom, A.P.; Wang, Q.; Beavers, D.L.; Reynolds, J.O.; Barreto-Torres, G.; Tran, V.; Showell, J.; Philippen, L.E.; Morris, S.A.; et al. Novel junctophilin-2 mutation A405S is associated with basal septal hypertrophy and diastolic dysfunction. JACC Basic Transl. Sci. 2017, 2, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Miura, A.; Kondo, H.; Yamamoto, T.; Okumura, Y.; Nishio, H. Sudden Unexpected Death of Infantile Dilated Cardiomyopathy with JPH2 and PKD1 Gene Variants. Int. Heart J. 2020, 61, 1079–1083. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, Y.; Furukawa, T.; Kasanuki, H.; Nishibatake, M.; Kurihara, Y.; Ikeda, A.; Kamatani, N.; Takeshima, H.; Matsuoka, R. Mutation of junctophilin type 2 associated with hypertrophic cardiomyopathy. J. Hum. Genet. 2007, 52, 543–548. [Google Scholar] [CrossRef]
- Gross, P.; Johnson, J.; Romero, C.M.; Eaton, D.M.; Poulet, C.; Sanchez-Alonso, J.; Lucarelli, C.; Ross, J.; Gibb, A.A.; Garbincius, J.F.; et al. Interaction of the Joining Region in Junctophilin-2 With the L-Type Ca2+ Channel Is Pivotal for Cardiac Dyad Assembly and Intracellular Ca2+ Dynamics. Circ. Res. 2021, 128, 92–114. [Google Scholar] [CrossRef]
- Murphy, R.M.; Dutka, T.L.; Horvath, D.; Bell, J.R.; Delbridge, L.M.; Lamb, G.D. Ca2+-dependent proteolysis of junctophilin-1 and junctophilin-2 in skeletal and cardiac muscle. J. Physiol. 2013, 591, 719–729. [Google Scholar] [CrossRef]
- Tammineni, E.R.; Figueroa, L.; Kraeva, N.; Manno, C.; Ibarra, C.A.; Klip, A.; Riazi, S.; Rios, E. Fragmentation and roles of junctophilin1 in muscle of patients with cytosolic leak of stored calcium. J. Gen. Physiol. 2022, 154, e2021ecc32. [Google Scholar] [CrossRef]
- Guo, A.; Wang, Y.; Chen, B.; Wang, Y.; Yuan, J.; Zhang, L.; Hall, D.; Wu, J.; Shi, Y.; Zhu, Q.; et al. E-C coupling structural protein junctophilin-2 encodes a stress-adaptive transcription regulator. Science 2018, 362, eaan3303. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Chen, B.; Jiang, Y.P.; Jia, Z.; Martin, D.W.; Liu, S.; Entcheva, E.; Song, L.S.; Lin, R.Z. Calpain-dependent cleavage of junctophilin-2 and T-tubule remodeling in a mouse model of reversible heart failure. J. Am. Heart Assoc. 2014, 3, e000527. [Google Scholar] [CrossRef] [Green Version]
- Lahiri, S.K.; Quick, A.P.; Samson-Couterie, B.; Hulsurkar, M.; Elzenaar, I.; van Oort, R.J.; Wehrens, X.H.T. Nuclear localization of a novel calpain-2 mediated junctophilin-2 C-terminal cleavage peptide promotes cardiomyocyte remodeling. Basic Res. Cardiol. 2020, 115, 49. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.Y.H.; Roczkowsky, A.; Cho, W.J.; Poirier, M.; Lee, T.Y.T.; Mahmud, Z.; Schulz, R. Junctophilin-2 is a target of matrix metalloproteinase-2 in myocardial ischemia-reperfusion injury. Basic Res. Cardiol. 2019, 114, 42. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. ‘It will change everything’: DeepMind’s AI makes gigantic leap in solving protein structures. Nature 2020, 588, 203–204. [Google Scholar] [CrossRef] [PubMed]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Zidek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef]
- Su, H.; Wang, W.; Du, Z.; Peng, Z.; Gao, S.H.; Cheng, M.M.; Yang, J. Improved Protein Structure Prediction Using a New Multi-Scale Network and Homologous Templates. Adv. Sci. (Weinh.) 2021, 8, 2102592. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
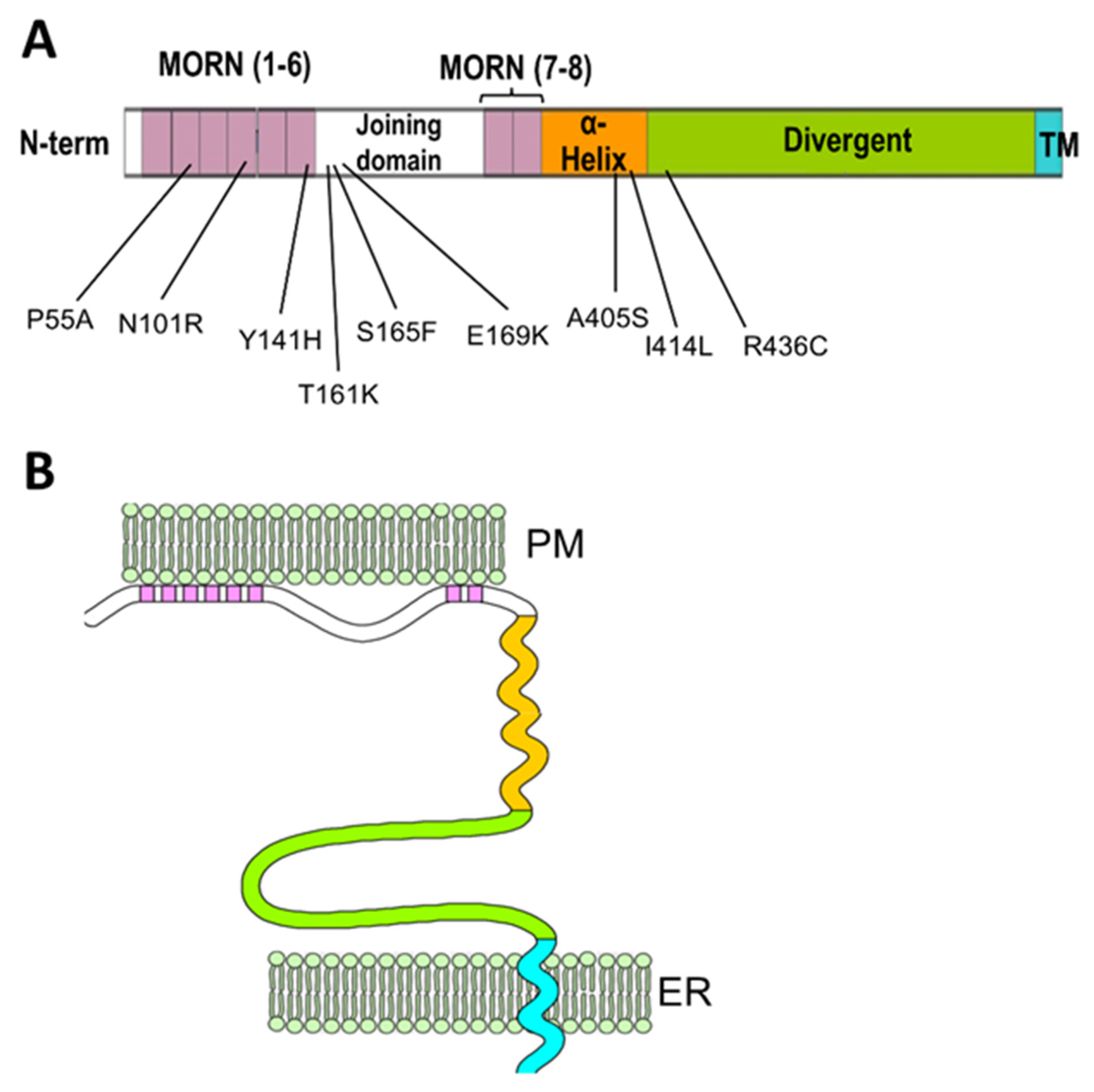
| Mutation | Position | Reference | Functional Characterization | Human Phenotype |
|---|---|---|---|---|
| P55A | MORN II | [58] | Uncharacterized | HCM |
| N101R | MORN IV | [59] | JPH2 mislocalization, disrupted Ca2+ signaling, cardiomyocytes hypertrophy. | HCM |
| Y141H | MORN VI | [59,60] | JPH2 mislocalization, disrupted Ca2+ signaling, cardiomyocytes hypertrophy. In skeletal muscle: abnormal ER-PM junctions, increased store-operated Ca2+ entry, decreased EC coupling gain, myotubes hypertrophy. | HCM |
| T161K | Joining domain | [61] | Uncharacterized | HCM |
| S165F | Joining domain | [59,62] | JPH2 mislocalization, disrupted Ca2+ signaling, cardiomyocytes hypertrophy. In skeletal muscle: Abnormal ER-PM junctions, decreased EC coupling gain, myotubes hypertrophy, | HCM |
| E169K | Joining domain | [52] | Reduced binding to RyR2, increases spontaneous Ca2+ release and Ca2+ sparks frequency | AF |
| A405S | α-helical domain | [52,63] | Irregular T-tubule pattern, mild effect on calcium signaling in the equivalent mutation (A399S) in mice. | HCM |
| I414L | α-helical domain | [64] | Uncharacterized | Dilated Cardiomyopathy |
| R436C | Divergent domain | [65] | Uncharacterized | HCM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perni, S. The Builders of the Junction: Roles of Junctophilin1 and Junctophilin2 in the Assembly of the Sarcoplasmic Reticulum–Plasma Membrane Junctions in Striated Muscle. Biomolecules 2022, 12, 109. https://doi.org/10.3390/biom12010109
Perni S. The Builders of the Junction: Roles of Junctophilin1 and Junctophilin2 in the Assembly of the Sarcoplasmic Reticulum–Plasma Membrane Junctions in Striated Muscle. Biomolecules. 2022; 12(1):109. https://doi.org/10.3390/biom12010109
Chicago/Turabian StylePerni, Stefano. 2022. "The Builders of the Junction: Roles of Junctophilin1 and Junctophilin2 in the Assembly of the Sarcoplasmic Reticulum–Plasma Membrane Junctions in Striated Muscle" Biomolecules 12, no. 1: 109. https://doi.org/10.3390/biom12010109
APA StylePerni, S. (2022). The Builders of the Junction: Roles of Junctophilin1 and Junctophilin2 in the Assembly of the Sarcoplasmic Reticulum–Plasma Membrane Junctions in Striated Muscle. Biomolecules, 12(1), 109. https://doi.org/10.3390/biom12010109