
Stress-Induced Epstein-Barr Virus Reactivation


 , and
, and
Abstract
1. Introduction
2. Overview of EBV Reactivation and the Lytic Cycle
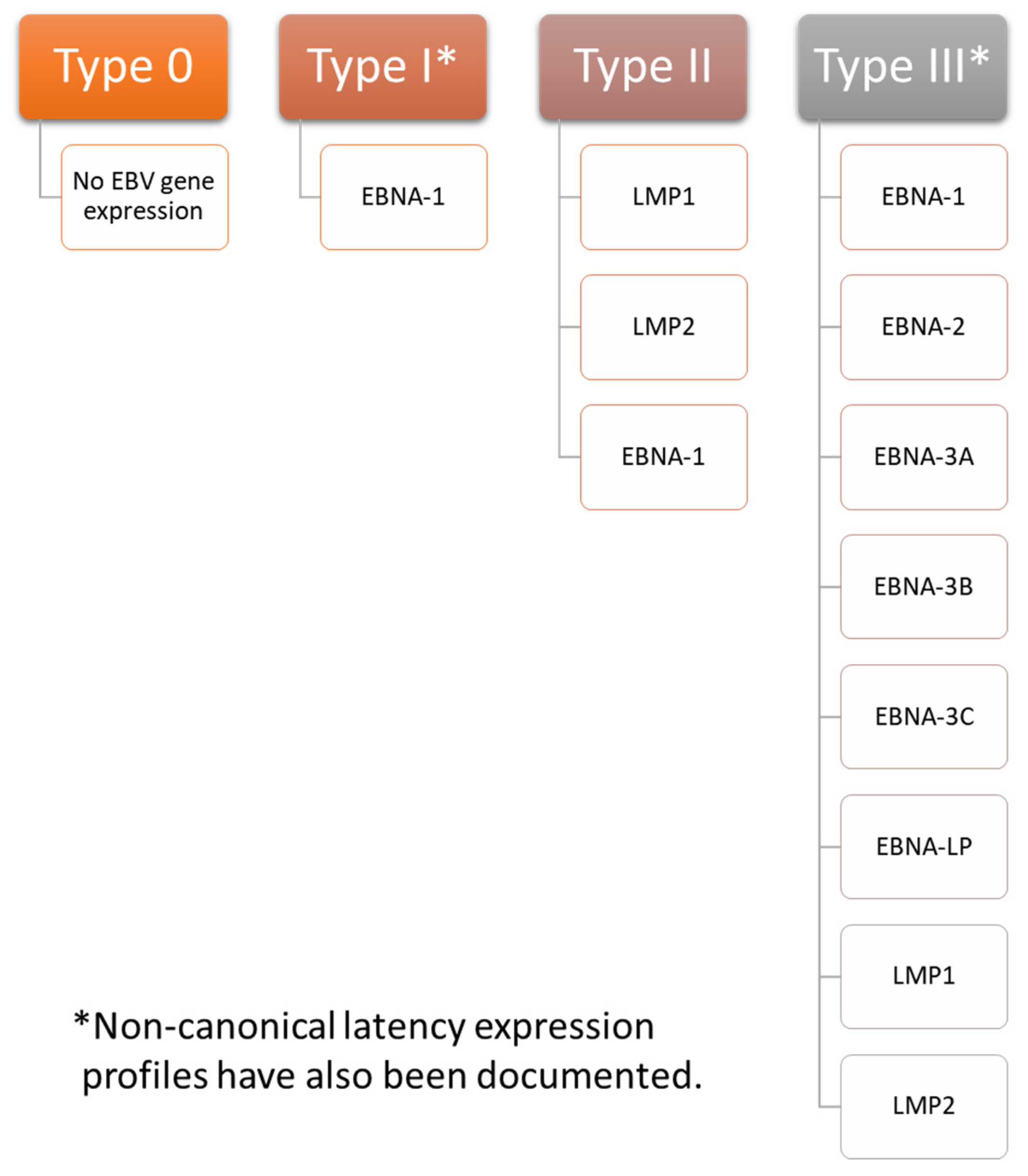
3. Overview of EBV Latency
Establishment and Maintenance of Latency in EBV Infection
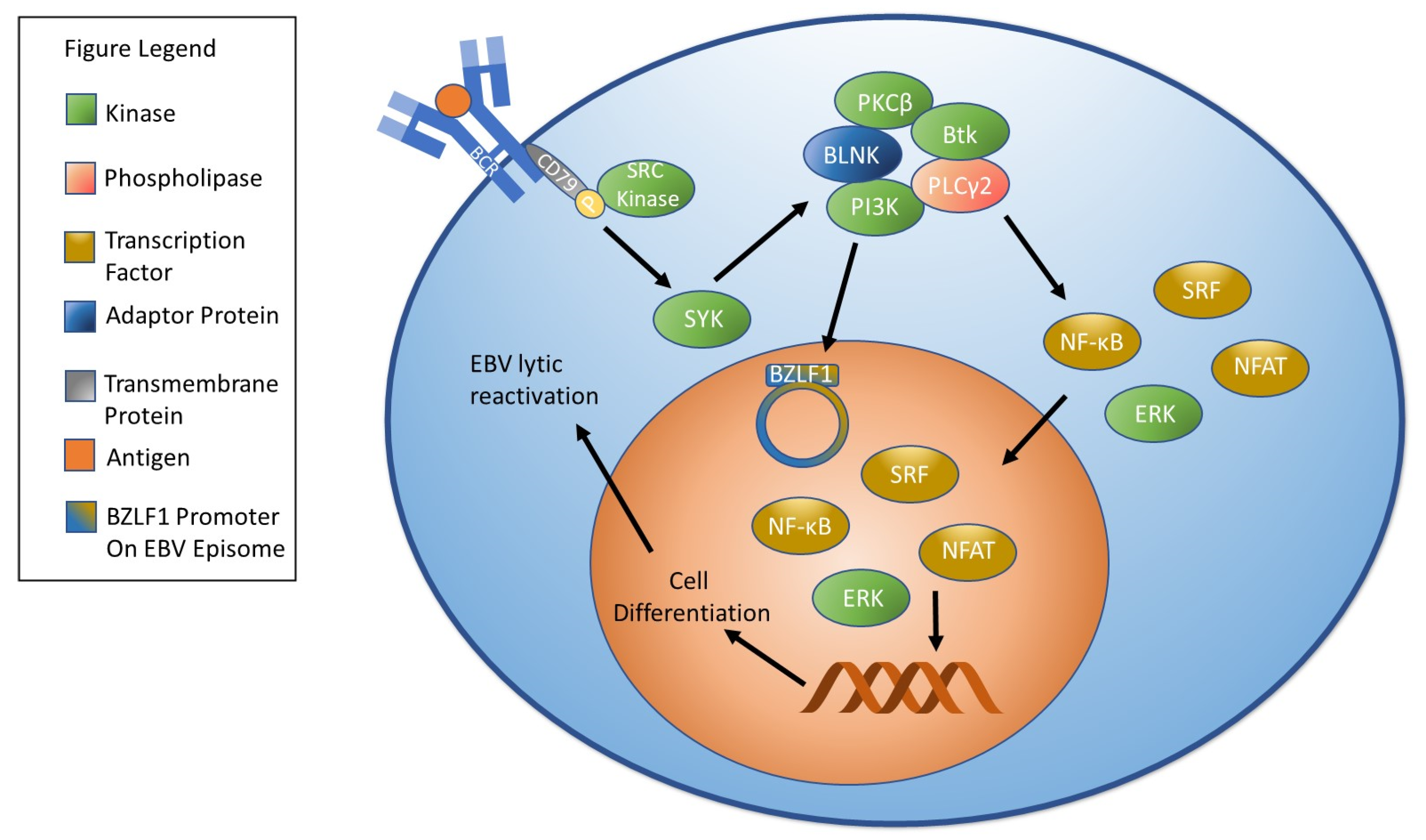
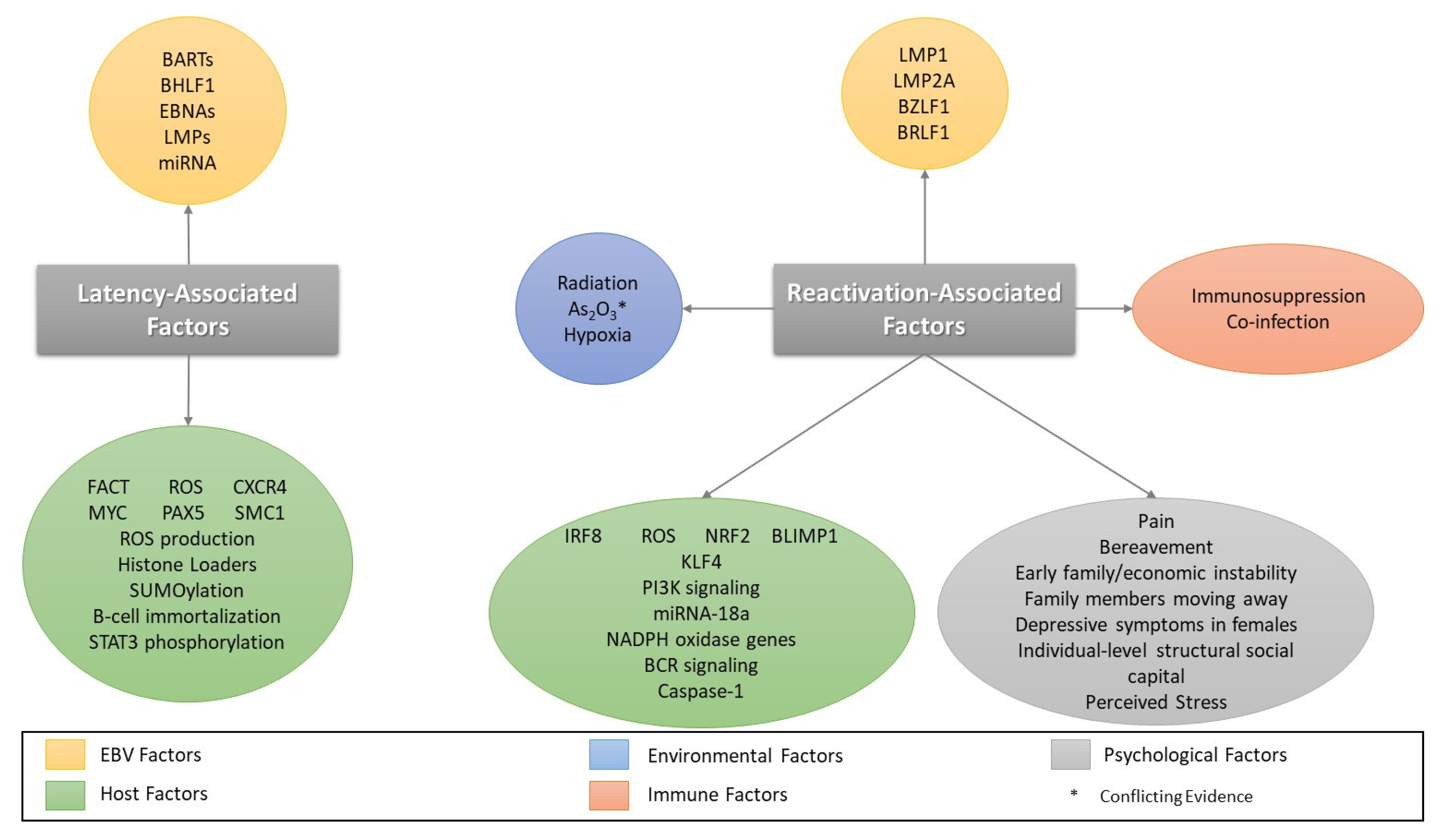
4. Factors Involved in EBV Reactivation
4.1. Oxidative Stress/Reactive Oxygen Species and EBV Reactivation
4.2. Co-Infection/Immunosuppression and Reactivation
4.3. Other Cellular Stressors and Reactivation
4.4. Psychological Stressors
5. Advances in EBV Antiviral Therapy
5.1. Advances in EBV Vaccines
5.2. Vaccination in EBV-Related Cancers
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BTdCPU | 1-(benzo[d][1–3]thiadiazol-6-yl)-3-(3,4-dichlorophenyl)urea |
| TPA | 12-0-tetradacanoyl-phorbol- 13-acetate |
| ATF4 | Activating transcription factor 4 |
| AKT | AKT serine/threonine kinase |
| ATRX | Alpha thalassemia/mental retardation syndrome X-linked chromatin remodeler |
| ATM | ATM serine/threonine kinase |
| BLNK | B cell linker protein |
| BCR | B cell receptor |
| BART | BamHI fragment A rightward transcript |
| BLIMP1 | B-lymphocyte-induced maturation protein 1 |
| BLK | B-lymphoid tyrosine kinase |
| Btk | Bruton’s tyrosine kinase |
| cP | C promoter |
| CTCF | CCCTC-binding factor |
| sgCHAF1B | CHAF1B single guide RNA |
| CAF | Chromatin assembly factor |
| CHAF1A | Chromatin assembly factor 1 subunit A |
| CHAF1B | Chromatin assembly factor 1 subunit B |
| 3C | Chromatin conformation capture |
| ChIP | Chromatin immunoprecipitation |
| CD21 | Complement receptor type 2 |
| COVID-19 | Coronavirus disease-19 |
| CBF | C-promoter binding factor |
| CXCR4 | C-X-C motif chemokine receptor 4 |
| CMV | Cytomegalovirus |
| DAXX | Death domain-associated protein |
| dATP | Deoxyadenosine triphosphate |
| DNMT3A | DNA methyltransferase 3 alpha |
| PKR | Double-stranded RNA-dependent protein kinase |
| EBV-LP | EBV-like particle |
| EGFRK | Epidermal growth factor receptor-associated tyrosine kinase |
| EBNA | Epstein-Barr nuclear antigen |
| EBV | Epstein-Barr Virus |
| EBER | Epstein-Barr virus-encoded small RNA |
| eIF2α | Eukaryotic translation initiation factor 2 alpha |
| ERK | Extracellular signal-related kinase |
| FACT | Facilitated chromatin transcription |
| FPAX5 | Flag epitope-tagged PAX5 |
| GCN2 | General control non-derepressible 2 |
| GSK-3β | Glycogen synthase kinase 3β |
| g | Glycoprotein |
| Gy | Gray |
| GRB2 | Growth factor receptor-bound protein 2 |
| IC50 | Half-maximal inhibitory concentration |
| HO-1 | Heme oxygenase-1 |
| HRI | Heme-regulated eIF2α kinase |
| HBV | Hepatitis B virus |
| H3K27me3 | Histone 3 lysine 27 trimethyl |
| H3K4me3 | Histone 3 lysine 4 trimethyl |
| H3K9me3 | Histone 3 lysine 9 trimethyl |
| HHV | Human herpesvirus |
| HPV | Human papillomavirus |
| H2O2 | Hydrogen peroxide |
| HPA | Hypothalamic-pituitary-adrenal |
| Ig | Immunoglobulin |
| ITAM | Immunoreceptor tyrosine-based activation motifs |
| IGF1 | Insulin like growth factor 1 |
| ISR | Integrated stress response |
| ICU | Intensive care unit |
| IRF | Interferon regulatory factor |
| IL | Interleukin |
| KSHV | Kaposi sarcoma-associated herpesvirus |
| KLF4 | Krüppel-like factor 4 |
| LMP | Latent membrane protein |
| oriP | Latent replication origin |
| lncRNA | long noncoding RNA |
| LS | Lumazine synthase |
| PCAF | Lysine acetyltransferase 2B |
| miRNA | microRNA |
| MAP | Mitogen-activated protein |
| ME/CFS | Myalgic encephalomyelitis/chronic fatigue syndrome |
| NACA | N-acetylcysteine amide |
| NQO-1 | NAD(P)H-quinone oxidoreductase 1 |
| NOX | NADPH oxidase |
| NPC | Nasopharyngeal carcinoma |
| Nrf2 | Nuclear factor erythroid 2-related-factor 2 |
| NF-κB | Nuclear factor kappa B |
| NFAT | Nuclear factor of activated T cells |
| NCL | Nucleolin |
| ORF | Open reading frame |
| oriLyt | Origin of lytic replication |
| GSSG | Oxidized glutathione |
| PAX | Paired box |
| PBMC | Peripheral blood mononuclear cell |
| PI3K | phosphatidylinositol 3-kinase |
| PLCγ2 | Phospholipase Cγ2 |
| PERK | PKR-like ER kinase |
| PDGFRK | Platelet-derived growth factor receptor-associated tyrosine kinase |
| PARP | Poly(ADP-ribose) polymerase |
| PTLD | Posttransplant lymphoproliferative disorder |
| PKC | Protein kinase C |
| Qp | Q promoter |
| RAC1 | Ras-related C3 botulinum toxin substrate 1 |
| RBBP4 | RB binding protein 4 |
| ROS | Reactive oxygen species |
| RT-PCR | Reverse transcription polymerase chain reaction |
| SRF | Serum response factor |
| shRNA | Short hairpin RNA |
| STAT3 | Signal transducer and activator of transcription 3 |
| siRNA | Small interfering RNA |
| SUMO | Small ubiquitin-related modifier |
| SOS1 | SOS Ras/rac guanine nucleotide Exchange factor |
| SYC | Spleen tyrosine kinase |
| SUPT16H | SPT16 homolog, facilitates chromatin remodeling subunit |
| STUB1 | STIP1 homology and U-box containing protein 1 |
| SMC1A | Structural maintenance of chromosomes 1A |
| SSRP1 | Structure specific recognition protein 1 |
| SIMs | SUMO interaction motifs |
| Th2 | T helper 2 cells |
| TAF | Tenofovir alafenamide |
| TDF | Tenofovir disoproxil fumarate |
| TET2 | Tet methylcytosine dioxygenase 2 |
| TLR | Toll-like receptor |
| GSH | Total glutathione |
| TRIB3 | Tribbles pseudokinase 3 |
| KAP1 | Tripartite motif containing 28 |
| UEV | Ubiquitin E2 variant |
| USP7 | Ubiquitin specific peptidase 7 |
| UV | Ultraviolet |
| VCA | Viral capsid antigen |
References
- Dunmire, S.K.; Verghese, P.S.; Balfour, H.H., Jr. Primary Epstein-Barr virus infection. J. Clin. Virol. 2018, 102, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Fugl, A.; Andersen, C.L. Epstein-Barr virus and its association with disease—A review of relevance to general practice. BMC Fam. Pract. 2019, 20, 62. [Google Scholar] [CrossRef]
- Womack, J.; Jimenez, M. Common questions about infectious mononucleosis. Am. Fam. Phys. 2015, 91, 372–376. [Google Scholar]
- Kerr, J.R. Epstein-Barr virus (EBV) reactivation and therapeutic inhibitors. J. Clin. Pathol. 2019, 72, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Dunmire, S.K.; Hogquist, K.A.; Balfour, H.H. Infectious Mononucleosis. Curr. Top. Microbiol. Immunol. 2015, 390, 211–240. [Google Scholar] [CrossRef] [PubMed]
- Houen, G.; Trier, N.H. Epstein-Barr Virus and Systemic Autoimmune Diseases. Front. Immunol. 2020, 11, 587380. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.; Asprusten, T.T.; Godang, K.; Leegaard, T.M.; Osnes, L.T.; Skovlund, E.; Tjade, T.; Oie, M.G.; Wyller, V.B.B. Predictors of chronic fatigue in adolescents six months after acute Epstein-Barr virus infection: A prospective cohort study. Brain Behav. Immun. 2019, 75, 94–100. [Google Scholar] [CrossRef]
- Nagata, K.; Hara, S.; Nakayama, Y.; Higaki, K.; Sugihara, H.; Kuwamoto, S.; Matsushita, M.; Kato, M.; Tanio, S.; Ishiguro, K.; et al. Epstein-Barr Virus Lytic Reactivation Induces IgG4 Production by Host B Lymphocytes in Graves’ Disease Patients and Controls: A Subset of Graves’ Disease Is an IgG4-Related Disease-Like Condition. Viral Immunol. 2018, 31, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Al Hamed, R.; Bazarbachi, A.H.; Mohty, M. Epstein-Barr virus-related post-transplant lymphoproliferative disease (EBV-PTLD) in the setting of allogeneic stem cell transplantation: A comprehensive review from pathogenesis to forthcoming treatment modalities. Bone Marrow Transpl. 2020, 55, 25–39. [Google Scholar] [CrossRef]
- Houen, G.; Trier, N.H.; Frederiksen, J.L. Epstein-Barr Virus and Multiple Sclerosis. Front. Immunol. 2020, 11, 587078. [Google Scholar] [CrossRef]
- Ou, Y.N.; Zhu, J.X.; Hou, X.H.; Shen, X.N.; Xu, W.; Dong, Q.; Tan, L.; Yu, J.T. Associations of Infectious Agents with Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Alzheimers Dis. 2020, 75, 299–309. [Google Scholar] [CrossRef]
- Robinson, T.J.; Glenn, M.S.; Temple, R.W.; Wyatt, D.; Connolly, J.H. Encephalitis and cerebellar ataxia associated with Epstein-Barr virus infections. Ulster Med. J. 1980, 49, 158–164. [Google Scholar]
- Abul-Kasim, K.; Palm, L.; Maly, P.; Sundgren, P.C. The neuroanatomic localization of Epstein-Barr virus encephalitis may be a predictive factor for its clinical outcome: A case report and review of 100 cases in 28 reports. J. Child. Neurol. 2009, 24, 720–726. [Google Scholar] [CrossRef]
- Greenspan, J.S.; Greenspan, D.; Lennette, E.T.; Abrams, D.I.; Conant, M.A.; Petersen, V.; Freese, U.K. Replication of Epstein-Barr virus within the epithelial cells of oral "hairy" leukoplakia, an AIDS-associated lesion. N. Engl. J. Med. 1985, 313, 1564–1571. [Google Scholar] [CrossRef]
- Ko, Y.H. EBV and human cancer. Exp. Mol. Med. 2015, 47, e130. [Google Scholar] [CrossRef] [PubMed]
- Neparidze, N.; Lacy, J. Malignancies associated with epstein-barr virus: Pathobiology, clinical features, and evolving treatments. Clin. Adv. Hematol. Oncol. 2014, 12, 358–371. [Google Scholar]
- Tsao, S.W.; Tsang, C.M.; To, K.F.; Lo, K.W. The role of Epstein-Barr virus in epithelial malignancies. J. Pathol. 2015, 235, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Utsuki, S.; Oka, H.; Miyajima, Y.; Kijima, C.; Yasui, Y.; Fujii, K. Epstein-Barr virus (EBV)-associated primary central nervous system lymphoma: Is incidence of EBV expression associated with median survival time? Brain Tumor Pathol. 2011, 28, 145–149. [Google Scholar] [CrossRef]
- Khan, G.; Hashim, M.J. Global burden of deaths from Epstein-Barr virus attributable malignancies 1990–2010. Infect. Agent Cancer 2014, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Khan, G.; Fitzmaurice, C.; Naghavi, M.; Ahmed, L.A. Global and regional incidence, mortality and disability-adjusted life-years for Epstein-Barr virus-attributable malignancies, 1990–2017. BMJ Open 2020, 10, e037505. [Google Scholar] [CrossRef] [PubMed]
- Drosu, N.C.; Edelman, E.R.; Housman, D.E. Tenofovir prodrugs potently inhibit Epstein-Barr virus lytic DNA replication by targeting the viral DNA polymerase. Proc. Natl. Acad. Sci. USA 2020, 117, 12368–12374. [Google Scholar] [CrossRef]
- Cohen, J.I. Vaccine Development for Epstein-Barr Virus. Adv. Exp. Med. Biol. 2018, 1045, 477–493. [Google Scholar] [CrossRef]
- Pellett, P.E.; Roizman, B. Herpesviridae. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1802–1822. [Google Scholar]
- Machon, C.; Fabrega-Ferrer, M.; Zhou, D.; Cuervo, A.; Carrascosa, J.L.; Stuart, D.I.; Coll, M. Atomic structure of the Epstein-Barr virus portal. Nat. Commun. 2019, 10, 3891. [Google Scholar] [CrossRef]
- Liu, X.; Cohen, J.I. Epstein-Barr Virus (EBV) Tegument Protein BGLF2 Promotes EBV Reactivation through Activation of the p38 Mitogen-Activated Protein Kinase. J. Virol. 2016, 90, 1129–1138. [Google Scholar] [CrossRef]
- He, H.P.; Luo, M.; Cao, Y.L.; Lin, Y.X.; Zhang, H.; Zhang, X.; Ou, J.Y.; Yu, B.; Chen, X.; Xu, M.; et al. Structure of Epstein-Barr virus tegument protein complex BBRF2-BSRF1 reveals its potential role in viral envelopment. Nat. Commun. 2020, 11, 5405. [Google Scholar] [CrossRef]
- van Gent, M.; Braem, S.G.; de Jong, A.; Delagic, N.; Peeters, J.G.; Boer, I.G.; Moynagh, P.N.; Kremmer, E.; Wiertz, E.J.; Ovaa, H.; et al. Epstein-Barr virus large tegument protein BPLF1 contributes to innate immune evasion through interference with toll-like receptor signaling. PLoS Pathog. 2014, 10, e1003960. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wang, Y.; Xu, Z.; Zou, X.; Wang, P.; Ou, X.; Li, Y.; Peng, T.; Chen, D.; Li, M.; et al. Epstein-Barr virus tegument protein BGLF2 inhibits NF-kappaB activity by preventing p65 Ser536 phosphorylation. FASEB J. 2019, 33, 10563–10576. [Google Scholar] [CrossRef]
- Hutt-Fletcher, L.M. EBV glycoproteins: Where are we now? Future Virol. 2015, 10, 1155–1162. [Google Scholar] [CrossRef]
- Tosato, G.; Cohen, J.I. Generation of Epstein-Barr Virus (EBV)-immortalized B cell lines. Curr. Protoc. Immunol. 2007, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Longnecker, R. Epithelial cell infection by Epstein-Barr virus. FEMS Microbiol. Rev. 2019, 43, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Connolly, S.A.; Jackson, J.O.; Jardetzky, T.S.; Longnecker, R. Fusing structure and function: A structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 2011, 9, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Heldwein, E.E. gH/gL supercomplexes at early stages of herpesvirus entry. Curr. Opin. Virol. 2016, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Young, K.A.; Chen, X.S.; Holers, V.M.; Hannan, J.P. Isolating the Epstein-Barr virus gp350/220 binding site on complement receptor type 2 (CR2/CD21). J. Biol. Chem. 2007, 282, 36614–36625. [Google Scholar] [CrossRef] [PubMed]
- Longnecker, R.M.; Kieff, E.; Cohen, J.I. Epstein-Barr Virus. In Field’s Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1898–1959. [Google Scholar]
- Wang, H.B.; Zhang, H.; Zhang, J.P.; Li, Y.; Zhao, B.; Feng, G.K.; Du, Y.; Xiong, D.; Zhong, Q.; Liu, W.L.; et al. Neuropilin 1 is an entry factor that promotes EBV infection of nasopharyngeal epithelial cells. Nat. Commun. 2015, 6, 6240. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, Y.; Wang, H.B.; Zhang, A.; Chen, M.L.; Fang, Z.X.; Dong, X.D.; Li, S.B.; Du, Y.; Xiong, D.; et al. Ephrin receptor A2 is an epithelial cell receptor for Epstein-Barr virus entry. Nat. Microbiol. 2018, 3, 1–8. [Google Scholar] [CrossRef]
- Xiong, D.; Du, Y.; Wang, H.B.; Zhao, B.; Zhang, H.; Li, Y.; Hu, L.J.; Cao, J.Y.; Zhong, Q.; Liu, W.L.; et al. Nonmuscle myosin heavy chain IIA mediates Epstein-Barr virus infection of nasopharyngeal epithelial cells. Proc. Natl. Acad. Sci. USA 2015, 112, 11036–11041. [Google Scholar] [CrossRef]
- Fingeroth, J.D.; Diamond, M.E.; Sage, D.R.; Hayman, J.; Yates, J.L. CD21-Dependent infection of an epithelial cell line, 293, by Epstein-Barr virus. J. Virol. 1999, 73, 2115–2125. [Google Scholar] [CrossRef]
- Turk, S.M.; Jiang, R.; Chesnokova, L.S.; Hutt-Fletcher, L.M. Antibodies to gp350/220 enhance the ability of Epstein-Barr virus to infect epithelial cells. J. Virol. 2006, 80, 9628–9633. [Google Scholar] [CrossRef]
- Janz, A.; Oezel, M.; Kurzeder, C.; Mautner, J.; Pich, D.; Kost, M.; Hammerschmidt, W.; Delecluse, H.J. Infectious Epstein-Barr virus lacking major glycoprotein BLLF1 (gp350/220) demonstrates the existence of additional viral ligands. J. Virol. 2000, 74, 10142–10152. [Google Scholar] [CrossRef]
- Smith, N.A.; Coleman, C.B.; Gewurz, B.E.; Rochford, R. CD21 (Complement Receptor 2) Is the Receptor for Epstein-Barr Virus Entry into T Cells. J. Virol. 2020, 94, e00428-20. [Google Scholar] [CrossRef]
- Lee, J.H.; Choi, J.; Ahn, Y.O.; Kim, T.M.; Heo, D.S. CD21-independent Epstein-Barr virus entry into NK cells. Cell Immunol. 2018, 327, 21–25. [Google Scholar] [CrossRef]
- Tabiasco, J.; Vercellone, A.; Meggetto, F.; Hudrisier, D.; Brousset, P.; Fournie, J.J. Acquisition of viral receptor by NK cells through immunological synapse. J. Immunol. 2003, 170, 5993–5998. [Google Scholar] [CrossRef]
- Mohl, B.S.; Chen, J.; Sathiyamoorthy, K.; Jardetzky, T.S.; Longnecker, R. Structural and Mechanistic Insights into the Tropism of Epstein-Barr Virus. Mol. Cells 2016, 39, 286–291. [Google Scholar] [CrossRef]
- Shannon-Lowe, C.; Rowe, M. Epstein Barr virus entry; kissing and conjugation. Curr. Opin. Virol. 2014, 4, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Hui-Yuen, J.; McAllister, S.; Koganti, S.; Hill, E.; Bhaduri-McIntosh, S. Establishment of Epstein-Barr virus growth-transformed lymphoblastoid cell lines. J. Vis. Exp. 2011, 57, 3321. [Google Scholar] [CrossRef]
- Jiang, R.; Kanamori, M.; Satoh, Y.; Fukuda, M.; Ikuta, K.; Murakami, M.; Sairenji, T. Contrasting effects of hydroxyurea on cell growth and reduction in Epstein-Barr virus genomes in EBV-infected epithelioid cell lines vs Burkitt’s lymphoma cell lines. J. Med. Virol. 2003, 70, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.H.; Dekroon, R.; Raab-Traub, N. Alterations in cellular expression in EBV infected epithelial cell lines and tumors. PLoS Pathog. 2019, 15, e1008071. [Google Scholar] [CrossRef] [PubMed]
- Callard, R.E.; Lau, Y.L.; Shields, J.G.; Smith, S.H.; Cairns, J.; Flores-Romo, L.; Gordon, J. The marmoset B-lymphoblastoid cell line (B95-8) produces and responds to B-cell growth and differentiation factors: Role of shed CD23 (sCD23). Immunology 1988, 65, 379–384. [Google Scholar] [PubMed]
- Isaksson, A.; Berggren, M.; Ekeland-Sjoberg, K.; Samuelsson, T.; Ricksten, A. Cell specific internal translation efficiency of Epstein-Barr virus present in solid organ transplant patients. J. Med. Virol. 2007, 79, 573–581. [Google Scholar] [CrossRef]
- Savard, M.; Belanger, C.; Tardif, M.; Gourde, P.; Flamand, L.; Gosselin, J. Infection of primary human monocytes by Epstein-Barr virus. J. Virol. 2000, 74, 2612–2619. [Google Scholar] [CrossRef]
- Masy, E.; Adriaenssens, E.; Montpellier, C.; Crepieux, P.; Mougel, A.; Quatannens, B.; Goormachtigh, G.; Faumont, N.; Meggetto, F.; Auriault, C.; et al. Human monocytic cell lines transformed in vitro by Epstein-Barr virus display a type II latency and LMP-1-dependent proliferation. J. Virol. 2002, 76, 6460–6472. [Google Scholar] [CrossRef][Green Version]
- Jha, H.C.; Mehta, D.; Lu, J.; El-Naccache, D.; Shukla, S.K.; Kovacsics, C.; Kolson, D.; Robertson, E.S. Gammaherpesvirus Infection of Human Neuronal Cells. mBio 2015, 6, e01844-15. [Google Scholar] [CrossRef]
- Tiwari, D.; Jakhmola, S.; Pathak, D.K.; Kumar, R.; Jha, H.C. Temporal In Vitro Raman Spectroscopy for Monitoring Replication Kinetics of Epstein-Barr Virus Infection in Glial Cells. ACS Omega 2020, 5, 29547–29560. [Google Scholar] [CrossRef] [PubMed]
- Gruffat, H.; Marchione, R.; Manet, E. Herpesvirus Late Gene Expression: A Viral-Specific Pre-initiation Complex Is Key. Front. Microbiol. 2016, 7, 869. [Google Scholar] [CrossRef]
- McKenzie, J.; El-Guindy, A. Epstein-Barr Virus Lytic Cycle Reactivation. Curr. Top. Microbiol. Immunol. 2015, 391, 237–261. [Google Scholar] [CrossRef]
- Feederle, R.; Kost, M.; Baumann, M.; Janz, A.; Drouet, E.; Hammerschmidt, W.; Delecluse, H.J. The Epstein-Barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J. 2000, 19, 3080–3089. [Google Scholar] [CrossRef]
- Ruvolo, V.; Wang, E.; Boyle, S.; Swaminathan, S. The Epstein-Barr virus nuclear protein SM is both a post-transcriptional inhibitor and activator of gene expression. Proc. Natl. Acad. Sci. USA 1998, 95, 8852–8857. [Google Scholar] [CrossRef]
- Semmes, O.J.; Chen, L.; Sarisky, R.T.; Gao, Z.; Zhong, L.; Hayward, S.D. Mta has properties of an RNA export protein and increases cytoplasmic accumulation of Epstein-Barr virus replication gene mRNA. J. Virol. 1998, 72, 9526–9534. [Google Scholar] [CrossRef] [PubMed]
- Tsurumi, T.; Daikoku, T.; Kurachi, R.; Nishiyama, Y. Functional interaction between Epstein-Barr virus DNA polymerase catalytic subunit and its accessory subunit in vitro. J. Virol. 1993, 67, 7648–7653. [Google Scholar] [CrossRef] [PubMed]
- Strockbine, L.D.; Cohen, J.I.; Farrah, T.; Lyman, S.D.; Wagener, F.; DuBose, R.F.; Armitage, R.J.; Spriggs, M.K. The Epstein-Barr virus BARF1 gene encodes a novel, soluble colony-stimulating factor-1 receptor. J. Virol. 1998, 72, 4015–4021. [Google Scholar] [CrossRef]
- Henderson, S.; Huen, D.; Rowe, M.; Dawson, C.; Johnson, G.; Rickinson, A. Epstein-Barr virus-coded BHRF1 protein, a viral homologue of Bcl-2, protects human B cells from programmed cell death. Proc. Natl. Acad. Sci. USA 1993, 90, 8479–8483. [Google Scholar] [CrossRef]
- Summers, W.C.; Klein, G. Inhibition of Epstein-Barr virus DNA synthesis and late gene expression by phosphonoacetic acid. J. Virol. 1976, 18, 151–155. [Google Scholar] [CrossRef]
- Young, L.S.; Arrand, J.R.; Murray, P.G. EBV gene expression and regulation. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Aubry, V.; Mure, F.; Mariame, B.; Deschamps, T.; Wyrwicz, L.S.; Manet, E.; Gruffat, H. Epstein-Barr virus late gene transcription depends on the assembly of a virus-specific preinitiation complex. J. Virol. 2014, 88, 12825–12838. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.L.; Camiolo, S.M.; Bashaw, J.M. The minimal replicator of Epstein-Barr virus oriP. J. Virol. 2000, 74, 4512–4522. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.E.; Corces, V.G. CTCF: Master weaver of the genome. Cell 2009, 137, 1194–1211. [Google Scholar] [CrossRef]
- Tempera, I.; Klichinsky, M.; Lieberman, P.M. EBV latency types adopt alternative chromatin conformations. PLoS Pathog. 2011, 7, e1002180. [Google Scholar] [CrossRef]
- Lupey-Green, L.N.; Caruso, L.B.; Madzo, J.; Martin, K.A.; Tan, Y.; Hulse, M.; Tempera, I. PARP1 Stabilizes CTCF Binding and Chromatin Structure to Maintain Epstein-Barr Virus Latency Type. J. Virol. 2018, 92, e00755-18. [Google Scholar] [CrossRef]
- Fejer, G.; Koroknai, A.; Banati, F.; Gyory, I.; Salamon, D.; Wolf, H.; Niller, H.H.; Minarovits, J. Latency type-specific distribution of epigenetic marks at the alternative promoters Cp and Qp of Epstein-Barr virus. J. Gen. Virol. 2008, 89, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Kis, L.L.; Takahara, M.; Nagy, N.; Klein, G.; Klein, E. IL-10 can induce the expression of EBV-encoded latent membrane protein-1 (LMP-1) in the absence of EBNA-2 in B lymphocytes and in Burkitt lymphoma- and NK lymphoma-derived cell lines. Blood 2006, 107, 2928–2935. [Google Scholar] [CrossRef] [PubMed]
- Kis, L.L.; Salamon, D.; Persson, E.K.; Nagy, N.; Scheeren, F.A.; Spits, H.; Klein, G.; Klein, E. IL-21 imposes a type II EBV gene expression on type III and type I B cells by the repression of C- and activation of LMP-1-promoter. Proc. Natl. Acad. Sci. USA 2010, 107, 872–877. [Google Scholar] [CrossRef]
- Choi, S.J.; Jung, S.W.; Huh, S.; Cho, H.; Kang, H. Phylogenetic comparison of Epstein-Barr virus genomes. J. Microbiol. 2018, 56, 525–533. [Google Scholar] [CrossRef]
- Murata, T. Regulation of Epstein-Barr virus reactivation from latency. Microbiol. Immunol. 2014, 58, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Granai, M.; Mundo, L.; Akarca, A.U.; Siciliano, M.C.; Rizvi, H.; Mancini, V.; Onyango, N.; Nyagol, J.; Abinya, N.O.; Maha, I.; et al. Immune landscape in Burkitt lymphoma reveals M2-macrophage polarization and correlation between PD-L1 expression and non-canonical EBV latency program. Infect. Agent Cancer 2020, 15, 28. [Google Scholar] [CrossRef]
- Touitou, R.; Arbach, H.; Cochet, C.; Feuillard, J.; Martin, A.; Raphael, M.; Joab, I. Heterogeneous Epstein-Barr virus latent gene expression in AIDS-associated lymphomas and in type I Burkitt’s lymphoma cell lines. J. Gen. Virol 2003, 84, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Frappier, L. Ebna1. Curr. Top. Microbiol. Immunol. 2015, 391, 3–34. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, P.M. Chromatin Structure of Epstein-Barr Virus Latent Episomes. Curr. Top. Microbiol. Immunol. 2015, 390, 71–102. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.D.; Tanizawa, H.; De Leo, A.; Vladimirova, O.; Kossenkov, A.; Lu, F.; Showe, L.C.; Noma, K.I.; Lieberman, P.M. Epigenetic specifications of host chromosome docking sites for latent Epstein-Barr virus. Nat. Commun. 2020, 11, 877. [Google Scholar] [CrossRef] [PubMed]
- Kempkes, B.; Ling, P.D. EBNA2 and Its Coactivator EBNA-LP. Curr. Top. Microbiol. Immunol. 2015, 391, 35–59. [Google Scholar] [CrossRef]
- Jin, X.W.; Speck, S.H. Identification of critical cis elements involved in mediating Epstein-Barr virus nuclear antigen 2-dependent activity of an enhancer located upstream of the viral BamHI C promoter. J. Virol. 1992, 66, 2846–2852. [Google Scholar] [CrossRef]
- Hammerschmidt, W.; Sugden, B. Genetic analysis of immortalizing functions of Epstein-Barr virus in human B lymphocytes. Nature 1989, 340, 393–397. [Google Scholar] [CrossRef]
- Harada, S.; Kieff, E. Epstein-Barr virus nuclear protein LP stimulates EBNA-2 acidic domain-mediated transcriptional activation. J. Virol. 1997, 71, 6611–6618. [Google Scholar] [CrossRef]
- Matsuda, G.; Nakajima, K.; Kawaguchi, Y.; Yamanashi, Y.; Hirai, K. Epstein-Barr virus (EBV) nuclear antigen leader protein (EBNA-LP) forms complexes with a cellular anti-apoptosis protein Bcl-2 or its EBV counterpart BHRF1 through HS1-associated protein X-1. Microbiol. Immunol. 2003, 47, 91–99. [Google Scholar] [CrossRef]
- Szymula, A.; Palermo, R.D.; Bayoumy, A.; Groves, I.J.; Ba Abdullah, M.; Holder, B.; White, R.E. Epstein-Barr virus nuclear antigen EBNA-LP is essential for transforming naive B cells, and facilitates recruitment of transcription factors to the viral genome. PLoS Pathog. 2018, 14, e1006890. [Google Scholar] [CrossRef] [PubMed]
- Styles, C.T.; Paschos, K.; White, R.E.; Farrell, P.J. The Cooperative Functions of the EBNA3 Proteins Are Central to EBV Persistence and Latency. Pathogens 2018, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Divisconte, M.; Jiang, X.; Quink, C.; Wang, F. Epstein-Barr virus with the latent infection nuclear antigen 3B completely deleted is still competent for B-cell growth transformation in vitro. J. Virol. 2005, 79, 4506–4509. [Google Scholar] [CrossRef]
- Tomkinson, B.; Robertson, E.; Kieff, E. Epstein-Barr virus nuclear proteins EBNA-3A and EBNA-3C are essential for B-lymphocyte growth transformation. J. Virol. 1993, 67, 2014–2025. [Google Scholar] [CrossRef]
- Kieser, A.; Sterz, K.R. The Latent Membrane Protein 1 (LMP1). Curr. Top. Microbiol. Immunol. 2015, 391, 119–149. [Google Scholar] [CrossRef] [PubMed]
- Cen, O.; Longnecker, R. Latent Membrane Protein 2 (LMP2). Curr. Top. Microbiol. Immunol. 2015, 391, 151–180. [Google Scholar] [CrossRef]
- Rovedo, M.; Longnecker, R. Epstein-barr virus latent membrane protein 2B (LMP2B) modulates LMP2A activity. J. Virol. 2007, 81, 84–94. [Google Scholar] [CrossRef]
- Merchant, M.; Caldwell, R.G.; Longnecker, R. The LMP2A ITAM is essential for providing B cells with development and survival signals in vivo. J. Virol. 2000, 74, 9115–9124. [Google Scholar] [CrossRef]
- Kang, D.; Skalsky, R.L.; Cullen, B.R. EBV BART MicroRNAs Target Multiple Pro-apoptotic Cellular Genes to Promote Epithelial Cell Survival. PLoS Pathog. 2015, 11, e1004979. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, Z.; Shu, Y.; Zhou, H.; Wang, H.; Zhang, W. BART miRNAs: An unimaginable force in the development of nasopharyngeal carcinoma. Eur. J. Cancer Prev. 2017, 26, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Takada, K. Role of EBER and BARF1 in nasopharyngeal carcinoma (NPC) tumorigenesis. Semin. Cancer Biol. 2012, 22, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, D. Epstein-Barr Virus-Encoded RNAs: Key Molecules in Viral Pathogenesis. Cancers 2014, 6, 1615–1630. [Google Scholar] [CrossRef]
- Dong, M.; Chen, J.N.; Huang, J.T.; Gong, L.P.; Shao, C.K. The roles of EBV-encoded microRNAs in EBV-associated tumors. Crit. Rev. Oncol. Hematol. 2019, 135, 30–38. [Google Scholar] [CrossRef]
- Swaminathan, S.; Tomkinson, B.; Kieff, E. Recombinant Epstein-Barr virus with small RNA (EBER) genes deleted transforms lymphocytes and replicates in vitro. Proc. Natl. Acad. Sci. USA 1991, 88, 1546–1550. [Google Scholar] [CrossRef] [PubMed]
- Herbert, K.M.; Pimienta, G. Consideration of Epstein-Barr Virus-Encoded Noncoding RNAs EBER1 and EBER2 as a Functional Backup of Viral Oncoprotein Latent Membrane Protein 1. mBio 2016, 7, e01926-15. [Google Scholar] [CrossRef]
- Verhoeven, R.J.A.; Tong, S.; Mok, B.W.; Liu, J.; He, S.; Zong, J.; Chen, Y.; Tsao, S.W.; Lung, M.L.; Chen, H. Epstein-Barr Virus BART Long Non-coding RNAs Function as Epigenetic Modulators in Nasopharyngeal Carcinoma. Front. Oncol. 2019, 9, 1120. [Google Scholar] [CrossRef]
- De Re, V.; Caggiari, L.; De Zorzi, M.; Fanotto, V.; Miolo, G.; Puglisi, F.; Cannizzaro, R.; Canzonieri, V.; Steffan, A.; Farruggia, P.; et al. Epstein-Barr virus BART microRNAs in EBV- associated Hodgkin lymphoma and gastric cancer. Infect. Agent Cancer 2020, 15, 42. [Google Scholar] [CrossRef]
- Leibold, W.; Flanagan, T.D.; Menezes, J.; Klein, G. Induction of Epstein-Barr virus-associated nuclear antigen during in vitro transformation of human lymphoid cells. J. Natl. Cancer Inst. 1975, 54, 65–68. [Google Scholar] [CrossRef]
- Gruhne, B.; Sompallae, R.; Marescotti, D.; Kamranvar, S.A.; Gastaldello, S.; Masucci, M.G. The Epstein-Barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2009, 106, 2313–2318. [Google Scholar] [CrossRef]
- Hu, J.; Li, H.; Luo, X.; Li, Y.; Bode, A.; Cao, Y. The role of oxidative stress in EBV lytic reactivation, radioresistance and the potential preventive and therapeutic implications. Int. J. Cancer 2017, 141, 1722–1729. [Google Scholar] [CrossRef]
- Chen, X.; Kamranvar, S.A.; Masucci, M.G. Oxidative stress enables Epstein-Barr virus-induced B-cell transformation by posttranscriptional regulation of viral and cellular growth-promoting factors. Oncogene 2016, 35, 3807–3816. [Google Scholar] [CrossRef] [PubMed]
- Koganti, S.; de la Paz, A.; Freeman, A.F.; Bhaduri-McIntosh, S. B lymphocytes from patients with a hypomorphic mutation in STAT3 resist Epstein-Barr virus-driven cell proliferation. J. Virol. 2014, 88, 516–524. [Google Scholar] [CrossRef]
- Yetming, K.D.; Lupey-Green, L.N.; Biryukov, S.; Hughes, D.J.; Marendy, E.M.; Miranda, J.L.; Sample, J.T. The BHLF1 Locus of Epstein-Barr Virus Contributes to Viral Latency and B-Cell Immortalization. J. Virol. 2020, 94, e01215-20. [Google Scholar] [CrossRef]
- Conacci-Sorrell, M.; McFerrin, L.; Eisenman, R.N. An overview of MYC and its interactome. Cold Spring Harb. Perspect. Med. 2014, 4, a014357. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Jiang, C.; Zhang, Y.; Govande, A.; Trudeau, S.J.; Chen, F.; Fry, C.J.; Puri, R.; Wolinsky, E.; Schineller, M.; et al. MYC Controls the Epstein-Barr Virus Lytic Switch. Mol. Cell 2020, 78, 653–669. [Google Scholar] [CrossRef] [PubMed]
- Carter, D.R.; Murray, J.; Cheung, B.B.; Gamble, L.; Koach, J.; Tsang, J.; Sutton, S.; Kalla, H.; Syed, S.; Gifford, A.J.; et al. Therapeutic targeting of the MYC signal by inhibition of histone chaperone FACT in neuroblastoma. Sci. Transl. Med. 2015, 7, 312ra176. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, Y.; Liu, W.; Zhang, X.; Xiao, H.; Zhao, M.; Luo, B. CXCR4 induces cell autophagy and maintains EBV latent infection in EBVaGC. Theranostics 2020, 10, 11549–11561. [Google Scholar] [CrossRef]
- Chang, P.C.; Campbell, M.; Robertson, E.S. Human Oncogenic Herpesvirus and Post-translational Modifications—Phosphorylation and SUMOylation. Front. Microbiol. 2016, 7, 962. [Google Scholar] [CrossRef]
- Cheng, J.; Kang, X.; Zhang, S.; Yeh, E.T. SUMO-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell 2007, 131, 584–595. [Google Scholar] [CrossRef]
- Chen, C.; Li, D.; Guo, N. Regulation of cellular and viral protein expression by the Epstein-Barr virus transcriptional regulator Zta: Implications for therapy of EBV associated tumors. Cancer Biol. Ther. 2009, 8, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Du, S.; Zhu, C.; Wang, C.; Yu, N.; Lin, Z.; Gan, J.; Guo, Y.; Huang, X.; He, Y.; et al. STUB1 is targeted by the SUMO-interacting motif of EBNA1 to maintain Epstein-Barr Virus latency. PLoS Pathog. 2020, 16, e1008447. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, P.; Morin, P., Jr.; Ouellette, R.J.; Robichaud, G.A. The Pax-5 gene: A pluripotent regulator of B-cell differentiation and cancer disease. Cancer Res. 2011, 71, 7345–7350. [Google Scholar] [CrossRef]
- Chen, Y.L.; Tsai, H.L.; Peng, C.W. EGCG debilitates the persistence of EBV latency by reducing the DNA binding potency of nuclear antigen 1. Biochem. Biophys. Res. Commun. 2012, 417, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.D.; Lee, H.L.; Peng, C.W. B Cell-Specific Transcription Activator PAX5 Recruits p300 To Support EBNA1-Driven Transcription. J. Virol. 2020, 94, e02028-19. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, C.; Trudeau, S.J.; Narita, Y.; Zhao, B.; Teng, M.; Guo, R.; Gewurz, B.E. Histone Loaders CAF1 and HIRA Restrict Epstein-Barr Virus B-Cell Lytic Reactivation. mBio 2020, 11, e01063-20. [Google Scholar] [CrossRef]
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications—Writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef]
- De Gorter, D.J.; Vos, J.C.; Pals, S.T.; Spaargaren, M. The B cell antigen receptor controls AP-1 and NFAT activity through Ras-mediated activation of Ral. J. Immunol. 2007, 178, 1405–1414. [Google Scholar] [CrossRef]
- Chen, Y.; Fachko, D.; Ivanov, N.S.; Skinner, C.M.; Skalsky, R.L. Epstein-Barr virus microRNAs regulate B cell receptor signal transduction and lytic reactivation. PLoS Pathog. 2019, 15, e1007535. [Google Scholar] [CrossRef]
- Crawford, D.H.; Ando, I. EB virus induction is associated with B-cell maturation. Immunology 1986, 59, 405–409. [Google Scholar]
- Laichalk, L.L.; Thorley-Lawson, D.A. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J. Virol. 2005, 79, 1296–1307. [Google Scholar] [CrossRef]
- Ghaleb, A.M.; Yang, V.W. Kruppel-like factor 4 (KLF4): What we currently know. Gene 2017, 611, 27–37. [Google Scholar] [CrossRef]
- Nawandar, D.M.; Wang, A.; Makielski, K.; Lee, D.; Ma, S.; Barlow, E.; Reusch, J.; Jiang, R.; Wille, C.K.; Greenspan, D.; et al. Differentiation-Dependent KLF4 Expression Promotes Lytic Epstein-Barr Virus Infection in Epithelial Cells. PLoS Pathog. 2015, 11, e1005195. [Google Scholar] [CrossRef]
- Reusch, J.A.; Nawandar, D.M.; Wright, K.L.; Kenney, S.C.; Mertz, J.E. Cellular differentiation regulator BLIMP1 induces Epstein-Barr virus lytic reactivation in epithelial and B cells by activating transcription from both the R and Z promoters. J. Virol. 2015, 89, 1731–1743. [Google Scholar] [CrossRef]
- Nawandar, D.M.; Ohashi, M.; Djavadian, R.; Barlow, E.; Makielski, K.; Ali, A.; Lee, D.; Lambert, P.F.; Johannsen, E.; Kenney, S.C. Differentiation-Dependent LMP1 Expression Is Required for Efficient Lytic Epstein-Barr Virus Reactivation in Epithelial Cells. J. Virol. 2017, 91, e02438-16. [Google Scholar] [CrossRef] [PubMed]
- Takada, K. Cross-linking of cell surface immunoglobulins induces Epstein-Barr virus in Burkitt lymphoma lines. Int. J. Cancer 1984, 33, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, Y.; Qin, M.; Li, D.; Odhiambo, W.O.; Yuan, M.; Lv, Z.; Liu, C.; Ma, Y.; Dong, Y.; et al. Involvement of Blnk and Foxo1 in tumor suppression in BCRABL1transformed proB cells. Oncol. Rep. 2021, 45, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Baba, Y. B Cell Receptor Signaling. Adv. Exp. Med. Biol. 2020, 1254, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, T.L.; Guo, B. Receptor crosstalk: Reprogramming B cell receptor signalling to an alternate pathway results in expression and secretion of the autoimmunity-associated cytokine, osteopontin. J. Intern. Med. 2009, 265, 632–643. [Google Scholar] [CrossRef]
- Rao, A.; Luo, C.; Hogan, P.G. Transcription factors of the NFAT family: Regulation and function. Annu. Rev. Immunol. 1997, 15, 707–747. [Google Scholar] [CrossRef]
- Hipp, L.; Beer, J.; Kuchler, O.; Reisser, M.; Sinske, D.; Michaelis, J.; Gebhardt, J.C.M.; Knoll, B. Single-molecule imaging of the transcription factor SRF reveals prolonged chromatin-binding kinetics upon cell stimulation. Proc. Natl. Acad. Sci. USA 2019, 116, 880–889. [Google Scholar] [CrossRef]
- Chang, F.; Steelman, L.S.; Lee, J.T.; Shelton, J.G.; Navolanic, P.M.; Blalock, W.L.; Franklin, R.A.; McCubrey, J.A. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: Potential targeting for therapeutic intervention. Leukemia 2003, 17, 1263–1293. [Google Scholar] [CrossRef] [PubMed]
- Pieper, K.; Grimbacher, B.; Eibel, H. B-cell biology and development. J. Allergy Clin. Immunol. 2013, 131, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Shapiro-Shelef, M.; Calame, K. Regulation of plasma-cell development. Nat. Rev. Immunol. 2005, 5, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, D.; Takada, K. Phosphatidylinositol 3-kinase is a determinant of responsiveness to B cell antigen receptor-mediated Epstein-Barr virus activation. J. Immunol. 2004, 172, 1561–1566. [Google Scholar] [CrossRef] [PubMed]
- Goswami, R.; Gershburg, S.; Satorius, A.; Gershburg, E. Protein kinase inhibitors that inhibit induction of lytic program and replication of Epstein-Barr virus. Antivir. Res. 2012, 96, 296–304. [Google Scholar] [CrossRef]
- Lv, D.W.; Zhang, K.; Li, R. Interferon regulatory factor 8 regulates caspase-1 expression to facilitate Epstein-Barr virus reactivation in response to B cell receptor stimulation and chemical induction. PLoS Pathog. 2018, 14, e1006868. [Google Scholar] [CrossRef]
- Han, X.; Han, Y.; Zheng, Y.; Sun, Q.; Ma, T.; Zhang, J.; Xu, L. Chaetocin induces apoptosis in human melanoma cells through the generation of reactive oxygen species and the intrinsic mitochondrial pathway, and exerts its anti-tumor activity in vivo. PLoS ONE 2017, 12, e0175950. [Google Scholar] [CrossRef]
- Zhang, S.; Yin, J.; Zhong, J. Chaetocin reactivates the lytic replication of Epstein-Barr virus from latency via reactive oxygen species. Sci. China Life Sci. 2017, 60, 66–71. [Google Scholar] [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Hu, J.; Li, Y.; Li, H.; Shi, F.; Xie, L.; Zhao, L.; Tang, M.; Luo, X.; Jia, W.; Fan, J.; et al. Targeting Epstein-Barr virus oncoprotein LMP1-mediated high oxidative stress suppresses EBV lytic reactivation and sensitizes tumors to radiation therapy. Theranostics 2020, 10, 11921–11937. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.M.; Kim, Y.S.; Hur, D.Y. LMP1 and 2A Induce the Expression of Nrf2 Through Akt Signaling Pathway in Epstein-Barr Virus-Transformed B Cells. Transl. Oncol. 2019, 12, 775–783. [Google Scholar] [CrossRef]
- Cao, P.; Zhang, M.; Wang, L.; Sai, B.; Tang, J.; Luo, Z.; Shuai, C.; Zhang, L.; Li, Z.; Wang, Y.; et al. miR-18a reactivates the Epstein-Barr virus through defective DNA damage response and promotes genomic instability in EBV-associated lymphomas. BMC Cancer 2018, 18, 1293. [Google Scholar] [CrossRef] [PubMed]
- Strycharz-Dudziak, M.; Kielczykowska, M.; Drop, B.; Swiatek, L.; Kliszczewska, E.; Musik, I.; Polz-Dacewicz, M. Total Antioxidant Status (TAS), Superoxide Dismutase (SOD), and Glutathione Peroxidase (GPx) in Oropharyngeal Cancer Associated with EBV Infection. Oxid. Med. Cell Longev. 2019, 2019, 5832410. [Google Scholar] [CrossRef] [PubMed]
- Martinez, O.M.; Krams, S.M. The Immune Response to Epstein Barr Virus and Implications for Posttransplant Lymphoproliferative Disorder. Transplantation 2017, 101, 2009–2016. [Google Scholar] [CrossRef] [PubMed]
- Shikova, E.; Reshkova, V.; Kumanova, A.C.; Raleva, S.; Alexandrova, D.; Capo, N.; Murovska, M.; European Network on ME/CFS. Cytomegalovirus, Epstein-Barr virus, and human herpesvirus-6 infections in patients with myalgic encephalomyelitis/chronic fatigue syndrome. J. Med. Virol. 2020, 92, 3682–3688. [Google Scholar] [CrossRef]
- Tan, E.M.; Sugiura, K.; Gupta, S. The case definition of chronic fatigue syndrome. J. Clin. Immunol. 2002, 22, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Fichtner, A.S.; Ravens, S.; Prinz, I. Human gammadelta TCR Repertoires in Health and Disease. Cells 2020, 9, 800. [Google Scholar] [CrossRef]
- Xiang, Z.; Liu, Y.; Zheng, J.; Liu, M.; Lv, A.; Gao, Y.; Hu, H.; Lam, K.T.; Chan, G.C.; Yang, Y.; et al. Targeted activation of human Vgamma9Vdelta2-T cells controls epstein-barr virus-induced B cell lymphoproliferative disease. Cancer Cell 2014, 26, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Gao, H.; Xu, L.P.; Mo, X.D.; Liu, R.; Liang, S.; Wu, N.; Wang, M.; Wang, Z.; Chang, Y.J.; et al. Immunosuppressant indulges EBV reactivation and related lymphoproliferative disease by inhibiting Vdelta2(+) T cells activities after hematopoietic transplantation for blood malignancies. J. Immunother. Cancer 2020, 8, 208. [Google Scholar] [CrossRef]
- Hatayama, Y.; Hashimoto, Y.; Motokura, T. Frequent co-reactivation of Epstein-Barr virus in patients with cytomegalovirus viremia under immunosuppressive therapy and/or chemotherapy. J. Int. Med. Res. 2020, 48, 300060520972880. [Google Scholar] [CrossRef] [PubMed]
- Hirsiger, J.R.; Fuchs, P.S.; Hausermann, P.; Muller-Durovic, B.; Daikeler, T.; Recher, M.; Hirsch, H.H.; Terracciano, L.; Berger, C.T. Syphilis Reactivates Latent Epstein-Barr Virus Reservoir via Toll-Like Receptor 2 and B-Cell Receptor Activation. Open Forum Infect. Dis. 2019, 6, ofz317. [Google Scholar] [CrossRef]
- Makielski, K.R.; Lee, D.; Lorenz, L.D.; Nawandar, D.M.; Chiu, Y.F.; Kenney, S.C.; Lambert, P.F. Human papillomavirus promotes Epstein-Barr virus maintenance and lytic reactivation in immortalized oral keratinocytes. Virology 2016, 495, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Guidry, J.T.; Myers, J.E.; Bienkowska-Haba, M.; Songock, W.K.; Ma, X.; Shi, M.; Nathan, C.O.; Bodily, J.M.; Sapp, M.J.; Scott, R.S. Inhibition of Epstein-Barr Virus Replication in Human Papillomavirus-Immortalized Keratinocytes. J. Virol. 2019, 93, e01216-18. [Google Scholar] [CrossRef]
- Zelazowska, M.A.; McBride, K.; Krug, L.T. Dangerous Liaisons: Gammaherpesvirus Subversion of the Immunoglobulin Repertoire. Viruses 2020, 12, 788. [Google Scholar] [CrossRef] [PubMed]
- McHugh, D.; Caduff, N.; Barros, M.H.M.; Ramer, P.C.; Raykova, A.; Murer, A.; Landtwing, V.; Quast, I.; Styles, C.T.; Spohn, M.; et al. Persistent KSHV Infection Increases EBV-Associated Tumor Formation In Vivo via Enhanced EBV Lytic Gene Expression. Cell Host Microbe 2017, 22, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Hayakawa, F.; Kiyoi, H. Biology and management of primary effusion lymphoma. Blood 2018, 132, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Simonnet, A.; Engelmann, I.; Moreau, A.S.; Garcia, B.; Six, S.; El Kalioubie, A.; Robriquet, L.; Hober, D.; Jourdain, M. High incidence of Epstein-Barr virus, cytomegalovirus, and human-herpes virus-6 reactivations in critically ill patients with COVID-19. Infect. Dis. Now 2021, 51, 296–299. [Google Scholar] [CrossRef]
- Gold, J.E.; Okyay, R.A.; Licht, W.E.; Hurley, D.J. Investigation of Long COVID Prevalence and Its Relationship to Epstein-Barr Virus Reactivation. Pathogens 2021, 10, 763. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef]
- Lee, J.; Stone, J.; Desai, P.; Kosowicz, J.G.; Liu, J.O.; Ambinder, R.F. Arsenicals, the Integrated Stress Response, and Epstein-Barr Virus Lytic Gene Expression. Viruses 2021, 13, 812. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Sides, M.; Parsons, C.H.; Flemington, E.K.; Lasky, J.A. Arsenic trioxide inhibits EBV reactivation and promotes cell death in EBV-positive lymphoma cells. Virol. J. 2017, 14, 121. [Google Scholar] [CrossRef]
- Mehta, S.K.; Bloom, D.C.; Plante, I.; Stowe, R.; Feiveson, A.H.; Renner, A.; Dhummakupt, A.; Markan, D.; Zhang, Y.; Wu, H.; et al. Reactivation of Latent Epstein-Barr Virus: A Comparison after Exposure to Gamma, Proton, Carbon, and Iron Radiation. Int. J. Mol. Sci. 2018, 19, 2961. [Google Scholar] [CrossRef] [PubMed]
- Dhabhar, F.S. Effects of stress on immune function: The good, the bad, and the beautiful. Immunol. Res. 2014, 58, 193–210. [Google Scholar] [CrossRef]
- Dhabhar, F.S. Enhancing versus suppressive effects of stress on immune function: Implications for immunoprotection and immunopathology. Neuroimmunomodulation 2009, 16, 300–317. [Google Scholar] [CrossRef] [PubMed]
- Schakel, L.; Veldhuijzen, D.S.; Crompvoets, P.I.; Bosch, J.A.; Cohen, S.; van Middendorp, H.; Joosten, S.A.; Ottenhoff, T.H.M.; Visser, L.G.; Evers, A.W.M. Effectiveness of Stress-Reducing Interventions on the Response to Challenges to the Immune System: A Meta-Analytic Review. Psychother. Psychosom. 2019, 88, 274–286. [Google Scholar] [CrossRef]
- Brook, M.J.; Christian, L.M.; Hade, E.M.; Ruffin, M.T. The Effect of Perceived Stress on Epstein-Barr Virus Antibody Titers in Appalachian Ohio Women. Neuroimmunomodulation 2017, 24, 67–73. [Google Scholar] [CrossRef]
- Schmeer, K.K.; Ford, J.L.; Browning, C.R. Early childhood family instability and immune system dysregulation in adolescence. Psychoneuroendocrinology 2019, 102, 189–195. [Google Scholar] [CrossRef]
- Yazawa, A.; Inoue, Y.; Cai, G.; Tu, R.; Huang, M.; He, F.; Chen, J.; Yamamoto, T.; Watanabe, C. Epstein-Barr virus antibody titer as a stress biomarker and its association with social capital in rural Fujian communities, China. Am. J. Hum. Biol. 2018, 30, e23135. [Google Scholar] [CrossRef] [PubMed]
- Hamano, T.; Fujisawa, Y.; Ishida, Y.; Subramanian, S.V.; Kawachi, I.; Shiwaku, K. Social capital and mental health in Japan: A multilevel analysis. PLoS ONE 2010, 5, e13214. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, C.U.; LaGory, M. Social Capital and Mental Distress in an Impoverished Community. City Community 2002, 1, 199–222. [Google Scholar] [CrossRef]
- Yazawa, A.; Inoue, Y.; Cai, G.; Tu, R.; Huang, M.; He, F.; Chen, J.; Yamamoto, T.; Watanabe, C. The association between family members’ migration and Epstein-Barr virus antibody titers among people left behind in rural Fujian, China. Am. J. Hum. Biol. 2020, 32, e23327. [Google Scholar] [CrossRef] [PubMed]
- Panerai, A.E. Pain stress and headache. Neurol. Sci. 2012, 33 (Suppl. 1), S1–S3. [Google Scholar] [CrossRef]
- Crofford, L.J. Chronic Pain: Where the Body Meets the Brain. Trans. Am. Clin. Climatol. Assoc. 2015, 126, 167–183. [Google Scholar]
- Seiler, A.; Murdock, K.; Stowe, R.; Fagundes, C. Pain in older individuals and its association with latent Epstein-Barr virus reactivation. Ann. Pain Med. 2019, 2, 1007. [Google Scholar]
- Ford, J.L.; Stowe, R.P. Depressive symptoms are associated with salivary shedding of Epstein-Barr virus in female adolescents: The role of sex differences. Psychoneuroendocrinology 2017, 86, 128–133. [Google Scholar] [CrossRef]
- Bale, T.L.; Epperson, C.N. Sex differences and stress across the lifespan. Nat. Neurosci. 2015, 18, 1413–1420. [Google Scholar] [CrossRef]
- Da Silva, J.A. Sex hormones and glucocorticoids: Interactions with the immune system. Ann. N. Y. Acad. Sci. 1999, 876, 102–117. [Google Scholar] [CrossRef]
- Guevara, J.E.; Gilbert, S.; Murdock, K.W.; Stowe, R.P.; Fagundes, C.P. Sex differences in executive functioning and latent herpesvirus reactivation among bereaved and nonbereaved individuals. Stress Health 2019, 35, 396–406. [Google Scholar] [CrossRef]
- Lau, G.; Yu, M.L.; Wong, G.; Thompson, A.; Ghazinian, H.; Hou, J.L.; Piratvisuth, T.; Jia, J.D.; Mizokami, M.; Cheng, G.; et al. APASL clinical practice guideline on hepatitis B reactivation related to the use of immunosuppressive therapy. Hepatol. Int. 2021. [Google Scholar] [CrossRef]
- Andrei, G.; Trompet, E.; Snoeck, R. Novel Therapeutics for Epstein(-)Barr Virus. Molecules 2019, 24, 997. [Google Scholar] [CrossRef]
- Thome, M.P.; Borde, C.; Larsen, A.K.; Henriques, J.A.P.; Lenz, G.; Escargueil, A.E.; Marechal, V. Dipyridamole as a new drug to prevent Epstein-Barr virus reactivation. Antivir. Res. 2019, 172, 104615. [Google Scholar] [CrossRef]
- Chiba, N.; De Gara, C.J.; Wilkinson, J.M.; Hunt, R.H. Speed of healing and symptom relief in grade II to IV gastroesophageal reflux disease: A meta-analysis. Gastroenterology 1997, 112, 1798–1810. [Google Scholar] [CrossRef]
- Watanabe, S.M.; Ehrlich, L.S.; Strickland, M.; Li, X.; Soloveva, V.; Goff, A.J.; Stauft, C.B.; Bhaduri-McIntosh, S.; Tjandra, N.; Carter, C. Selective Targeting of Virus Replication by Proton Pump Inhibitors. Sci Rep. 2020, 10, 4003. [Google Scholar] [CrossRef]
- Christ, L.; Raiborg, C.; Wenzel, E.M.; Campsteijn, C.; Stenmark, H. Cellular Functions and Molecular Mechanisms of the ESCRT Membrane-Scission Machinery. Trends Biochem. Sci. 2017, 42, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.P.; Liu, G.T.; Kung, H.N.; Liu, P.T.; Liao, Y.T.; Chow, L.P.; Chang, L.S.; Chang, Y.H.; Chang, C.W.; Shu, W.C.; et al. The Ubiquitin Ligase Itch and Ubiquitination Regulate BFRF1-Mediated Nuclear Envelope Modification for Epstein-Barr Virus Maturation. J. Virol. 2016, 90, 8994–9007. [Google Scholar] [CrossRef] [PubMed]
- Mannemuddhu, S.S.; Xu, H.; Bleck, C.K.E.; Tjandra, N.; Carter, C.; Bhaduri-McIntosh, S. Prazoles Targeting Tsg101 Inhibit Release of Epstein-Barr Virus following Reactivation from Latency. J. Virol. 2021, 95, e0246620. [Google Scholar] [CrossRef] [PubMed]
- Onozawa, M.; Hashino, S.; Darmanin, S.; Okada, K.; Morita, R.; Takahata, M.; Shigematsu, A.; Kahata, K.; Kondo, T.; Tanaka, J.; et al. HB vaccination in the prevention of viral reactivation in allogeneic hematopoietic stem cell transplantation recipients with previous HBV infection. Biol. Blood Marrow Transpl. 2008, 14, 1226–1230. [Google Scholar] [CrossRef] [PubMed]
- Takahata, M.; Hashino, S.; Onozawa, M.; Shigematsu, A.; Sugita, J.; Fujimoto, K.; Endo, T.; Kondo, T.; Tanaka, J.; Imamura, M.; et al. Hepatitis B virus (HBV) reverse seroconversion (RS) can be prevented even in non-responders to hepatitis B vaccine after allogeneic stem cell transplantation: Long-term analysis of intervention in RS with vaccine for patients with previous HBV infection. Transpl. Infect. Dis. 2014, 16, 797–801. [Google Scholar] [CrossRef]
- Schmid, D.S.; Miao, C.; Leung, J.; Johnson, M.; Weinberg, A.; Levin, M.J. Comparative Antibody Responses to the Live-Attenuated and Recombinant Herpes Zoster Vaccines. J. Virol. 2021, 95, e00240-21. [Google Scholar] [CrossRef] [PubMed]
- Aldoss, I.; La Rosa, C.; Baden, L.R.; Longmate, J.; Ariza-Heredia, E.J.; Rida, W.N.; Lingaraju, C.R.; Zhou, Q.; Martinez, J.; Kaltcheva, T.; et al. Poxvirus Vectored Cytomegalovirus Vaccine to Prevent Cytomegalovirus Viremia in Transplant Recipients: A Phase 2, Randomized Clinical Trial. Ann. Intern. Med. 2020, 172, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Tanner, J.; Whang, Y.; Sample, J.; Sears, A.; Kieff, E. Soluble gp350/220 and deletion mutant glycoproteins block Epstein-Barr virus adsorption to lymphocytes. J. Virol. 1988, 62, 4452–4464. [Google Scholar] [CrossRef] [PubMed]
- Weiss, E.R.; Alter, G.; Ogembo, J.G.; Henderson, J.L.; Tabak, B.; Bakis, Y.; Somasundaran, M.; Garber, M.; Selin, L.; Luzuriaga, K. High Epstein-Barr Virus Load and Genomic Diversity Are Associated with Generation of gp350-Specific Neutralizing Antibodies following Acute Infectious Mononucleosis. J. Virol. 2017, 91, e01562-16. [Google Scholar] [CrossRef] [PubMed]
- Thorley-Lawson, D.A.; Geilinger, K. Monoclonal antibodies against the major glycoprotein (gp350/220) of Epstein-Barr virus neutralize infectivity. Proc. Natl. Acad. Sci. USA 1980, 77, 5307–5311. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, B.; Ding, M.; Song, S.; Kang, Y.; Yu, Y.; Xu, M.; Xiang, T.; Gao, L.; Feng, Q.; et al. A novel vaccine candidate based on chimeric virus-like particle displaying multiple conserved epitope peptides induced neutralizing antibodies against EBV infection. Theranostics 2020, 10, 5704–5718. [Google Scholar] [CrossRef]
- Escalante, G.M.; Foley, J.; Mutsvunguma, L.Z.; Rodriguez, E.; Mulama, D.H.; Muniraju, M.; Ye, P.; Barasa, A.K.; Ogembo, J.G. A Pentavalent Epstein-Barr Virus-Like Particle Vaccine Elicits High Titers of Neutralizing Antibodies against Epstein-Barr Virus Infection in Immunized Rabbits. Vaccines 2020, 8, 169. [Google Scholar] [CrossRef]
- Lopez-Sagaseta, J.; Malito, E.; Rappuoli, R.; Bottomley, M.J. Self-assembling protein nanoparticles in the design of vaccines. Comput. Struct. Biotechnol. J. 2016, 14, 58–68. [Google Scholar] [CrossRef]
- Kang, Y.F.; Zhang, X.; Yu, X.H.; Zheng, Q.; Liu, Z.; Li, J.P.; Sun, C.; Kong, X.W.; Zhu, Q.Y.; Chen, H.W.; et al. Immunization with a Self-Assembled Nanoparticle Vaccine Elicits Potent Neutralizing Antibody Responses against EBV Infection. Nano Lett. 2021, 21, 2476–2486. [Google Scholar] [CrossRef]
- Zeng, Y.; Si, Y.F.; Lan, G.P.; Wang, Z.; Zhou, L.; Tang, M.Z.; Sj, O.B.; Lan, J.; Zhou, X.Y.; Wang, Y.L.; et al. LMP2-DC Vaccine Elicits Specific EBV-LMP2 Response to Effectively Improve Immunotherapy in Patients with Nasopharyngeal Cancer. Biomed. Environ. Sci. 2020, 33, 849–856. [Google Scholar] [CrossRef]
- Valdes, I.; Lazo, L.; Hermida, L.; Guillen, G.; Gil, L. Can Complementary Prime-Boost Immunization Strategies Be an Alternative and Promising Vaccine Approach Against Dengue Virus? Front. Immunol. 2019, 10, 1956. [Google Scholar] [CrossRef] [PubMed]
- Ruhl, J.; Citterio, C.; Engelmann, C.; Haigh, T.; Dzionek, A.; Dreyer, J.; Khanna, R.; Taylor, G.S.; Wilson, J.B.; Leung, C.S.; et al. Heterologous prime-boost vaccination protects against EBV antigen-expressing lymphomas. J. Clin. Investig. 2019, 129, 2071–2087. [Google Scholar] [CrossRef] [PubMed]
- Dawson, C.W.; Port, R.J.; Young, L.S. The role of the EBV-encoded latent membrane proteins LMP1 and LMP2 in the pathogenesis of nasopharyngeal carcinoma (NPC). Semin. Cancer Biol. 2012, 22, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Fukayama, M.; Ushiku, T. Epstein-Barr virus-associated gastric carcinoma. Pathol. Res. Pract. 2011, 207, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Pang, M.F.; Lin, K.W.; Peh, S.C. The signaling pathways of Epstein-Barr virus-encoded latent membrane protein 2A (LMP2A) in latency and cancer. Cell Mol. Biol. Lett. 2009, 14, 222–247. [Google Scholar] [CrossRef] [PubMed]
- Wojtak, K.; Perales-Puchalt, A.; Weiner, D.B. Novel Synthetic DNA Immunogens Targeting Latent Expressed Antigens of Epstein-Barr Virus Elicit Potent Cellular Responses and Inhibit Tumor Growth. Vaccines 2019, 7, 44. [Google Scholar] [CrossRef]
- Singavi, A.K.; Harrington, A.M.; Fenske, T.S. Post-transplant lymphoproliferative disorders. Cancer Treat. Res. 2015, 165, 305–327. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, X.; Yu, G.; Cai, H.; Gu, J.; Hu, M.; Xiang, D.; Lian, J.; Yu, L.; Jia, H.; et al. Epstein-Barr virus infection is associated with a higher Child-Pugh score and may predict poor prognoses for patients with liver cirrhosis. BMC Gastroenterol. 2019, 19, 94. [Google Scholar] [CrossRef]
- Gao, L.; Han, H.; Wang, H.; Cao, L.; Feng, W.H. IL-10 knockdown with siRNA enhances the efficacy of Doxorubicin chemotherapy in EBV-positive tumors by inducing lytic cycle via PI3K/p38 MAPK/NF-kB pathway. Cancer Lett. 2019, 462, 12–22. [Google Scholar] [CrossRef]
- Wildeman, M.A.; Novalic, Z.; Verkuijlen, S.A.; Juwana, H.; Huitema, A.D.; Tan, I.B.; Middeldorp, J.M.; de Boer, J.P.; Greijer, A.E. Cytolytic virus activation therapy for Epstein-Barr virus-driven tumors. Clin. Cancer Res. 2012, 18, 5061–5070. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.F.; Ho, D.N.; Tsang, C.M.; Middeldorp, J.M.; Tsao, G.S.; Chiang, A.K. Activation of lytic cycle of Epstein-Barr virus by suberoylanilide hydroxamic acid leads to apoptosis and tumor growth suppression of nasopharyngeal carcinoma. Int. J. Cancer 2012, 131, 1930–1940. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.H.; Kenney, S.C. Valproic acid enhances the efficacy of chemotherapy in EBV-positive tumors by increasing lytic viral gene expression. Cancer Res. 2006, 66, 8762–8769. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.G.; Kim, H.; Kim, E.J.; Park, P.G.; Dong, S.M.; Choi, T.H.; Kim, H.; Chong, C.R.; Liu, J.O.; Chen, J.; et al. Targeted therapy for Epstein-Barr virus-associated gastric carcinoma using low-dose gemcitabine-induced lytic activation. Oncotarget 2015, 6, 31018–31029. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Mechanism | Effect | References |
|---|---|---|
| ROS Expression | B cell immortalization | [104,105,106] |
| - | Required for normal LMP1 expression | [106] |
| - | STAT3 phosphorylation | [106,107] |
| BHLF1 | Maintenance of type III latency | [108] |
| PAX5 | EBNA-1 localization to oriP and TR-DNA | [119] |
| - | Association of transcription enhancers from oriP and TR-DNA | [119] |
| CAF1 | Inhibits lytic gene expression and increases histone presence at multiple points on the EBV genome. | [120] |
| HIRA | Histone loader involved in maintaining latency | [120] |
| ATRX | Histone loader involved in maintaining latency | [120] |
| DAXX | Histone loader involved in maintaining latency | [120] |
| MYC | Acts on BZLF1 promoter to prevent oriLyt and TR-DNA from looping | [110] |
| SMC1A | Contributes to latency by promoting MYC expression | [110] |
| Facilitated Chromatic Transcription Complex | Contributes to latency by promoting MYC expression | [110] |
| CXCR4 | Maintenance of latency; stimulates LMP2A and EBNA-1 | [112] |
| SUMOylation/SIM-interacting motifs | Facilitates oriP mini genome maintenance and the binding of EBNA-1 to His-tagged SUMO1 and SUMO2 proteins | [116] |
| - | EBNA-1 targets proteins with SUMO2 modifications for degradation | [116] |
| - | Inhibits BZLF1 expression | [116] |
| miRNAs | Inhibition of B cell receptor activation by diminishing NF-κB and/or AP-1 signaling | [123] |
| Factor | Mechanism | Reference |
|---|---|---|
| KLF4, BLIMP1 | Stimulates LMP1, then works with LMP1 to activate BZLF1 and BRLF1 expression | [127,128,129] |
| BCR signaling | Stimulates EBV reactivation | [130] |
| PI3K signaling | Stimulates BZLF1 promoter | [139] |
| IRF8 | Influences caspase activity and KAP1 cleavage | [141] |
| Chaetocin | Stimulates ROS production | [143] |
| EBV upregulation of NADPH oxidase genes | Stimulates ROS production | [146] |
| NRF2 upregulation | Prevents ROS-mediated cell death | [146] |
| LMP1 | Stimulates ROS production | [146] |
| LMP1 and LMP2A | Promote NRF2 production and translocation to nucleus | [147] |
| AKT | Involved in LMP1/LMP2A/NRF2 signaling | [148] |
| miRNA-18a | Stimulates transition from G1 to S phase | [148] |
| Hypoxia | Stimulates reactivation | [148] |
| Mycophenolate mofetil (immunosuppressant) | Impairs Vδ2+ T cell recovery | [155] |
| Syphilis | Cross-links TLR2 and BCR | [157] |
| HPV | E6 and E7 ** stimulates reactivation | [158,159] |
| As2O3 * | Stimulates BZLF1 transcription | [166,167] |
| Radiation | Induces BZLF1 and BLLF | [168] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sausen, D.G.; Bhutta, M.S.; Gallo, E.S.; Dahari, H.; Borenstein, R. Stress-Induced Epstein-Barr Virus Reactivation. Biomolecules 2021, 11, 1380. https://doi.org/10.3390/biom11091380
Sausen DG, Bhutta MS, Gallo ES, Dahari H, Borenstein R. Stress-Induced Epstein-Barr Virus Reactivation. Biomolecules. 2021; 11(9):1380. https://doi.org/10.3390/biom11091380
Chicago/Turabian StyleSausen, Daniel G., Maimoona S. Bhutta, Elisa S. Gallo, Harel Dahari, and Ronen Borenstein. 2021. "Stress-Induced Epstein-Barr Virus Reactivation" Biomolecules 11, no. 9: 1380. https://doi.org/10.3390/biom11091380
APA StyleSausen, D. G., Bhutta, M. S., Gallo, E. S., Dahari, H., & Borenstein, R. (2021). Stress-Induced Epstein-Barr Virus Reactivation. Biomolecules, 11(9), 1380. https://doi.org/10.3390/biom11091380