
The Iron Maiden. Cytosolic Aconitase/IRP1 Conformational Transition in the Regulation of Ferritin Translation and Iron Hemostasis

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Structure of IRE mRNA
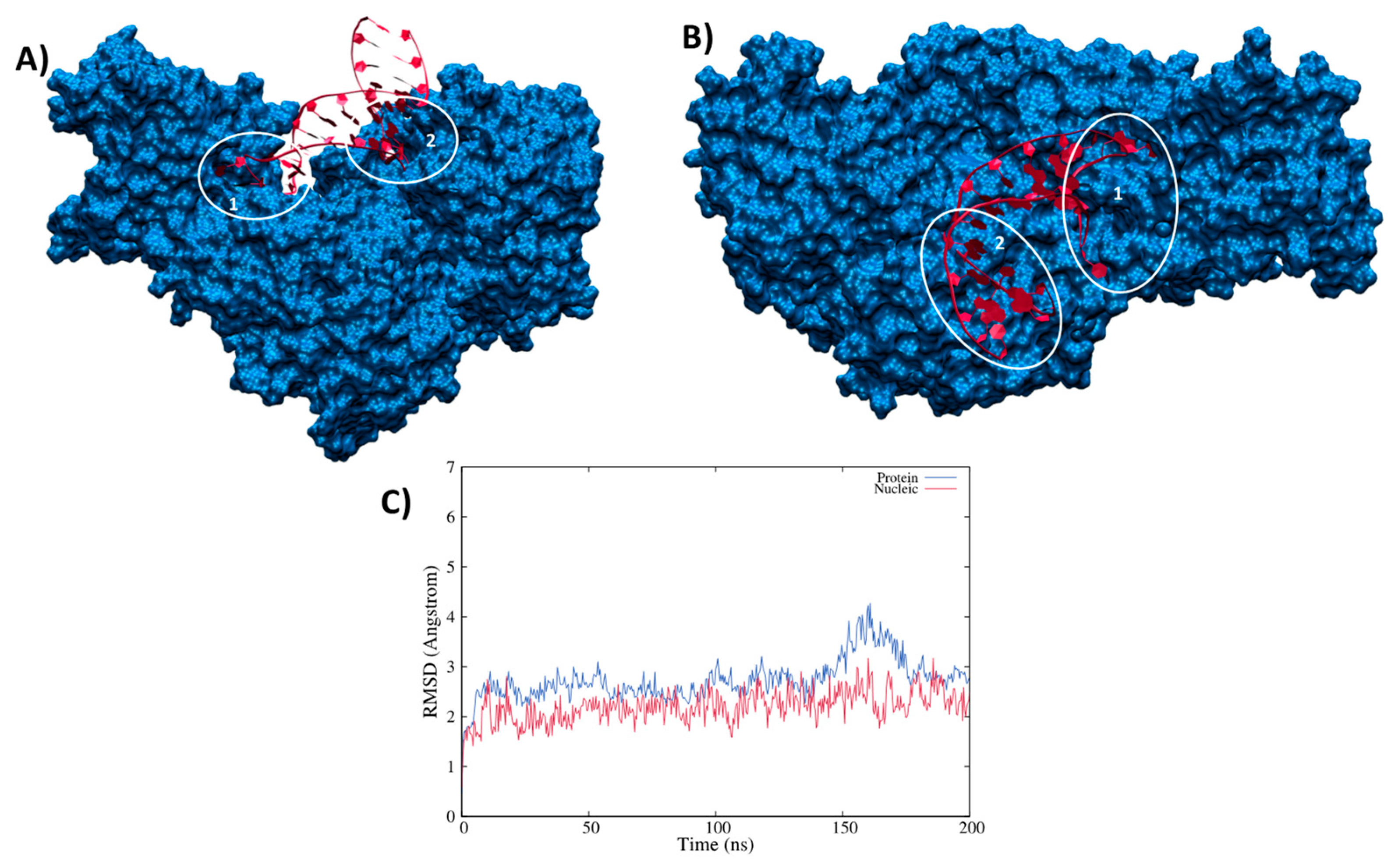
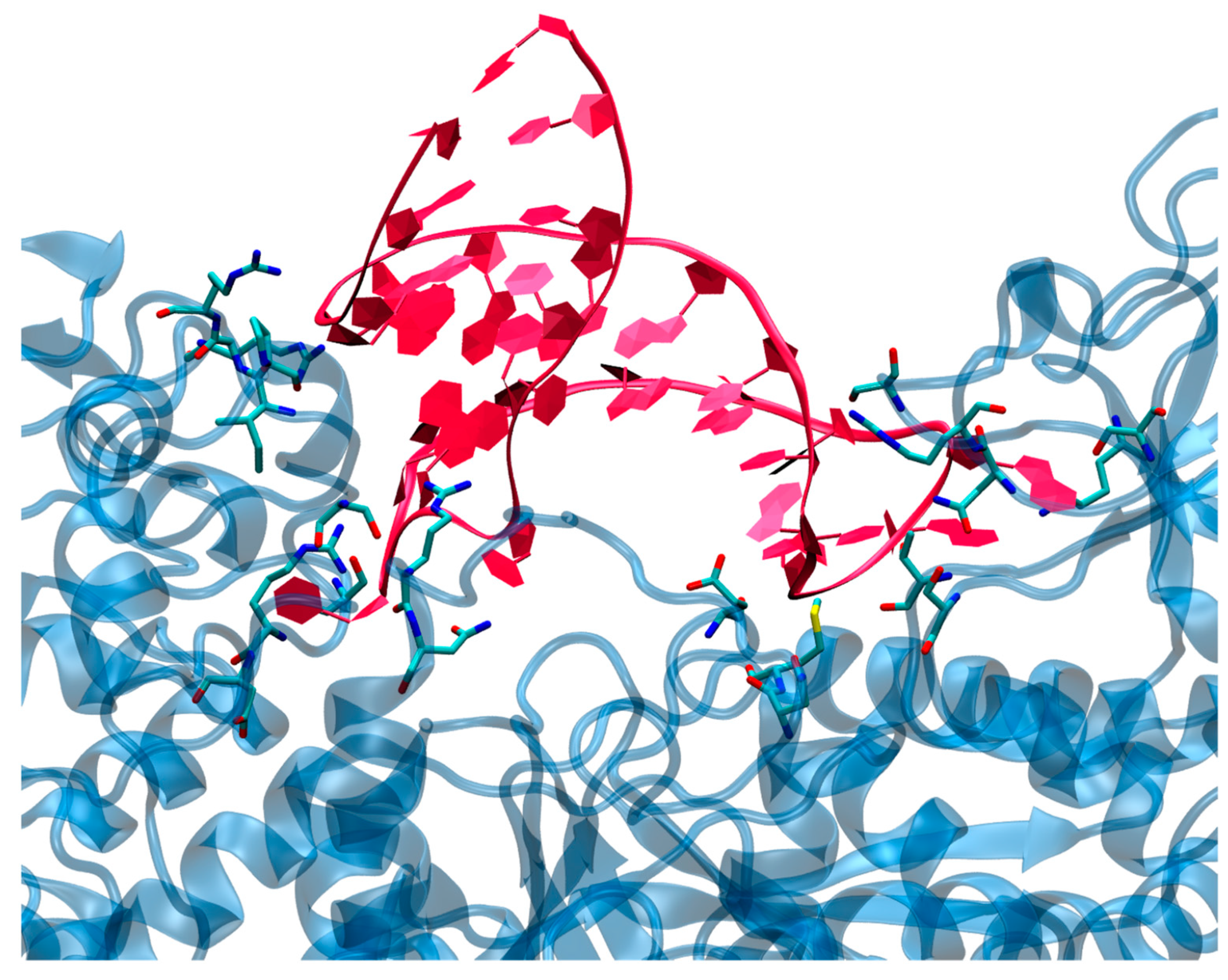
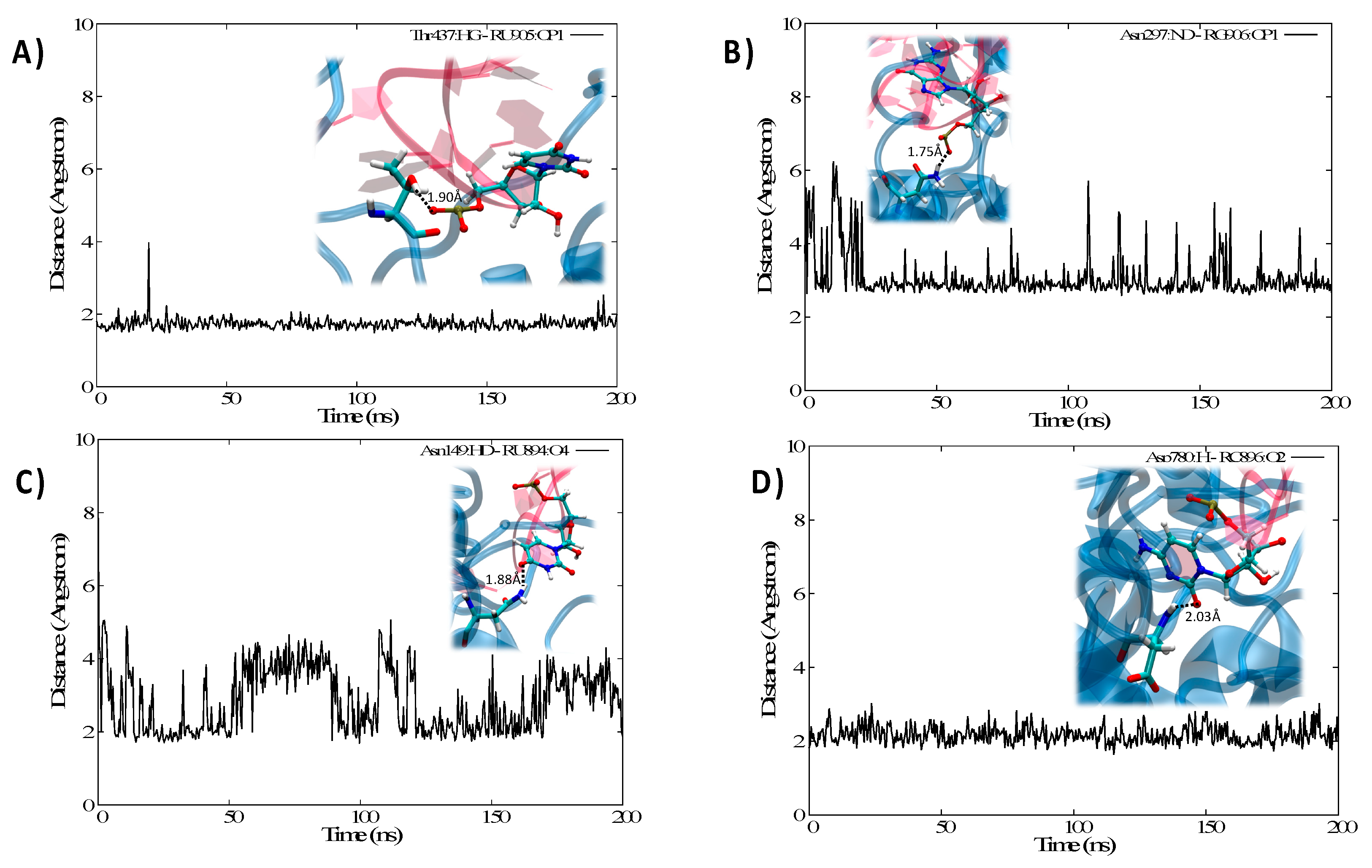
3.2. Structure of the IRP/IRE Complex
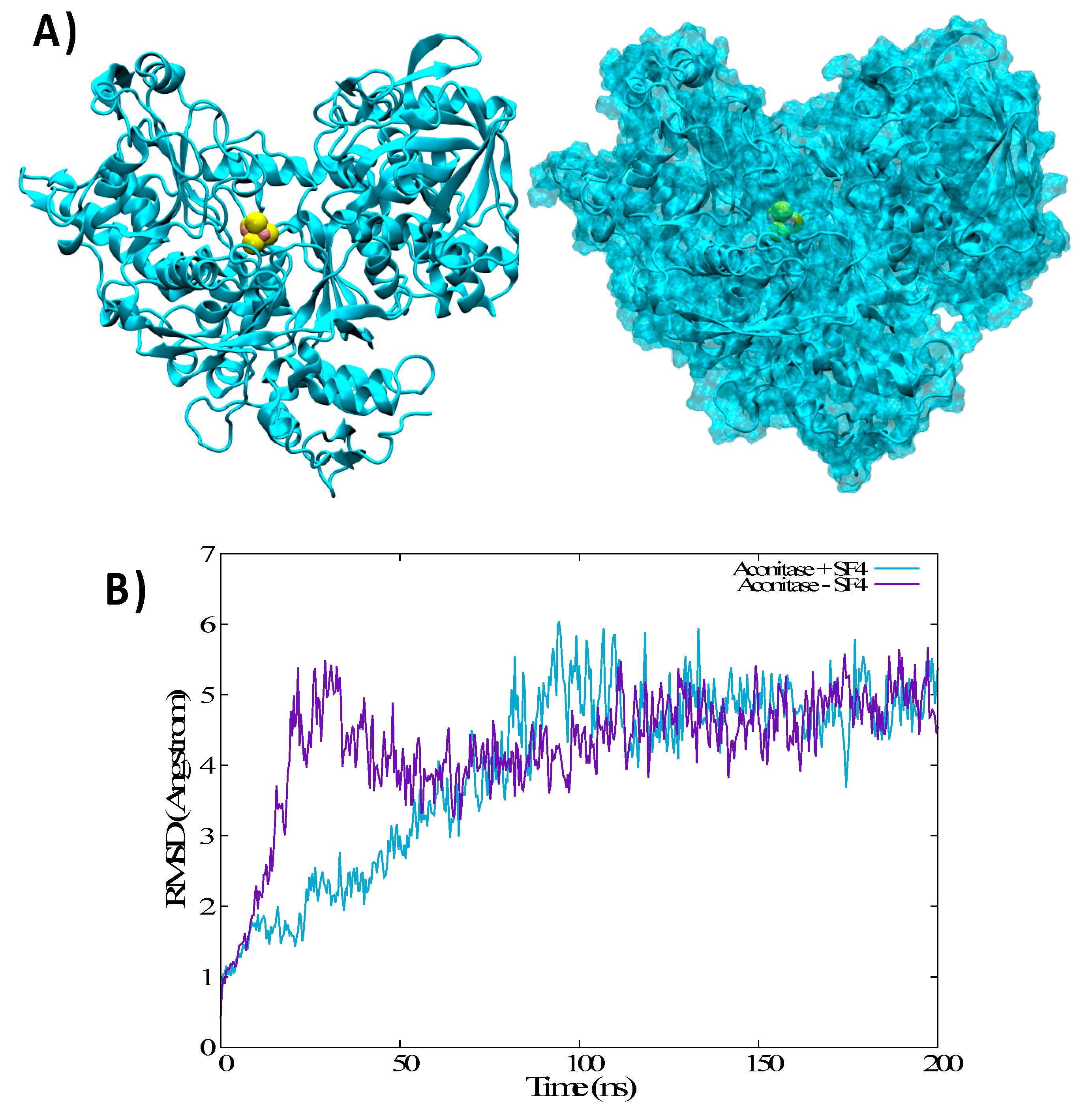
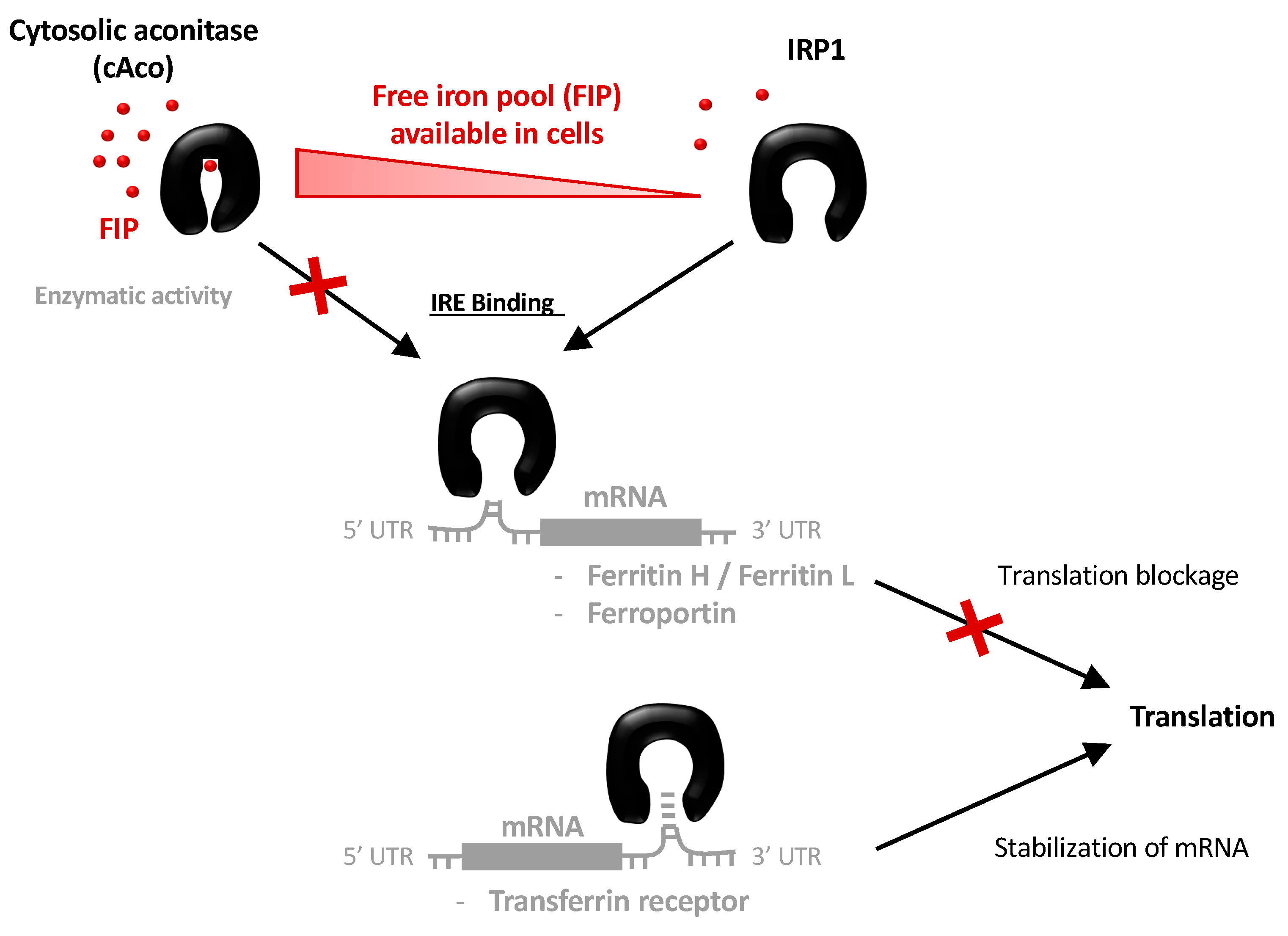
3.3. Structural Features of Unbound IRP1 and cAco
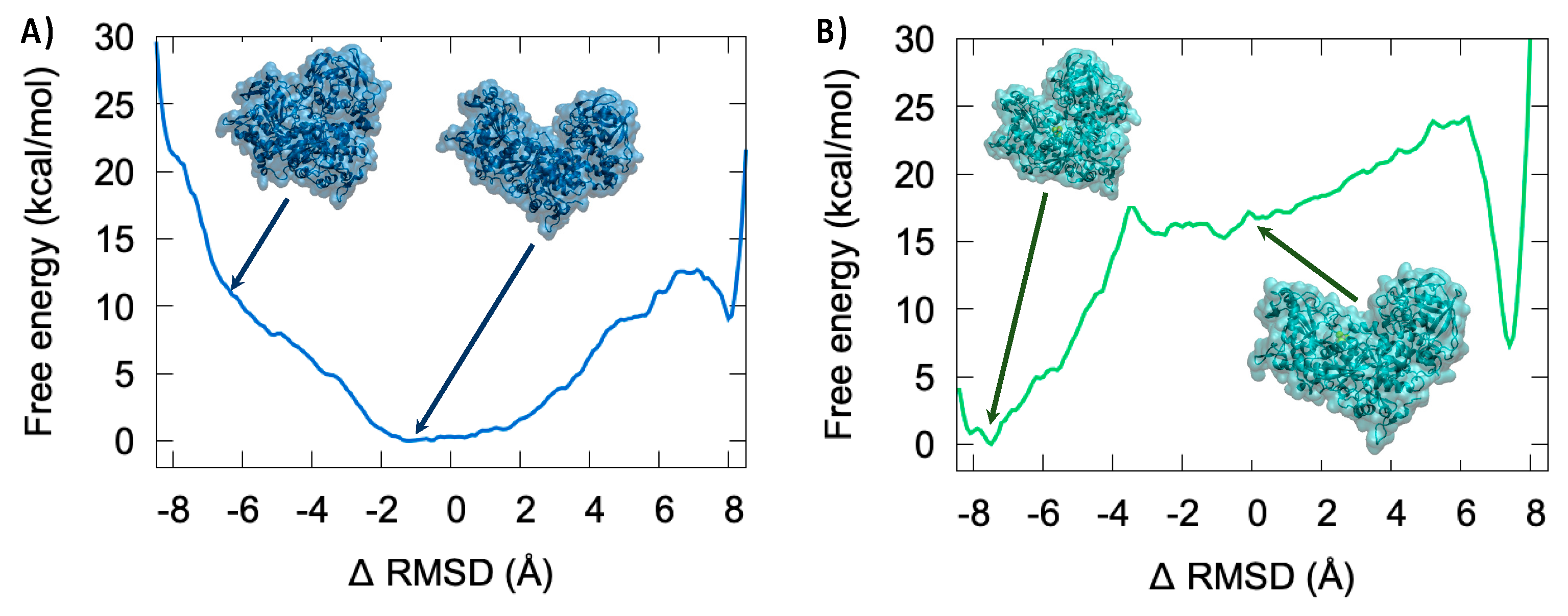
3.4. Thermodynamics of the IRP/Aco Transition
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abbate, V.; Hider, R. Iron in biology. Metallomics 2017, 9, 1467–1469. [Google Scholar] [CrossRef]
- Lindley, P.F. Iron in biology: A structural viewpoint. Rep. Prog. Phys. 1996, 59, 867–933. [Google Scholar] [CrossRef]
- Andrews, N.C. Forging a field: The golden age of iron biology. Blood 2008, 112, 219–230. [Google Scholar] [CrossRef]
- Beard, J.L. Iron biology in immune function, muscle metabolism and neuronal functioning. J. Nutr. 2001, 131, 568S–580S. [Google Scholar] [CrossRef]
- Roat-Malone, R.M. Iron-Containing Proteins and Enzymes. In Bioinorganic Chemistry; John Wiley & Sons, Ltd.: New York, NY, USA, 2007; pp. 343–476. [Google Scholar]
- Fuss, J.O.; Tsai, C.L.; Ishida, J.P.; Tainer, J.A. Emerging critical roles of Fe-S clusters in DNA replication and repair. Biochim. Biophys. Acta-Mol. Cell Res. 2015, 1853, 1253–1271. [Google Scholar] [CrossRef] [PubMed]
- Walden, W.E.; Selezneva, A.; Volz, K. Accommodating variety in iron-responsive elements: Crystal structure of transferrin receptor 1 B IRE bound to iron regulatory protein 1. FEBS Lett. 2012, 586, 32–35. [Google Scholar] [CrossRef] [PubMed]
- Chanet, R.; Baïlle, D.; Golinelli-Cohen, M.-P.; Riquier, S.; Guittet, O.; Lepoivre, M.; Huang, M.-E.; Vernis, L. Fe-S coordination defects in the replicative DNA polymerase delta cause deleterious DNA replication in vivo and subsequent DNA damage in the yeast Saccharomyces cerevisiae. G3 Genes Genomes Genet. 2021, 11, jkab124. [Google Scholar]
- Kruszewski, M. Labile iron pool: The main determinant of cellular response to oxidative stress. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2003, 531, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Chutvanichkul, B.; Vattanaviboon, P.; Mas-oodi, S.; U-pratya, Y.; Wanachiwanawin, W. Labile iron pool as a parameter to monitor iron overload and oxidative stress status in β-thalassemic erythrocytes. Cytom. Part B Clin. Cytom. 2018, 94, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Kakhlon, O.; Cabantchik, Z.I. The labile iron pool: Characterization, measurement, and participation in cellular processes. Free Radic. Biol. Med. 2002, 33, 1037–1046. [Google Scholar] [CrossRef]
- Cabantchik, Z.I. Labile iron in cells and body fluids: Physiology, pathology, and pharmacology. Front. Pharmacol. 2014, 5, 45. [Google Scholar] [CrossRef]
- Bauckman, K.; Haller, E.; Taran, N.; Rockfield, S.; Ruiz-Rivera, A.; Nanjundan, M. Iron alters cell survival in a mitochondria-dependent pathway in ovarian cancer cells. Biochem. J. 2015, 466, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fan, Z.; Yang, Y.; Gu, C. Iron metabolism and its contribution to cancer (Review). Int. J. Oncol. 2019, 54, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.A.M.; Richardson, K.L.; Kabir, T.D.; Trinder, D.; Ganss, R.; Leedman, P.J. Altered Iron Metabolism and Impact in Cancer Biology, Metastasis, and Immunology. Front. Oncol. 2020, 10, 476. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, F. Iron homeostasis and tumorigenesis: Molecular mechanisms and therapeutic opportunities. Protein Cell 2015, 6, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.S.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zhou, Q.; Wu, D.; Chen, L. Mitochondrial iron metabolism and its role in diseases. Clin. Chim. Acta 2021, 513, 6–12. [Google Scholar] [CrossRef]
- Thomas, C.; Mackey, M.M.; Diaz, A.A.; Cox, D.P. Hydroxyl radical is produced via the Fenton reaction in submitochondrial particles under oxidative stress: Implications for diseases associated with iron accumulation. Redox Rep. 2009, 14, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Eisenstein, R.S.; Blemings, K.P. Iron regulatory proteins, iron responsive elements and iron homeostasis. J. Nutr. 1998, 128, 2295–2298. [Google Scholar] [CrossRef]
- Peyssonnaux, C.; Zinkernagel, A.S.; Schuepbach, R.A.; Rankin, E.; Vaulont, S.; Haase, V.H.; Nizet, V.; Johnson, R.S. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J. Clin. Investig. 2007, 117, 1926–1932. [Google Scholar] [CrossRef]
- Winter, W.E.; Bazydlo, L.A.L.; Harris, N.S. The molecular biology of human iron metabolism. Lab. Med. 2014, 45, 92–102. [Google Scholar] [CrossRef]
- Wallace, D.F. The Regulation of Iron Absorption and Homeostasis. Clin. Biochem. Rev. 2016, 37, 51–62. [Google Scholar]
- Zhou, Z.D.; Tan, E.-K. Iron regulatory protein (IRP)-iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Mol. Neurodegener. 2017, 12, 1–12. [Google Scholar] [CrossRef]
- Wilkinson, N.; Pantopoulos, K. The IRP/IRE system in vivo: Insights from mouse models. Front. Pharmacol. 2014, 5, 176. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.L.; Ghosh, M.C.; Rouault, T.A. The physiological functions of iron regulatory proteins in iron homeostasis—An update. Front. Pharmacol. 2014, 5, 124. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, N.; Pantopoulos, K. IRP1 regulates erythropoiesis and systemic iron homeostasis by controlling HIF2α mRNA translation. Blood 2013, 122, 1658–1669. [Google Scholar] [CrossRef] [PubMed]
- Pantopoulos, K. Iron metabolism and the IRE/IRP regulatory system: An update. Ann. N. Y. Acad. Sci. 2004, 1012, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.H.; Rouault, T.A. Metabolic regulation of citrate and iron by aconitases: Role of iron-sulfur cluster biogenesis. BioMetals 2007, 20, 549–564. [Google Scholar] [CrossRef]
- Gourley, B.L.; Parker, S.B.; Jones, B.J.; Zumbrennen, K.B.; Leibold, E.A. Cytosolic aconitase and ferritin are regulated by iron in Caenorhabditis elegans. J. Biol. Chem. 2003, 278, 3227–3234. [Google Scholar] [CrossRef]
- Khan, M.A.; Walden, W.E.; Theil, E.C.; Goss, D.J. Thermodynamic and Kinetic Analyses of Iron Response Element (IRE)-mRNA Binding to Iron Regulatory Protein, IRP1. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Theil, E.C.; McKenzie, R.A.; Sierzputowska-Gracz, H. Structure and function of IREs, the noncoding mRNA sequences regulating synthesis of ferritin, transferrin receptor and (erythroid) 5-aminolevulinate synthase. Adv. Exp. Med. Biol. 1994, 356, 111–118. [Google Scholar] [PubMed]
- Hall, K.B.; Williams, D.J. Dynamics of the IRE RNA hairpin loop probed by 2-aminopurine fluorescence and stochastic dynamics simulations. RNA 2004, 10, 34–47. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cairo, G.; Recalcati, S. Iron-regulatory proteins: Molecular biology and pathophysiological implications. Expert Rev. Mol. Med. 2007, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.C.; Olsthoorn, R.C.L. Relevance of the iron-responsive element (IRE) pseudotriloop structure for IRP1/2 binding and validation of IRE-like structures using the yeast three-hybrid system. Gene 2019, 710, 399–405. [Google Scholar] [CrossRef]
- Dupuy, J.; Volbeda, A.; Carpentier, P.; Darnault, C.; Moulis, J.M.; Fontecilla-Camps, J.C. Crystal structure of human iron regulatory protein 1 as cytosolic aconitase. Structure 2006, 14, 129–139. [Google Scholar] [CrossRef]
- Autenrieth, F.; Tajkhorshid, E.; Baudry, J.; Luthey-Schulten, Z. Classical force field parameters for the heme prosthetic group of cytochrome c. J. Comput. Chem. 2004, 25, 1613–1622. [Google Scholar] [CrossRef]
- Zgarbová, M.; Šponer, J.; Otyepka, M.; Cheatham, T.E.; Galindo-Murillo, R.; Jurečka, P. Refinement of the Sugar-Phosphate Backbone Torsion Beta for AMBER Force Fields Improves the Description of Z- and B-DNA. J. Chem. Theory Comput. 2015, 11, 5723–5736. [Google Scholar] [CrossRef]
- Dans, P.D.; Ivani, I.; Hospital, A.; Portella, G.; González, C.; Orozco, M. How accurate are accurate force-fields for B-DNA? Nucleic Acids Res. 2017, 45, 4217–4230. [Google Scholar] [CrossRef]
- Carvalho, A.T.P.; Swart, M. Electronic structure investigation and parametrization of biologically relevant iron-sulfur clusters. J. Chem. Inf. Model. 2014, 54, 613–620, Correction in 2015, 55, 1508. [Google Scholar] [CrossRef] [PubMed]
- Sarre, A.; Stelter, M.; Rollo, F.; De Bonis, S.; Seck, A.; Hognon, C.; Ravanat, J.L.J.-L.; Monari, A.; Dehez, F.; Moe, E.; et al. The three Endonuclease III variants of Deinococcus radiodurans possess distinct and complementary DNA repair activities. DNA Repair 2019, 78, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Davidchack, R.L.; Handel, R.; Tretyakov, M.V. Langevin thermostat for rigid body dynamics. J. Chem. Phys. 2009, 130, 234101. [Google Scholar] [CrossRef] [PubMed]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Hopkins, C.W.; Le Grand, S.; Walker, R.C.; Roitberg, A.E. Long-time-step molecular dynamics through hydrogen mass repartitioning. J. Chem. Theory Comput. 2015, 11, 1864–1874. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N ⋅log( N ) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Zhao, T.; Fu, H.; Lelièvre, T.; Shao, X.; Chipot, C.; Cai, W. The Extended Generalized Adaptive Biasing Force Algorithm for Multidimensional Free-Energy Calculations. J. Chem. Theory Comput. 2017, 13, 1566–1576. [Google Scholar] [CrossRef]
- Barducci, A.; Bonomi, M.; Parrinello, M. Metadynamics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 826–843. [Google Scholar] [CrossRef]
- Fu, H.; Zhang, H.; Chen, H.; Shao, X.; Chipot, C.; Cai, W. Zooming across the Free-Energy Landscape: Shaving Barriers, and Flooding Valleys. J. Phys. Chem. Lett. 2018, 9, 4738–4745. [Google Scholar] [CrossRef]
- Fu, H.; Shao, X.; Cai, W.; Chipot, C. Taming Rugged Free Energy Landscapes Using an Average Force. Acc. Chem. Res. 2019, 52, 3254–3264. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Piccinelli, P.; Samuelsson, T. Evolution of the iron-responsive element. RNA 2007, 13, 952–966. [Google Scholar] [CrossRef]
- Sierzputowska-gracz, H.; Mckenzie, R.A.; Theil, E.C. The importance of a single G in the hairpin loop of the iron responsive element (IRE) in ferritin mRNA for structure: An NMR spectroscopy study. Nucleic Acids Res. 1995, 23, 146–153. [Google Scholar] [CrossRef][Green Version]
- Hognon, C.; Garaude, S.; Timmins, J.; Chipot, C.; Dehez, F.F.; Monari, A. Molecular Bases of DNA Packaging in Bacteria Revealed by All-Atom Molecular Dynamics Simulations: The Case of Histone-Like Proteins in Borrelia burgdorferi. J. Phys. Chem. Lett. 2019, 10, 7200–7207. [Google Scholar] [CrossRef]
- Matoušková, E.; Bignon, E.; Claerbout, V.E.P.; Dršata, T.; Gillet, N.; Monari, A.; Dumont, E.; Lankaš, F. Impact of the Nucleosome Histone Core on the Structure and Dynamics of DNA-Containing Pyrimidine-Pyrimidone (6-4) Photoproduct. J. Chem. Theory Comput. 2020, 16, 5972–5981. [Google Scholar] [CrossRef]
- Hognon, C.; Monari, A. Staring at the Naked Goddess: Unraveling the Structure and Reactivity of Artemis Endonuclease Interacting with a DNA Double Strand. Molecules 2021, 26, 3986. [Google Scholar] [CrossRef]
- Gattuso, H.; Durand, E.; Bignon, E.; Morell, C.; Georgakilas, A.G.; Dumont, E.; Chipot, C.; Dehez, F.; Monari, A. Repair Rate of Clustered Abasic DNA Lesions by Human Endonuclease: Molecular Bases of Sequence Specificity. J. Phys. Chem. Lett. 2016, 7, 3760–3765. [Google Scholar] [CrossRef]
- Bignon, E.; Claerbout, V.E.P.; Jiang, T.; Morell, C.; Gillet, N.; Dumont, E. Nucleosomal embedding reshapes the dynamics of abasic sites. Sci. Rep. 2020, 10, 17314. [Google Scholar] [CrossRef]
- Bignon, E.; Gillet, N.; Jiang, T.; Morell, C.; Dumont, E. A Dynamic View of the Interaction of Histone Tails with Clustered Abasic Sites in a Nucleosome Core Particle. J. Phys. Chem. Lett. 2021, 12, 6014–6019. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hognon, C.; Bignon, E.; Harle, G.; Touche, N.; Grandemange, S.; Monari, A. The Iron Maiden. Cytosolic Aconitase/IRP1 Conformational Transition in the Regulation of Ferritin Translation and Iron Hemostasis. Biomolecules 2021, 11, 1329. https://doi.org/10.3390/biom11091329
Hognon C, Bignon E, Harle G, Touche N, Grandemange S, Monari A. The Iron Maiden. Cytosolic Aconitase/IRP1 Conformational Transition in the Regulation of Ferritin Translation and Iron Hemostasis. Biomolecules. 2021; 11(9):1329. https://doi.org/10.3390/biom11091329
Chicago/Turabian StyleHognon, Cécilia, Emmanuelle Bignon, Guillaume Harle, Nadège Touche, Stéphanie Grandemange, and Antonio Monari. 2021. "The Iron Maiden. Cytosolic Aconitase/IRP1 Conformational Transition in the Regulation of Ferritin Translation and Iron Hemostasis" Biomolecules 11, no. 9: 1329. https://doi.org/10.3390/biom11091329
APA StyleHognon, C., Bignon, E., Harle, G., Touche, N., Grandemange, S., & Monari, A. (2021). The Iron Maiden. Cytosolic Aconitase/IRP1 Conformational Transition in the Regulation of Ferritin Translation and Iron Hemostasis. Biomolecules, 11(9), 1329. https://doi.org/10.3390/biom11091329