
Coagulation Abnormalities in Renal Pathology of Chronic Kidney Disease: The Interplay between Blood Cells and Soluble Factors

 ,
,  , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Material Supplies
2.3. Hematological and Serum Biochemical Analysis
2.4. Measurement of Intracellular ROS Levels
2.5. Measurement of Intracellular Free Calcium Levels ([iCa2+])
2.6. P-Selectin Expression and Phospatidylserine Exposure
2.7. Platelet Function Assays
2.8. Screening Test and Coagulation Factors Analysis
2.9. ADAMTS Activity and Inhibitor Test
2.10. Statistical and Network Analysis
3. Results
3.1. Baseline Hematological and Serum Biochemical Profile
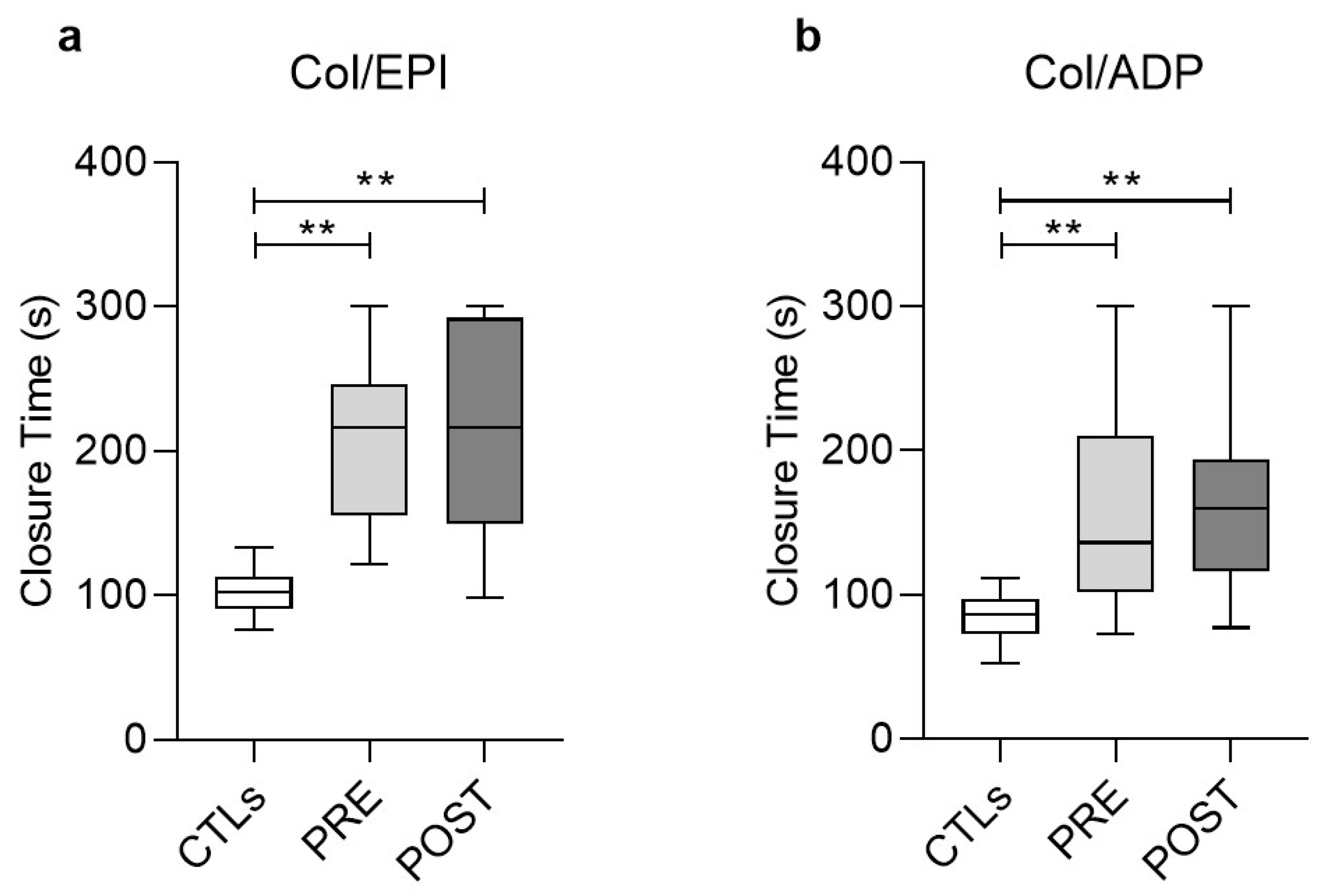
3.2. Functional Analysis of Platelets (PFA/EPI, PFA/ADP)
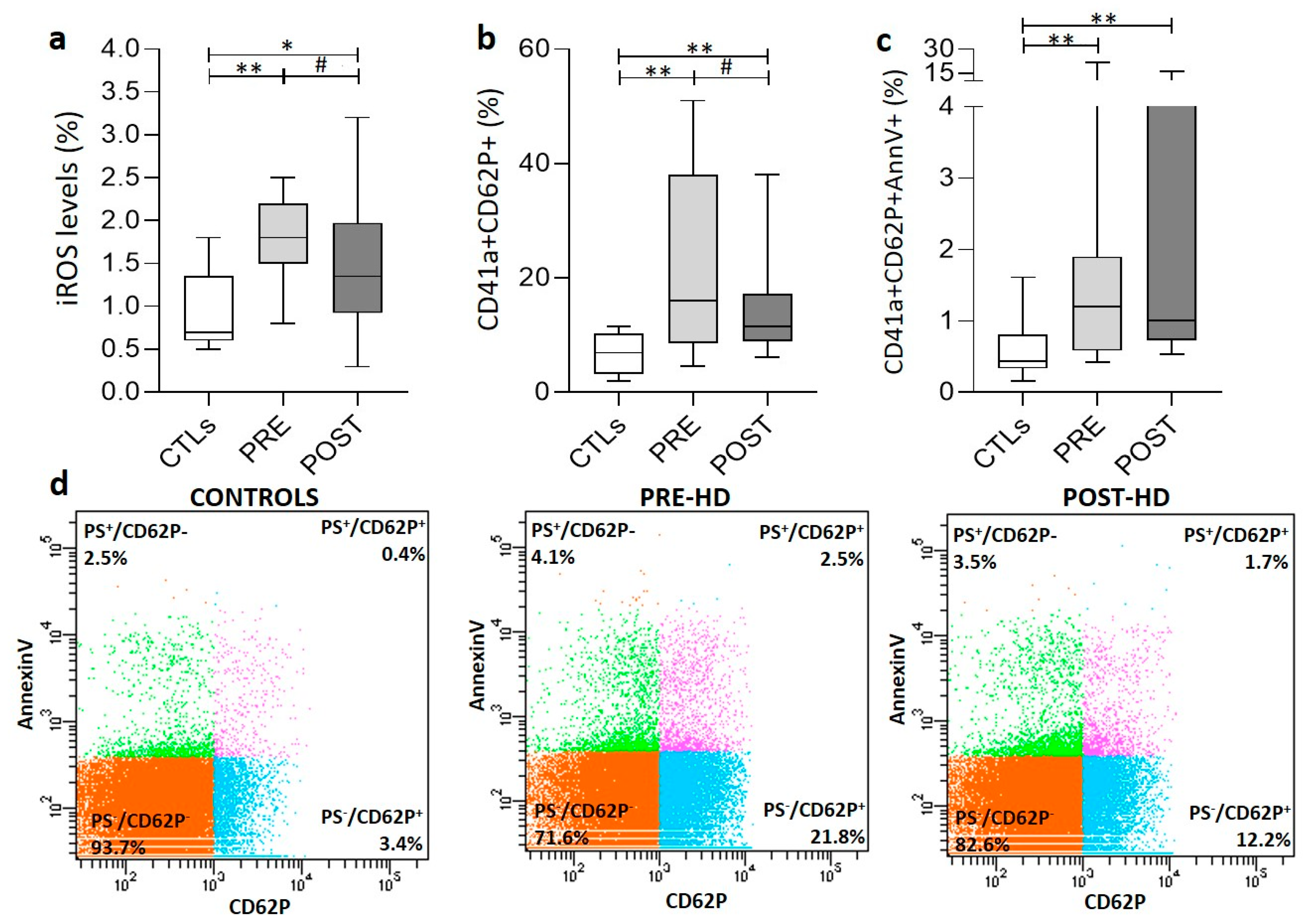
3.3. PLT Activation Signaling and Oxidative Stress
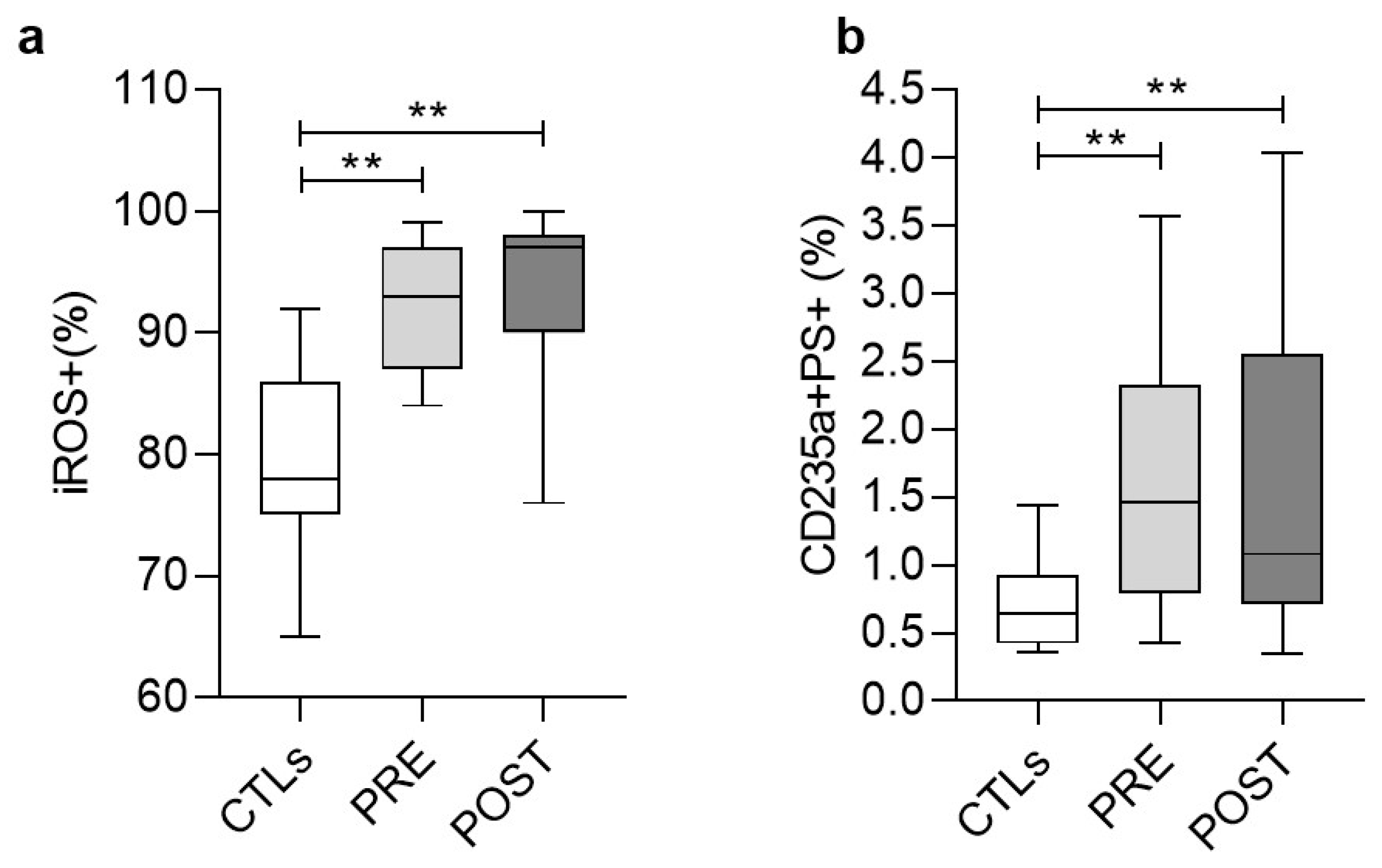
3.4. Stress Signaling in Red Blood Cells (RBCs)
3.5. Coagulation (Secondary Hemostasis) Screening Test
3.6. Functional Analysis of Factors Involved in the Coagulation Cascade and in Fibrinolysis
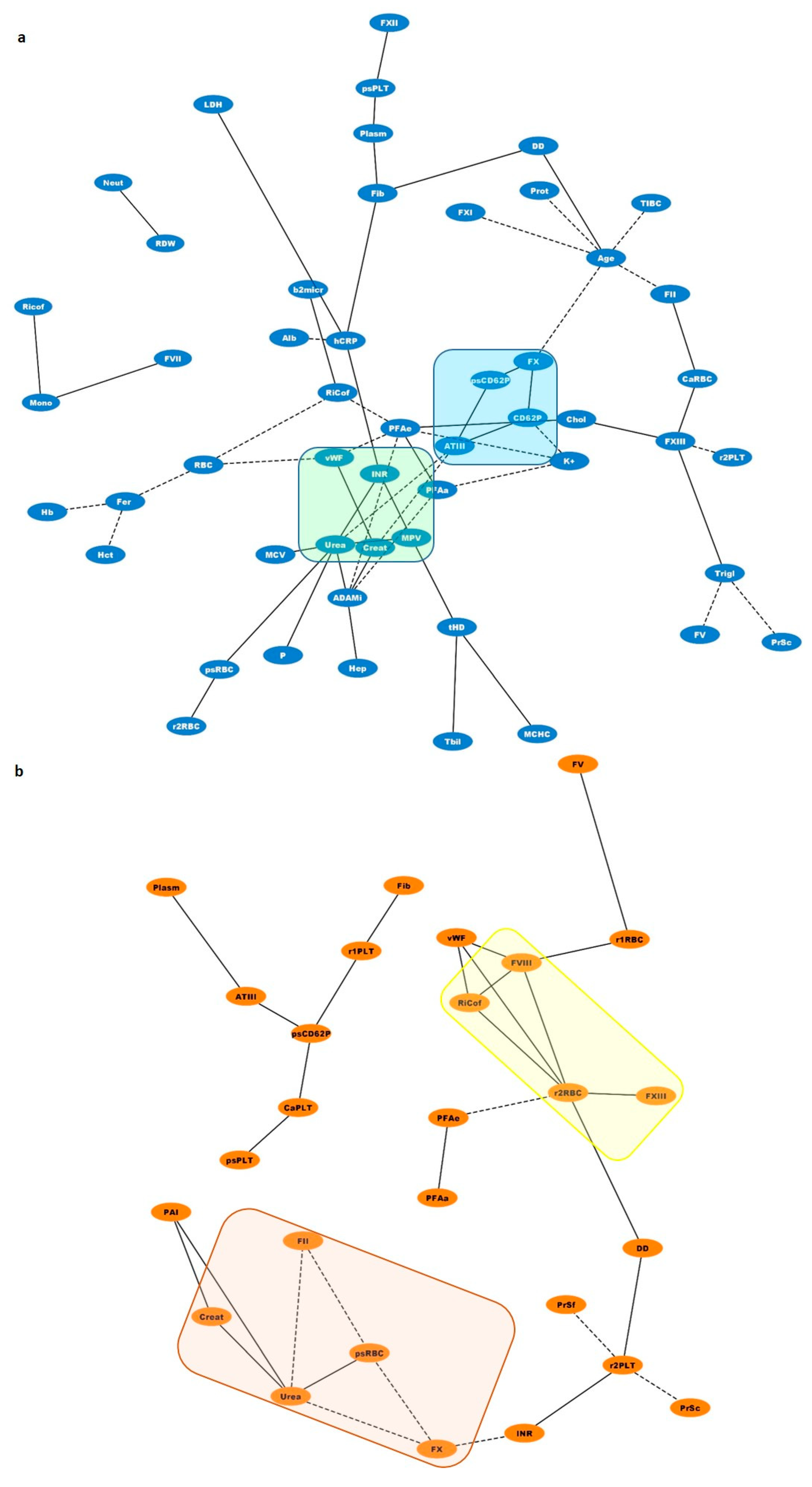
3.7. Biological Networks
4. Discussion
5. Conclusions
Author Contributions
Funding

Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gäckler, A.; Rohn, H.; Lisman, T.; Benkö, T.; Witzke, O.; Kribben, A.; Saner, F.H. Evaluation of hemostasis in patients with end-stage renal disease. PLoS ONE 2019, 14, e0212237. [Google Scholar] [CrossRef]
- Kaw, D.; Malhotra, D. Hematology: Issues in the Dialysis Patient: Platelet Dysfunction and End-Stage Renal Disease. Semin. Dial. 2006, 19, 317–322. [Google Scholar] [CrossRef]
- Lu, H.-Y.; Liao, K.-M. Increased risk of deep vein thrombosis in end-stage renal disease patients. BMC Nephrol. 2018, 19, 1–9. [Google Scholar] [CrossRef]
- Ocak, G.; Vossen, C.Y.; Rotmans, J.I.; Lijfering, W.M.; Rosendaal, F.R.; Parlevliet, K.J.; Krediet, R.T.; Boeschoten, E.W.; Verduijn, M.; Dekker, F.W. Venous and arterial thrombosis in dialysis patients. Thromb. Haemost. 2011, 106, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Harboe, M. A Method for Determination of Hemoglobin in Plasma by Near-Ultraviolet Spectrophotometry. Scand. J. Clin. Lab. Investig. 1959, 11, 66–70. [Google Scholar] [CrossRef]
- Tzounakas, V.L.; Georgatzakou, H.T.; Kriebardis, A.G.; Voulgaridou, A.I.; Stamoulis, K.E.; Foudoulaki-Paparizos, L.E.; Antonelou, M.H.; Papassideri, I.S. Donor variation effect on red blood cell storage lesion: A multivariable, yet consistent, story. Transfusion 2016, 56, 1274–1286. [Google Scholar] [CrossRef]
- Assenov, Y.; Ramírez, F.; Schelhorn, S.-E.; Lengauer, T.; Albrecht, M. Computing topological parameters of biological networks. Bioinformatics 2008, 24, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Weisel, J.W.; Litvinov, R.I. Red blood cells: The forgotten player in hemostasis and thrombosis. J. Thromb. Haemost. 2019, 17, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh, K.; Rodriguez, R.A.; Humphreys, M.H. Association between serum ferritin and measures of inflammation, nutrition and iron in haemodialysis patients. Nephrol. Dial. Transplant. 2004, 19, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Nazy, I.; Elliott, T.D.; Arnold, D.M. Platelet factor 4 inhibits ADAMTS13 activity and regulates the multimeric distribution of von Willebrand factor. Br. J. Haematol. 2020, 190, 594–598. [Google Scholar] [CrossRef]
- Stenvinkel, P. The role of inflammation in the anaemia of end-stage renal disease. Nephrol. Dial. Transplant. 2001, 16 (Suppl. 7), 36–40. [Google Scholar] [CrossRef] [PubMed]
- Van Bladel, E.R.; de Jager, R.L.; Walter, D.; Cornelissen, L.; Gaillard, C.A.; Boven, L.A.; Roest, M.; Fijnheer, R. Platelets of patients with chronic kidney disease demonstrate deficient platelet reactivity in vitro. BMC Nephrol. 2012, 13, 127. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, H.I.; Stein, I.M.; Cohen, B.D.; White, J.G. Further studies on the platelet-inhibitory effect of guanidinosuccinic acid and its role in uremic bleeding. Am. J. Med. 1970, 49, 336–345. [Google Scholar] [CrossRef]
- Linthorst, G.E.; Avis, H.J.; Levi, M. Uremic Thrombocytopathy Is not about Urea: Table 1. J. Am. Soc. Nephrol. 2010, 21, 753–755. [Google Scholar] [CrossRef] [PubMed]
- Mekawy, M.A.; Habashy, D.M.; Abd El-Mohsen, W.A. Effect of hemodialysis on platelet function in end-stage renal disease Egyptian patients usingin vitroclosure time test (PFA-100 analyzer). Platelets 2014, 26, 443–447. [Google Scholar] [CrossRef]
- Gawaz, M.P.; Dobos, G.; Späth, M.; Schollmeyer, P.; Gurland, H.J.; Mujais, S.K. Impaired function of platelet membrane glycoprotein IIb-IIIa in end-stage renal disease. J. Am. Soc. Nephrol. 1994, 5, 36–46. [Google Scholar] [CrossRef]
- Favaloro, E.J. Utility of the von Willebrand factor collagen binding assay in the diagnosis of von Willebrand disease. Am. J. Hematol. 2017, 92, 114–118. [Google Scholar] [CrossRef]
- Jubelirer, S.J. Hemostatic Abnormalities in Renal Disease. Am. J. Kidney Dis. 1985, 5, 219–225. [Google Scholar] [CrossRef]
- Spann, A.P.; Campbell, J.E.; FitzGibbon, S.R.; Rodriguez, A.; Cap, A.P.; Blackbourne, L.H.; Shaqfeh, E.S. The Effect of Hematocrit on Platelet Adhesion: Experiments and Simulations. Biophys. J. 2016, 111, 577–588. [Google Scholar] [CrossRef]
- Casonato, A.; Pontara, E.; Vertolli, U.; Steffan, A.; Durante, C.; de Marco, L.; Sartorello, F.; Girolami, A. Plasma and Platelet von Willebrand Factor Abnormalities in Patients with Uremia: Lack of Correlation with Uremic Bleeding. Clin. Appl. Thromb. Hemost. 2001, 7, 81–86. [Google Scholar] [CrossRef]
- Huang, M.-J.; Wei, R.-B.; Wang, Y.; Su, T.-Y.; Di, P.; Li, Q.-P.; Yang, X.; Li, P.; Chen, X.-M. Blood coagulation system in patients with chronic kidney disease: A prospective observational study. BMJ Open 2017, 7, e014294. [Google Scholar] [CrossRef] [PubMed]
- Holden, R.M.; Tuttle, A.; Burbidge, T.; Hegadorn, C.; Grabell, J.; Pruss, C.; James, P.D. Quantitative and qualitative changes of von Willebrand factor and their impact on mortality in patients with end-stage kidney disease. Blood Coagul. Fibrinolysis 2013, 24, 719–726. [Google Scholar] [CrossRef]
- Boneu, B.; Abbal, M.; Plante, J.; Bierme, R. Factor-VIII Complex and Endothelial Damage. Lancet 1975, 305, 1430. [Google Scholar] [CrossRef]
- Turner, N.A.; Moake, J.L. Factor VIII Is Synthesized in Human Endothelial Cells, Packaged in Weibel-Palade Bodies and Secreted Bound to ULVWF Strings. PLoS ONE 2015, 10, e0140740. [Google Scholar] [CrossRef]
- Van der Vorm, L.N.; Visser, R.; Huskens, D.; Veninga, A.; Adams, D.L.; Remijn, J.A.; Hemker, H.C.; Rensma, P.L.; van Horssen, R.; de Laat, B. Circulating active von Willebrand factor levels are increased in chronic kidney disease and end-stage renal disease. Clin. Kidney J. 2020, 13, 72–74. [Google Scholar] [CrossRef]
- Bernardo, A.; Ball, C.; Nolasco, L.; Moake, J.F.; Dong, J.-F. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell–derived ultralarge von Willebrand factor multimers under flow. Blood 2004, 104, 100–106. [Google Scholar] [CrossRef]
- Ocak, G.; Roest, M.; Verhaar, M.C.; Rookmaaker, M.B.; Blankestijn, P.J.; Bos, W.J.W.; Fijnheer, R.; Péquériaux, N.C.; Dekker, F.W. Von Willebrand factor, ADAMTS13 and mortality in dialysis patients. BMC Nephrol. 2021, 22, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Peyvandi, F.; Ferrari, S.; Lavoretano, S.; Canciani, M.T.; Mannucci, P.M. von Willebrand factor cleaving protease (ADAMTS-13) and ADAMTS-13 neutralizing autoantibodies in 100 patients with thrombotic thrombocytopenic purpura. Br. J. Haematol. 2004, 127, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.C.; Liu, A.; Liu, L.; Ball, C.; Choi, H.; May, W.S.; Aboulfatova, K.; Bergeron, A.L.; Dong, J.-F. Acquired ADAMTS-13 deficiency in pediatric patients with severe sepsis. Haematologica 2007, 92, 121–124. [Google Scholar] [CrossRef]
- Pillai, V.G.; Bao, J.; Zander, C.B.; McDaniel, J.K.; Chetty, P.S.; Seeholzer, S.H.; Bdeir, K.; Cines, D.B.; Zheng, X.L. Human neutrophil peptides inhibit cleavage of von Willebrand factor by ADAMTS13: A potential link of inflammation to TTP. Blood 2016, 128, 110–119. [Google Scholar] [CrossRef]
- Novelli, E.M.; Kato, G.; Hildesheim, M.E.; Barge, S.; Meyer, M.P.; Lozier, J.; Hassett, A.C.; Ragni, M.V.; Isenberg, J.S.; Gladwin, M.T. Thrombospondin-1 inhibits ADAMTS13 activity in sickle cell disease. Haematologica 2013, 98, e132–e134. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Simurda, T.; Zolkova, J.; Kolkova, Z.; Loderer, D.; Dobrotova, M.; Skornova, I.; Brunclíkova, M.; Grendar, M.; Lasabova, Z.; Stasko, J.; et al. Comparison of clinical phenotype with genetic and laboratory results in 31 patients with congenital dysfibrinogenemia in northern Slovakia. Int. J. Hematol. 2020, 111, 795–802. [Google Scholar] [CrossRef]
- Taslimi, R.; Golshani, K. Thrombotic and hemorrhagic presentation of congenital hypo/afibrinogenemia. Am. J. Emerg. Med. 2011, 29, 573.e3. [Google Scholar] [CrossRef] [PubMed]
- Hayes, D.T. Dysfibrinogenemia and Thrombosis. Arch. Pathol. Lab. Med. 2002, 126, 1387–1390. [Google Scholar] [CrossRef]
- Simurda, T.; Vilar, R.; Zolkova, J.; Ceznerova, E.; Kolkova, Z.; Loderer, D.; Neerman-Arbez, M.; Casini, A.; Brunclikova, M.; Skornova, I.; et al. A Novel Nonsense Mutation in FGB (c.1421G>A; p.Trp474Ter) in the Beta Chain of Fibrinogen Causing Hypofibrinogenemia with Bleeding Phenotype. Biomedicines 2020, 8, 605. [Google Scholar] [CrossRef]
- Chapin, J.C.; Hajjar, K.A. Fibrinolysis and the control of blood coagulation. Blood Rev. 2015, 29, 17–24. [Google Scholar] [CrossRef]
- Shlipak, M.G.; Fried, L.F.; Crump, C.; Bleyer, A.J.; Manolio, T.A.; Tracy, R.P.; Furberg, C.D.; Psaty, B.M. Elevations of Inflammatory and Procoagulant Biomarkers in Elderly Persons with Renal Insufficiency. Circulation 2003, 107, 87–92. [Google Scholar] [CrossRef]
- Machlus, K.R.; Cardenas, J.C.; Church, F.C.; Wolberg, A.S. Causal relationship between hyperfibrinogenemia, thrombosis, and resistance to thrombolysis in mice. Blood 2011, 117, 4953–4963. [Google Scholar] [CrossRef]
- Merten, M.; Thiagarajan, P. P-Selectin Expression on Platelets Determines Size and Stability of Platelet Aggregates. Circulation 2000, 102, 1931–1936. [Google Scholar] [CrossRef]
- Théorêt, J.-F.; Chahrour, W.; Yacoub, D.; Merhi, Y. Recombinant P-selectin glycoprotein-ligand-1 delays thrombin-induced platelet aggregation: A new role for P-selectin in early aggregation. Br. J. Pharmacol. 2006, 148, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Arya, M.; Anvari, B.; Romo, G.M.; Cruz, M.A.; Dong, J.-F.; McIntire, L.V.; Moake, J.L.; López, J. Ultralarge multimers of von Willebrand factor form spontaneous high-strength bonds with the platelet glycoprotein Ib-IX complex: Studies using optical tweezers. Blood 2002, 99, 3971–3977. [Google Scholar] [CrossRef]
- Tracy, P.B.; Mann, K.G. Prothrombinase complex assembly on the platelet surface is mediated through the 74,000-dalton component of factor Va. Proc. Natl. Acad. Sci. USA 1983, 80, 2380–2384. [Google Scholar] [CrossRef]
- Bonomini, M.; Dottori, S.; Amoroso, L.; Arduini, A.; Sirolli, V. Increased platelet phosphatidylserine exposure and caspase activation in chronic uremia. J. Thromb. Haemost. 2004, 2, 1275–1281. [Google Scholar] [CrossRef]
- Aggarwal, A.; Kabbani, S.S.; Rimmer, J.M.; Gennari, F.J.; Taatjes, D.J.; Sobel, B.E.; Schneider, D.J. Biphasic effects of hemodialysis on platelet reactivity in patients with end-stage renal disease: A potential contributor to cardiovascular risk. Am. J. Kidney Dis. 2002, 40, 315–322. [Google Scholar] [CrossRef]
- Mourikis, P.; Helten, C.; Dannenberg, L.; Hohlfeld, T.; Stegbauer, J.; Petzold, T.; Levkau, B.; Zeus, T.; Kelm, M.; Polzin, A. Platelet reactivity in patients with chronic kidney disease and hemodialysis. J. Thromb. Thrombolysis 2020, 49, 168–172. [Google Scholar] [CrossRef]
- Sobol, A.B.; Kamińska, M.; Walczynska, M.; Walkowiak, B. Effect of Uremia and Hemodialysis on Platelet Apoptosis. Clin. Appl. Thromb. Hemost. 2012, 19, 320–323. [Google Scholar] [CrossRef]
- De Laval, P.; Mobarrez, F.; Almquist, T.; Vassil, L.; Fellström, B.; Soveri, I. Acute effects of haemodialysis on circulating microparticles. Clin. Kidney J. 2018, 12, 456–462. [Google Scholar] [CrossRef]
- Yldrm, Z.K.; Orhan, M.F.; Büyükavc, M. Platelet function alterations and their relation to P-selectin (CD62P) expression in children with iron deficiency anemia. Blood Coagul. Fibrinolysis 2011, 22, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Schoorl, M.; Grooteman, M.P.; Bartels, P.C.; Nubé, M.J. Aspects of platelet disturbances in haemodialysis patients. Clin. Kidney J. 2013, 6, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Arthur, J.F.; Gardiner, E.E.; Andrews, R.K.; Zeng, L.; Xu, K. Regulation of platelet activation and thrombus formation by reactive oxygen species. Redox Biol. 2018, 14, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Du, C.; Wang, X.; Li, F.; Xu, Y.; Wang, S.; Chen, S.; Chen, F.; Shen, M.; Chen, M.; et al. Indoxyl sulfate induces platelet hyperactivity and contributes to chronic kidney disease–associated thrombosis in mice. Blood 2017, 129, 2667–2679. [Google Scholar] [CrossRef] [PubMed]
- Wachowicz, B.; Olas, B.; Zbikowska, H.M.; Buczyński, A. Generation of reactive oxygen species in blood platelets. Platelets 2002, 13, 175–182. [Google Scholar] [CrossRef]
- Ghasemzadeh, M.; Hosseini, E. Platelet granule release is associated with reactive oxygen species generation during platelet storage: A direct link between platelet pro-inflammatory and oxidation states. Thromb. Res. 2017, 156, 101–104. [Google Scholar] [CrossRef]
- Georgatzakou, H.T.; Tzounakas, V.L.; Kriebardis, A.G.; Velentzas, A.D.; Kokkalis, A.C.; Antonelou, M.H.; Papassideri, I.S. Short-term effects of hemodiafiltration versus conventional hemodialysis on erythrocyte performance. Can. J. Physiol. Pharmacol. 2018, 96, 249–257. [Google Scholar] [CrossRef]
- Burton, J.O.; Hamali, H.A.; Singh, R.; Abbasian, N.; Parsons, R.; Patel, A.K.; Goodall, A.H.; Brunskill, N.J. Elevated Levels of Procoagulant Plasma Microvesicles in Dialysis Patients. PLoS ONE 2013, 8, e72663. [Google Scholar] [CrossRef] [PubMed]
- Klatt, C.; Krüger, I.; Zey, S.; Krott, K.-J.; Spelleken, M.; Gowert, N.S.; Oberhuber, A.; Pfaff, L.; Lückstädt, W.; Jurk, K.; et al. Platelet-RBC interaction mediated by FasL/FasR induces procoagulant activity important for thrombosis. J. Clin. Investig. 2018, 128, 3906–3925. [Google Scholar] [CrossRef]
- Smeets, M.W.J.; Mourik, M.J.; Niessen, H.W.M.; Hordijk, P.L. Stasis Promotes Erythrocyte Adhesion to von Willebrand Factor. Arter. Thromb. Vasc. Biol. 2017, 37, 1618–1627. [Google Scholar] [CrossRef]
- Sudnitsyna, J.; Skverchinskaya, E.; Dobrylko, I.; Nikitina, E.; Gambaryan, S.; Mindukshev, I. Microvesicle Formation Induced by Oxidative Stress in Human Erythrocytes. Antioxidants 2020, 9, 929. [Google Scholar] [CrossRef] [PubMed]
- Georgatzakou, H.T.; Tzounakas, V.L.; Kriebardis, A.G.; Velentzas, A.D.; Papageorgiou, E.G.; Voulgaridou, A.I.; Kokkalis, A.C.; Antonelou, M.H.; Papassideri, I.S. Pathophysiological aspects of red blood cells in end-stage renal disease patients resistant to recombinant human erythropoietin therapy. Eur. J. Haematol. 2017, 98, 590–600. [Google Scholar] [CrossRef]
- Maharana, P.K.; Goel, S.; Sahay, P.; Singhal, D.; Titiyal, J.S.; Sharma, N. Intraoperative optical coherence tomography-guided release of lenticulo-corneal adhesion and lens aspiration in anterior dislocation of lens with corneal edema. Indian J. Ophthalmol. 2020, 68, 510–512. [Google Scholar] [CrossRef]
- Semenov, A.N.; Lugovtsov, A.E.; Shirshin, E.A.; Yakimov, B.P.; Ermolinskiy, P.B.; Bikmulina, P.Y.; Kudryavtsev, D.S.; Timashev, P.S.; Muravyov, A.V.; Wagner, C.; et al. Assessment of Fibrinogen Macromolecules Interaction with Red Blood Cells Membrane by Means of Laser Aggregometry, Flow Cytometry, and Optical Tweezers Combined with Microfluidics. Biomolecules 2020, 10, 1448. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Patients (n = 32) | Controls (n = 15) | Normal Range | |
|---|---|---|---|---|
| Pre-HD | Post-HD | |||
| HD treatment (months) | 41.3 ± 21.4 | - | - | |
| Age (years) | 59 ± 11.4 | 50 ± 9.8 | - | |
| EpO dose (IU/wk) | 6833.3 ± 3588.7 | - | - | |
| Weight (kg) | 82.2 ± 11.1 | 80.2 ± 11.8 | 85.6 ± 13.2 | - |
| General blood test | ||||
| WBC (×103/µL) | 7.60 ± 2.70 | N/D | 7.10 ± 1.80 | 5.2–12.4 |
| Neutrophils (%) | 64.7 ± 8.3 * | N/D | 59.0 ± 8.0 | 40.0–74.0 |
| Lymphocytes (%) | 20.7 ± 6.3 ** | N/D | 28.7 ± 6.6 | 19.0–48.0 |
| Monocytes (%) | 7.4 ± 1.6 ** | N/D | 6.1 ± 1.6 | 3.4–9.0 |
| Eosinophils (%) | 4.9 ± 3.9 | N/D | 2.9 ± 1.9 | 0.0–7.0 |
| Basophils (%) | 0.7 ± 0.4 | N/D | 0.8 ± 0.3 | 0.0–1.5 |
| Neutrophils/Lymphocytes ratio | 3.5 ± 1.6 ** | N/D | 1.9 ± 0.6 | 1.3–3.5 |
| RBC (×106/µL) | 3.80 ± 0.59 ** | N/D | 4.60 ± 0.46 | 4.2–6.1 |
| Hb (gr/dL) | 11.1 ± 1.3 ** | N/D | 13.5 ± 1.6 | 12.0–18.0 |
| Hct (%) | 34.3 ± 4.3 ** | N/D | 40.6 ± 4.4 | 37.0–52.0 |
| MCV (fL) | 91.1 ± 9.7 | N/D | 88.7 ± 5.3 | 80.0–99.0 |
| MCH (pg) | 29.4 ± 3.4 | N/D | 30.5 ± 2.0 | 27.0–31.0 |
| MCHC (gr/dL) | 32.3 ± 0.9 * | N/D | 34.4 ± 0.7 | 33.0–37.0 |
| RDW (%) | 15.8 ± 1.1 | N/D | 13.0 ± 3.0 | 11.5–14.5 |
| PLT (×103/µL) | 240.8 ± 82.0 | N/D | 250.1 ± 40.6 | 130.0–400.0 |
| MPV (fL) | 10.0 ± 0.6 ** | N/D | 7.8 ± 1.3 | 7.2–11.1 |
| Serum biochemical analysis | ||||
| Glucose | 86.8 ± 22.2 | N/D | 89.5 ± 12.1 | 70–105 |
| Urea (mg/dL) | 129.3 ± 27.9 ** | 39.1 ± 12.7 # | 33.9 ± 9.4 | 18.0–55.0 |
| Creatinine (mg/dL) | 10.42 ± 3.10 ** | 3.90 ± 1.42 **# | 0.92 ± 0.16 | 0.72–1.25 |
| Uric Acid (mg/dL) | 6.1 ± 1.0 ** | N/D | 4.4 ± 1.5 | 3.5–7.2 |
| Cholesterol (mg/dL) | 170 ± 42 | N/D | 178 ± 42 | 0.0–200.0 |
| Triglycerides (mg/dL) | 126.7 ± 52.8 | N/D | 115.2 ± 61.1 | 0.0–150.0 |
| PTH (pg/mL) | 388.3 ± 216.2 ** | N/D | 49.7 ± 15.5 | 15.0–68.0 |
| Calcium (mg/dL) | 8.5 ± 0.7 ** | N/D | 9.5 ± 0.2 | 8.4–10.2 |
| Phosphorus (mg/dL) | 4.5 ± 1.3 ** | N/D | 3.1 ± 0.5 | 2.4–4.7 |
| Potassium (mmol/L) | 4.95 ± 0.61 ** | N/D | 4.47 ± 0.39 | 3.5–5.1 |
| Sodium (mmol/L) | 136.6 ± 2.3 ** | N/D | 139.3 ± 2.3 | 136.0–145.0 |
| Chlorine(mmol/L) | 102.2 ± 2.9 | N/D | 102.1 ± 2.8 | 98.0–107.0 |
| Magnesium (mg/dL) | 2.28 ± 0.37 * | N/D | 2.02 ± 0.02 | 1.60–2.60 |
| Iron (mg/dL) | 69.2 ± 25.0 | N/D | 88.0 ± 39.8 | 50–150 (females); 60–160 (males) |
| Ferritin (ng/mL) | 263.3 ± 153.3 ** | N/D | 53.8 ± 29.1 | 14.0–233.0 (females); 16.4–293.3 (males) |
| TIBC (mg/dL) | 251.6 ± 53.3 ** | N/D | 342.0 ± 50.7 | 225.0–480.0 |
| B12 (pg/mL) | 602.6 ± 265.5 | N/D | 414.3 ± 179.5 | 179.0–1162.0 |
| Folate (ng/mL) | 14.60 ± 12.50 | N/D | 5.30 ± 2.60 | 2.5–17.0 |
| Proteins (mg/dL) | 6.90 ± 0.60 | N/D | 6.80 ± 0.60 | 6.40–8.30 |
| Albumin (gr/dL) | 4.05 ± 0.50 ** | N/D | 4.47 ± 0.33 | 3.50–5.00 |
| b2-microglobulin (mg/L) | 33.3 ± 11.4 ** | N/D | 1.4 ± 0.5 | 0.71.8 |
| SGOT (U/L) | 11.4 ± 5.0 ** | N/D | 18.1 ± 5.9 | 5.0–34.0 |
| SGPT (U/L) | 12.00 ± 4.06 ** | N/D | 22.00 ± 15.00 | 0.0–55.0 |
| γGT (U/L) | 25.4 ± 15.3 | N/D | 17.9 ± 8.0 | 12.0–64.0 |
| ALP (U/L) | 89. ± 42.90 | N/D | 80.1 ± 28.9 | 40.0–150.0 |
| Total Bilirubin (mg/dL) | 0.50 ± 0.20 | N/D | 0.40 ± 0.30 | 0.20–1.20 |
| Indirect Bilirubin (mg/dL) | 0.23 ± 0.09 ** | N/D | 0.15 ± 0.06 | 0.01–0.90 |
| Direct Bilirubin (mg/dL) | 0.31 ± 0.13 ** | N/D | 0.13 ± 0.10 | 0.00–0.30 |
| LDH (IU/L) | 210.6 ± 67.6 | N/D | 203.3 ± 54.3 | 125.0–220.0 |
| CPK total (IU/L) | 84.4 ± 58.0 | N/D | 80.2 ± 63.1 | 30.0–200.0 |
| Amylase (IU/L) | 120.0 ± 39.6 | N/D | 63.9 ± 18.5 | 20.0–160.0 |
| Vitamin-D (ng/mL) | 11.6 ± 6.5 * | N/D | 20.5 ± 4.9 | 30.0–100.0 |
| hs CRP (mg/L) | 12.5 ± 10.2 ** | N/D | 1.8 ± 1.0 | 0.0–5.0 |
| Characteristics | Patients (n = 32) | Controls (n = 15) | Normal Range | |
|---|---|---|---|---|
| Pre-HD | Post-HD | |||
| Secondary hemostasis screening test | ||||
| aPTT (s) | 56.7 ± 32.1 ** | 35.5 ± 7.0 **# | 29.7 ± 3.7 | <36 |
| D-Dimers (µg/L) | 661.2 ± 438.6 ** | 694.8 ± 425.3 ** | 272.7 ± 140.2 | <500 |
| Fibrinogen (mg/dL) | 411.8 ± 92.4 ** | 464.5 ± 112.7 **# | 328.4 ± 79.6 | 180–350 |
| PT/INR | 1.4 ± 0.7 ** | 1.2 ± 0.4 **# | 0.98 ± 0.04 | <1.2 |
| Coagulation system | ||||
| Factor II (%) | 79.0 ± 19.3 ** | 81.4 ± 21.4 ** | 107.9 ± 13.2 | 60–120 |
| Factor V (%) | 85.7 ± 23.6 ** | 106.0 ± 19.2 # | 104.9 ± 17.2 | 60–120 |
| Factor VII (%) | 105.9 ± 37.0 | 110.8 ± 39.2 | 116.0 ± 19.8 | 60–120 |
| Factor VIII (%) | 148.8 ± 79.9 | 247.1 ± 85.5 **# | 123.3 ± 17.6 | 60–140 |
| Factor IX (%) | 115.2 ± 43.4 | 123.0 ± 39.9 | 115.9 ± 17.9 | 60–120 |
| Factor X (%) | 77.1 ± 27.0 | 82.0 ± 35.1 | 89.1 ± 15.8 | 60–120 |
| Factor XI (%) | 100.3 ± 49.6 | 121.3 ± 50.6 | 112.6 ± 16.1 | 60–120 |
| Factor XII (%) | 87.1 ± 24.2 ** | 108.1 ± 22.3 # | 109.9 ± 26.9 | 60–120 |
| Factor XIII (%) | 145.9 ± 12.1 ** | 149.8 ± 11.0 ** | 101.1 ± 11.9 | 60–140 |
| RiCof (%) | 160.6 ± 53.6 | 191.8 ± 72.4 **# | 122.7 ± 34.1 | 60–140 |
| vWF (%) | 170.4 ± 58.0 ** | 216.8 ± 74.7 **# | 110.0 ± 20.8 | 60–140 |
| Fibrinolytic system | ||||
| PAI (U/mL) | 1.9 ± 1.2 ** | 1.6 ± 1.1 ** | 0.8 ± 1.1 | <4 |
| Plasminogen (ng/L) | 100.5 ± 15.3 | 112.8 ± 17.4 **# | 108.1 ± 8.8 | 80–120 |
| Inhibitory system | ||||
| Antithrombin III (%) | 96.9 ± 13.8 ** | 105.6 ± 14.5 # | 122.3 ± 10.1 | 80–120 |
| Protein C (%) | 80.8 ± 18.8 ** | 91.6 ± 23.1 **# | 110.3 ± 14.5 | 70–140 |
| Protein S activity (%) | 89.6 ± 23.3 | 99.3 ± 33.9 | 91.9 ± 16.5 | 60–130 |
| Protein Sf (%) | 95.6 ± 69.9 ** | 92.6 ± 27.4 **# | 109.1 ± 26.3 | 60–140 |
| ADAMTS-13 | ||||
| ADAMTS13 activity (%) | <5 ** | <5 ** | 57.3 ± 4.8 | 17–63 |
| ADAMTS13 Inhibitor (%) | 81.4 ± 25.1 ** | 86.6 ± 11.5 **# | 12.0 ± 5.0 | <30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavlou, E.G.; Georgatzakou, H.T.; Fortis, S.P.; Tsante, K.A.; Tsantes, A.G.; Nomikou, E.G.; Kapota, A.I.; Petras, D.I.; Venetikou, M.S.; Papageorgiou, E.G.; et al. Coagulation Abnormalities in Renal Pathology of Chronic Kidney Disease: The Interplay between Blood Cells and Soluble Factors. Biomolecules 2021, 11, 1309. https://doi.org/10.3390/biom11091309
Pavlou EG, Georgatzakou HT, Fortis SP, Tsante KA, Tsantes AG, Nomikou EG, Kapota AI, Petras DI, Venetikou MS, Papageorgiou EG, et al. Coagulation Abnormalities in Renal Pathology of Chronic Kidney Disease: The Interplay between Blood Cells and Soluble Factors. Biomolecules. 2021; 11(9):1309. https://doi.org/10.3390/biom11091309
Chicago/Turabian StylePavlou, Efthimia G., Hara T. Georgatzakou, Sotirios P. Fortis, Konstantina A. Tsante, Andreas G. Tsantes, Efrosyni G. Nomikou, Athanasia I. Kapota, Dimitrios I. Petras, Maria S. Venetikou, Effie G. Papageorgiou, and et al. 2021. "Coagulation Abnormalities in Renal Pathology of Chronic Kidney Disease: The Interplay between Blood Cells and Soluble Factors" Biomolecules 11, no. 9: 1309. https://doi.org/10.3390/biom11091309
APA StylePavlou, E. G., Georgatzakou, H. T., Fortis, S. P., Tsante, K. A., Tsantes, A. G., Nomikou, E. G., Kapota, A. I., Petras, D. I., Venetikou, M. S., Papageorgiou, E. G., Antonelou, M. H., & Kriebardis, A. G. (2021). Coagulation Abnormalities in Renal Pathology of Chronic Kidney Disease: The Interplay between Blood Cells and Soluble Factors. Biomolecules, 11(9), 1309. https://doi.org/10.3390/biom11091309