
Structural and Functional Insights into the Biofilm-Associated BceF Tyrosine Kinase Domain from Burkholderia cepacia

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. BceF Cloning
2.2. BceF Protein Expression and Purification for Crystallization
2.3. BceF Protein Purification for Biochemical Measurements
2.4. BceF Protein Purification for the Steady-State Kinetics
2.5. TEV Protease Expression and Purification
2.6. BceF Crystallization Experiments
2.7. X-ray Data Collection and Structure Determination
2.8. Evolutionary Conservation Analysis
2.9. Crystal Structure Calculations and Visualization
2.10. Steady-State Enzymatic Kinetics Analysis of BceF
2.11. MST Measurement of the Affinity of BceF for the ATP Analog
2.12. Size Exclusion Chromatography-Multi-Angle Light Scattering (SEC-MALS)
2.13. Tandem Mass Spectrometry (MS/MS) Analysis
3. Results and Discussion
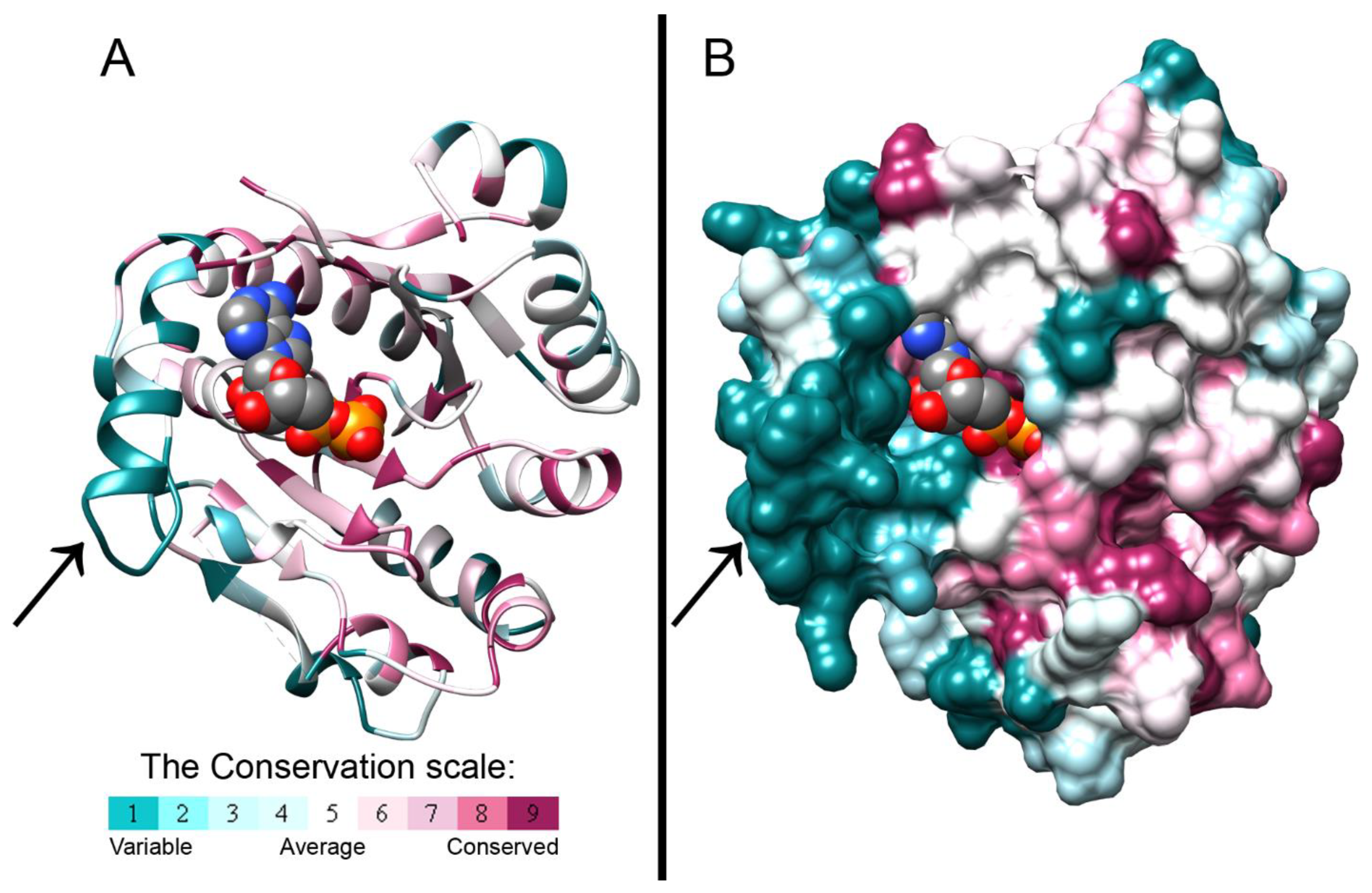
3.1. The Crystal Structure of the BceF Kinase Domain
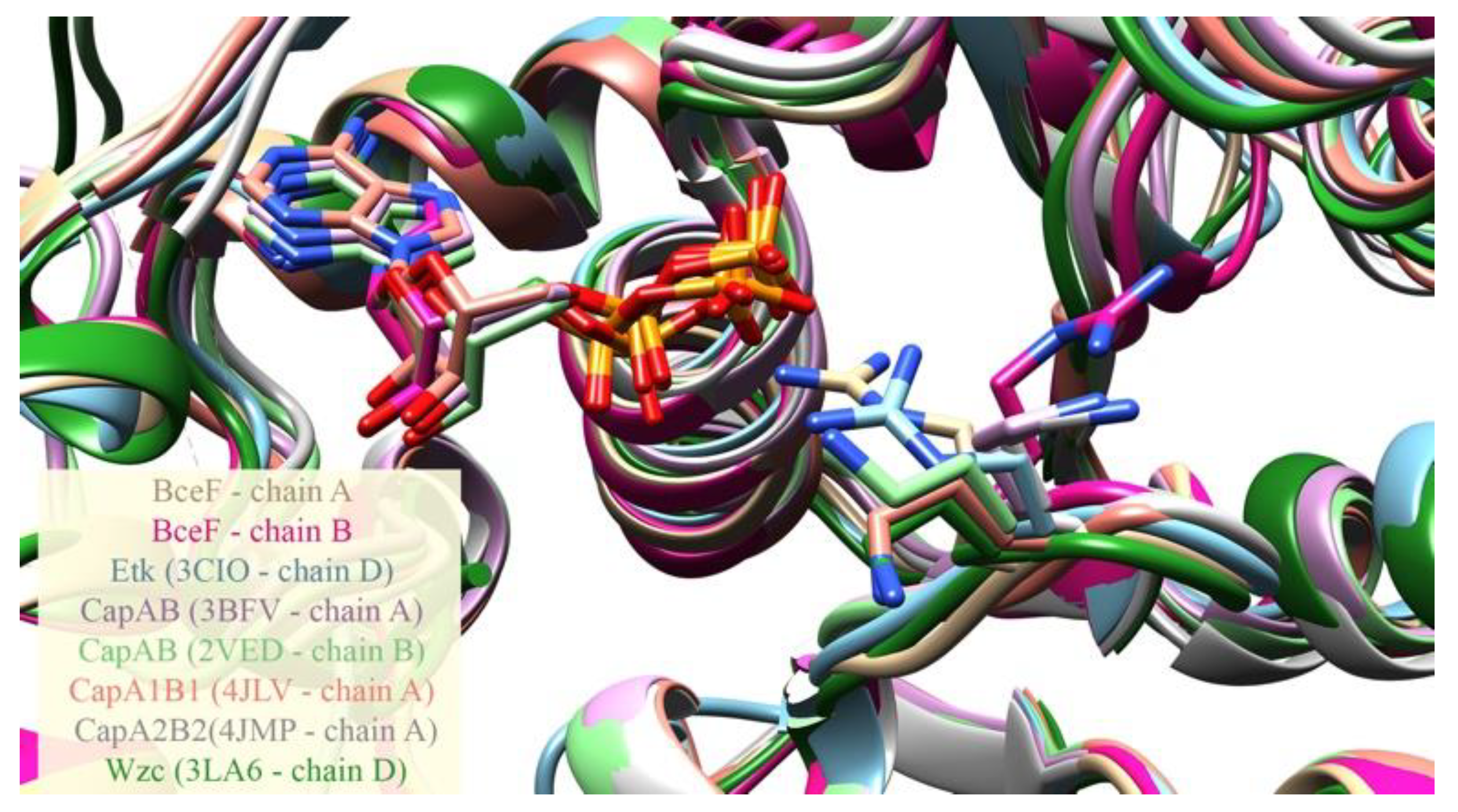
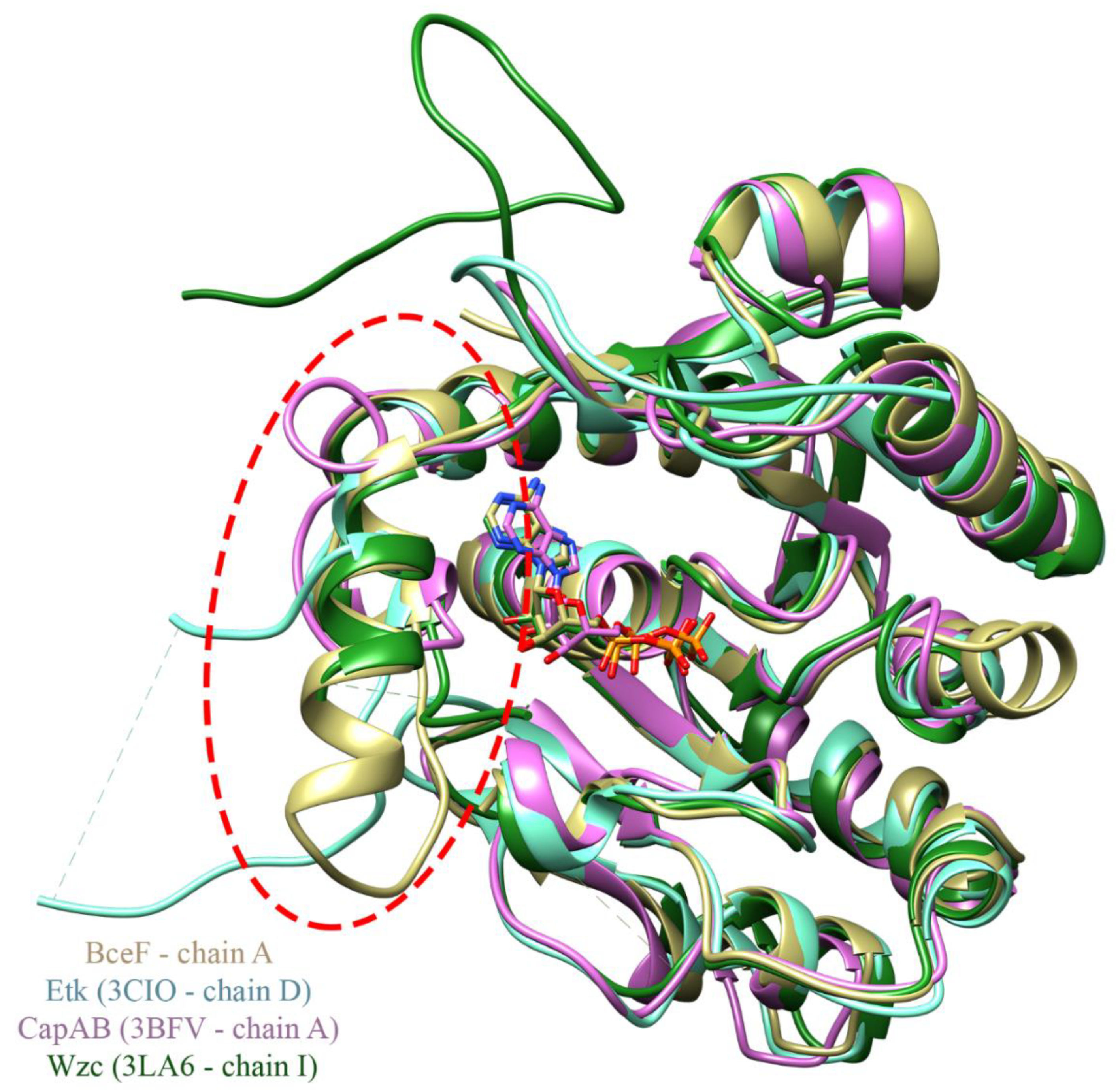
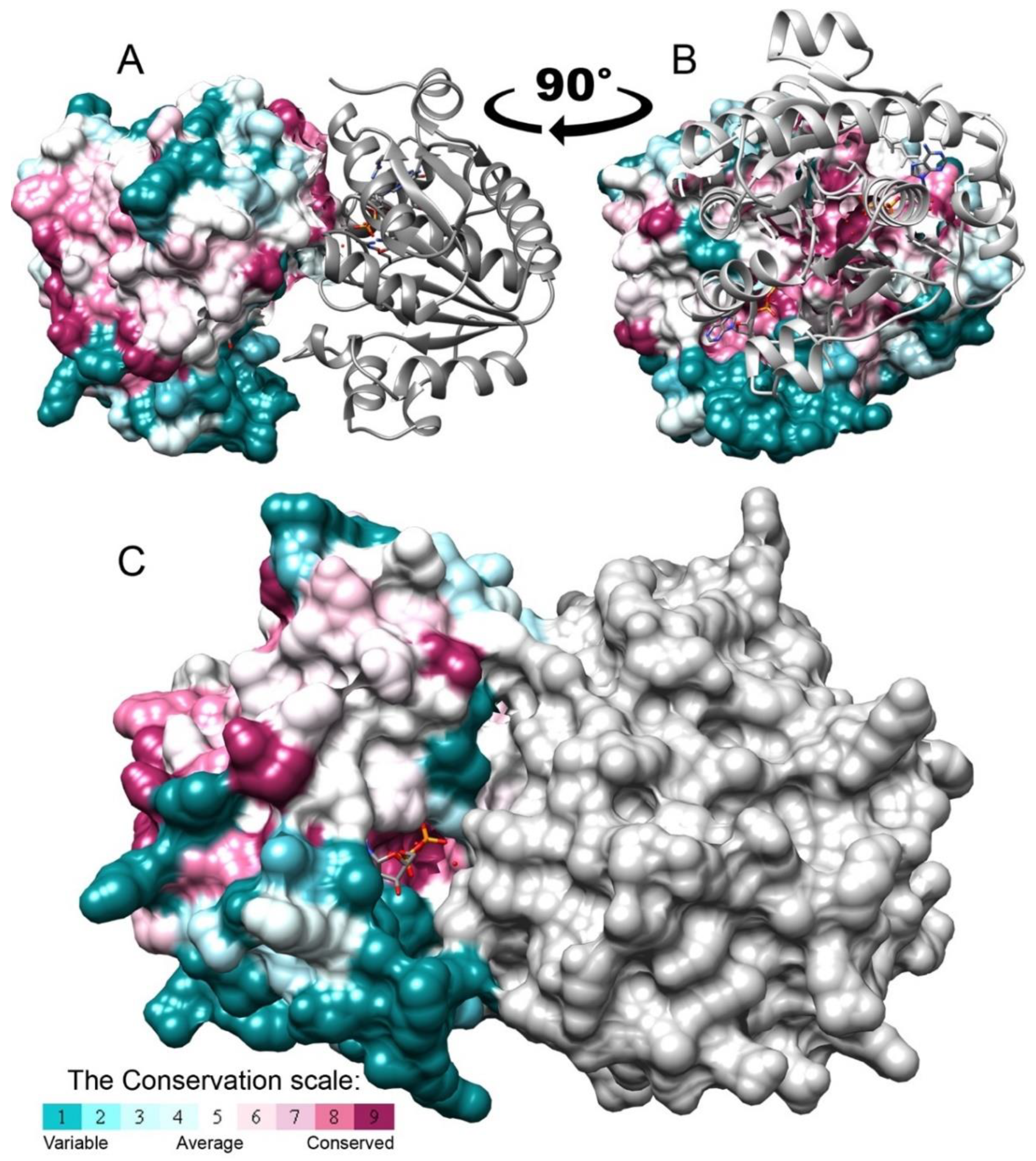
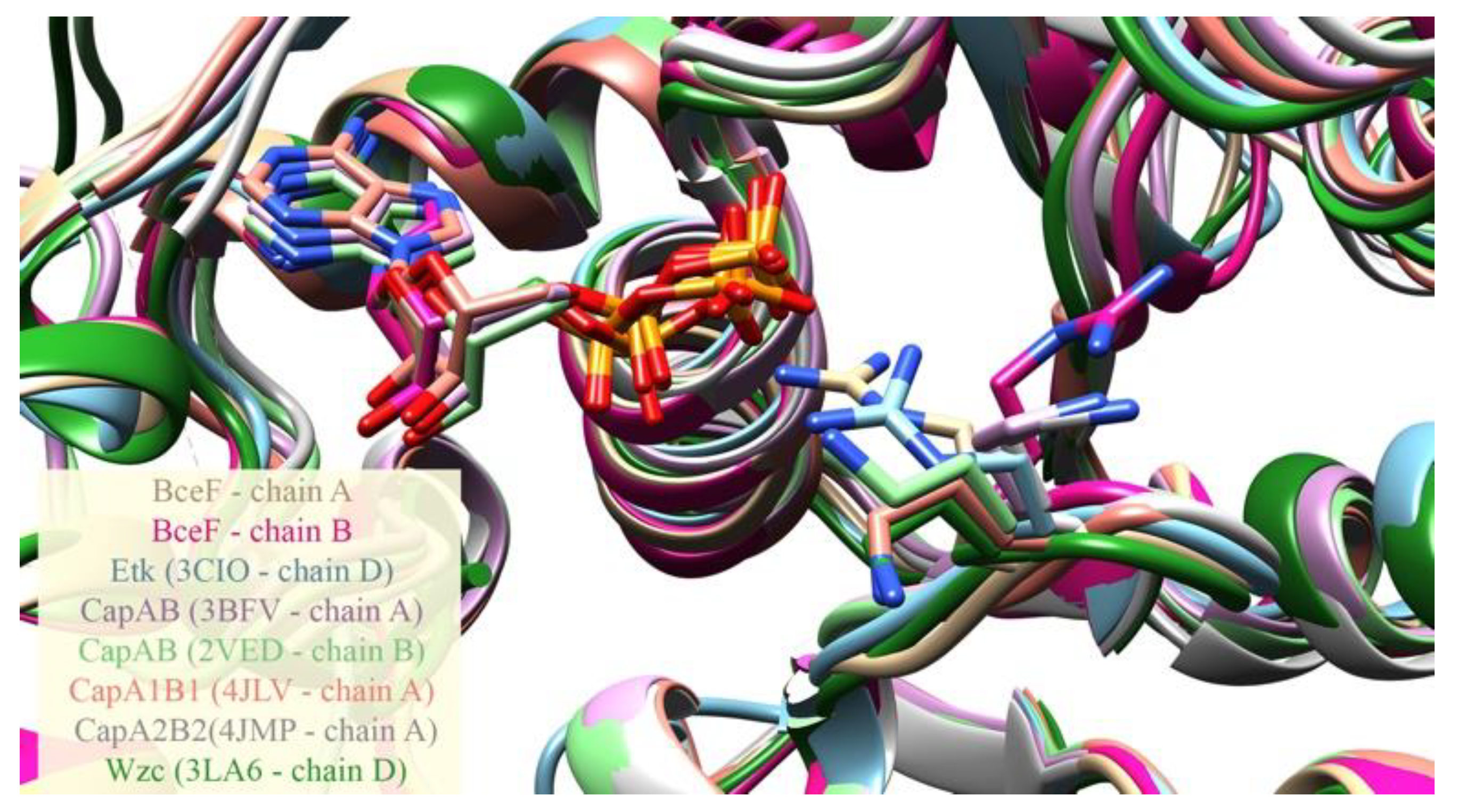
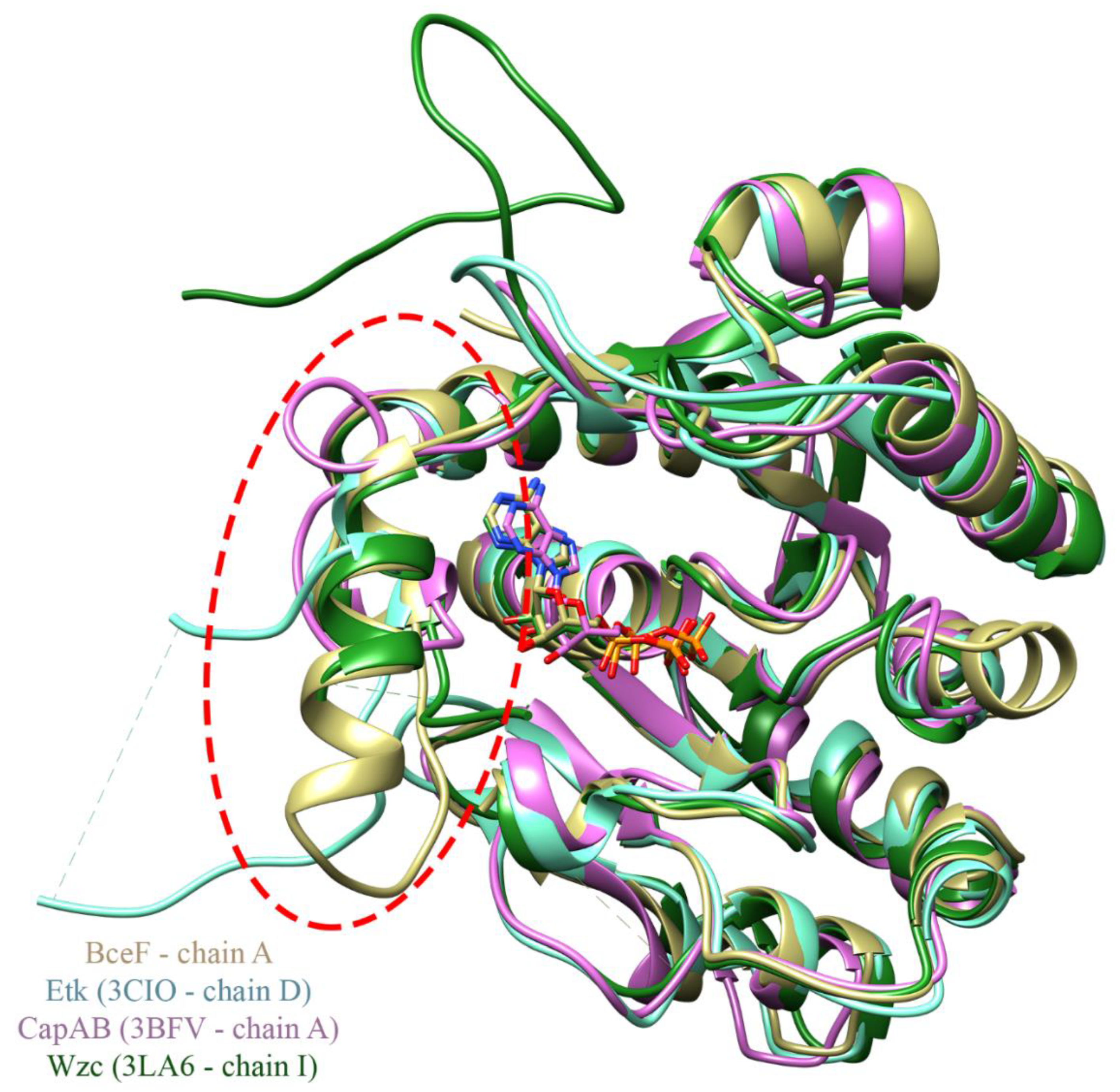
3.2. Evolutionary Conservation Analysis and Structural Comparison of BY-Kinases
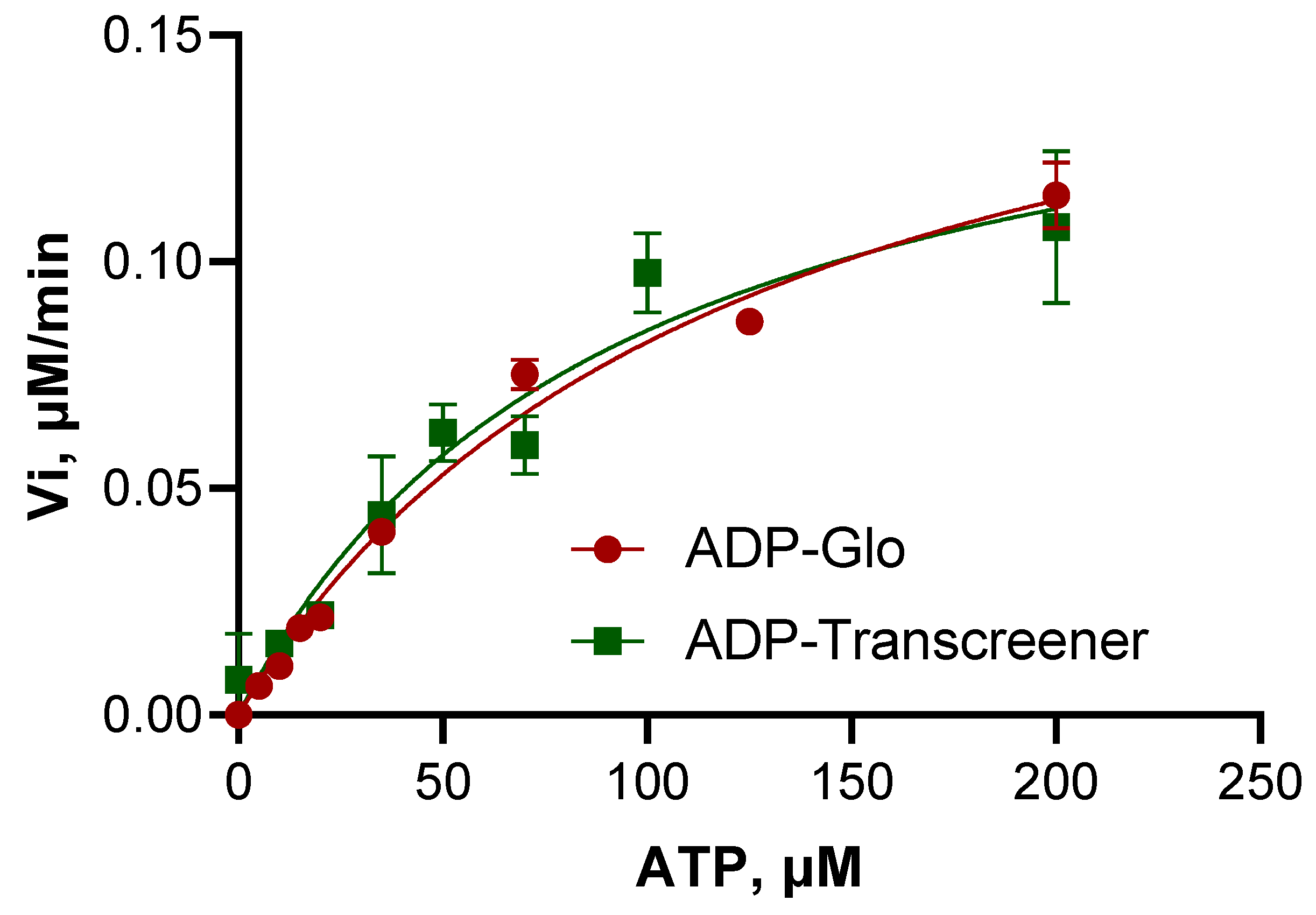
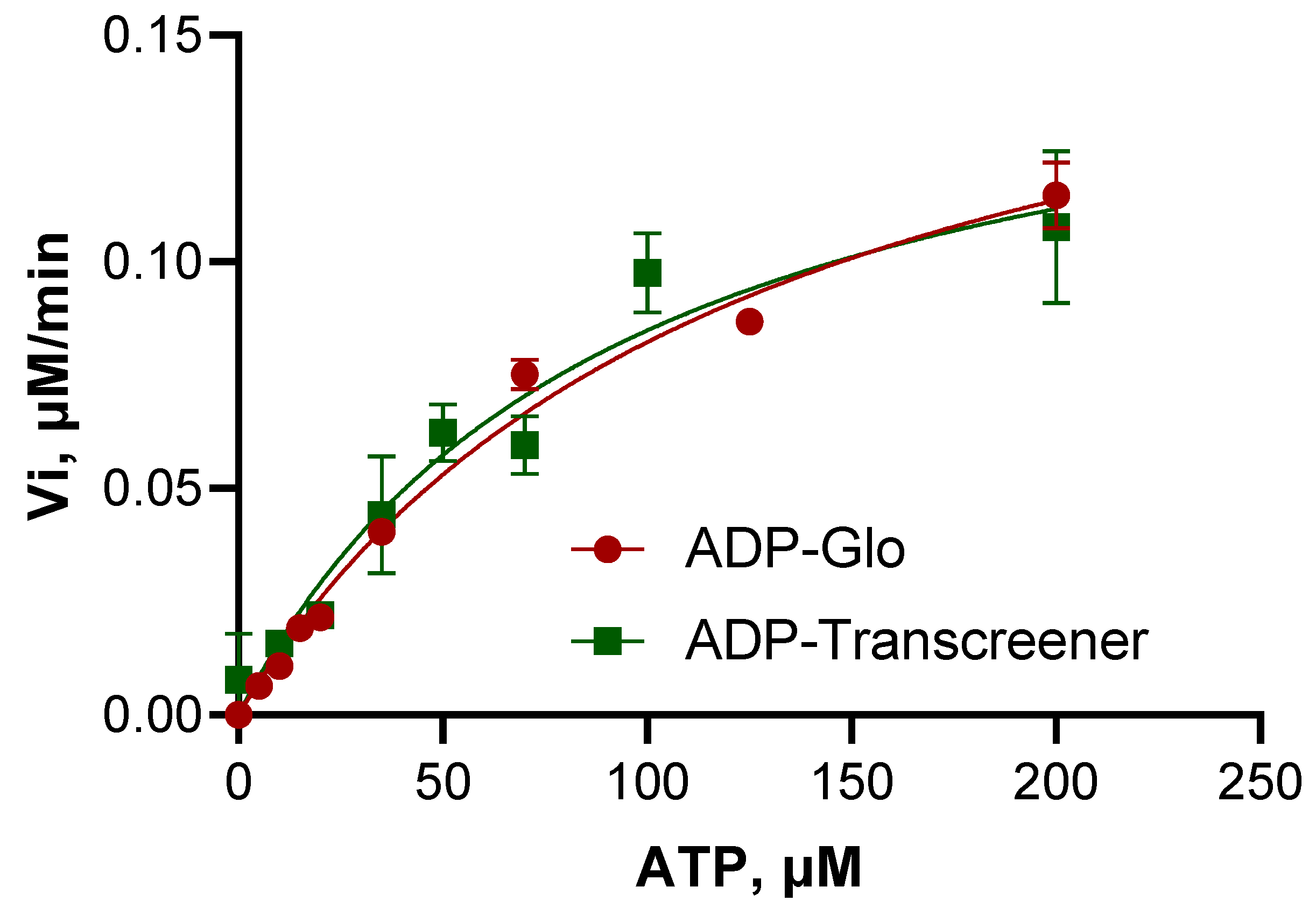
3.3. Steady-State Kinetics Reveals Low Enzymatic Efficiency of the BceF Kinase Domain
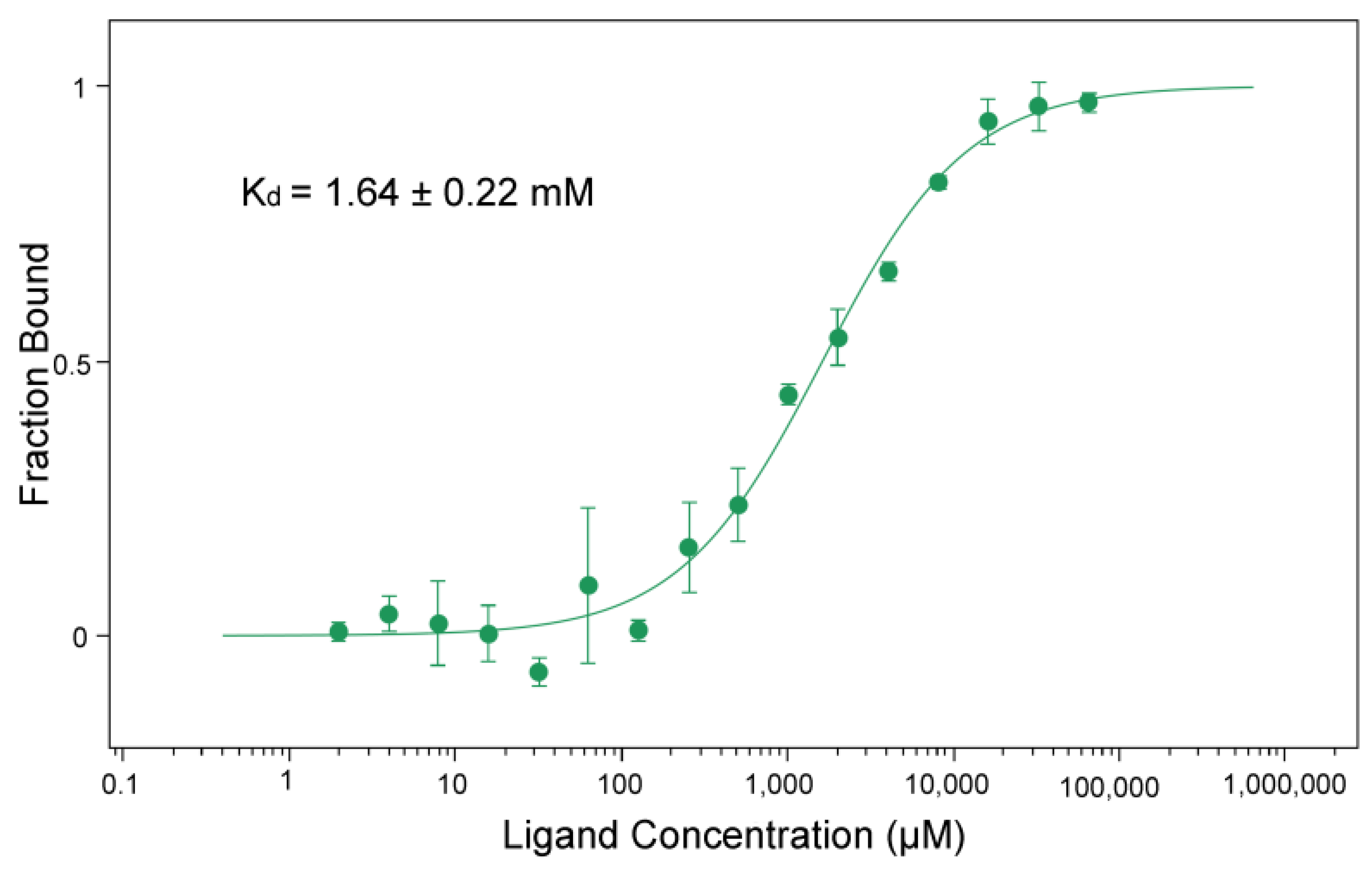
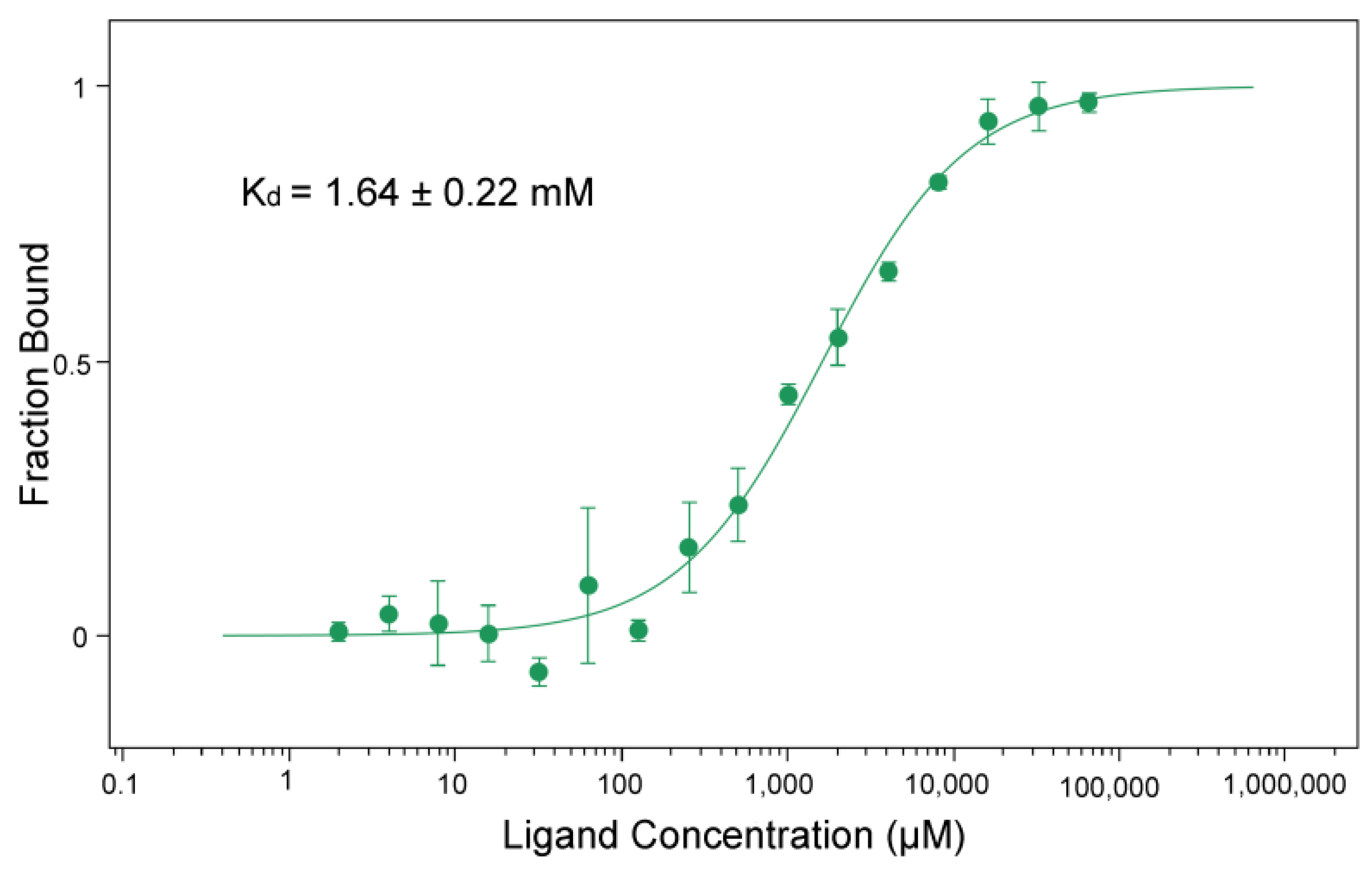
3.4. BceF Kinase Domain Displays a Low Binding Affinity for the Non-Hydrolyzable ATP Analog AMP-PNP
3.5. BceF Kinase Domain Exists as a Dimer in Solution and in the Crystals
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shommu, N.S.; Vogel, H.J.; Storey, D.G. Potential of metabolomics to reveal Burkholderia cepacia complex pathogenesis and antibiotic resistance. Front. Microbiol. 2015, 6, 668. [Google Scholar] [CrossRef]
- Ferreira, A.S.; Leitão, J.H.; Sousa, S.A.; Cosme, A.M.; Sá-Correia, I.; Moreira, L.M. Functional Analysis of Burkholderia cepacia Genes bceD and bceF, Encoding a Phosphotyrosine Phosphatase and a Tyrosine Autokinase, Respectively: Role in Exopolysaccharide Biosynthesis and Biofilm Formation. Appl. Environ. Microbiol. 2007, 73, 524–534. [Google Scholar] [CrossRef] [Green Version]
- Cozzone, A.J. Bacterial tyrosine kinases: Novel targets for antibacterial therapy? Trends Microbiol. 2009, 17, 536–543. [Google Scholar] [CrossRef]
- Cegelski, L.; Marshall, G.R.; Eldridge, G.R.; Hultgren, S.J. The biology and future prospects of antivirulence therapies. Nat. Rev. Microbiol. 2008, 6, 17–27. [Google Scholar] [CrossRef]
- Grangeasse, C.; Nessler, S.; Mijakovic, I. Bacterial tyrosine kinases: Evolution, biological function and structural insights. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 2640–2655. [Google Scholar] [CrossRef] [Green Version]
- Grangeasse, C.; Cozzone, A.J.; Deutscher, J.; Mijakovic, I. Tyrosine phosphorylation: An emerging regulatory device of bacterial physiology. Trends Biochem. Sci. 2007, 32, 86–94. [Google Scholar] [CrossRef]
- Shi, L.; Cavagnino, A.; Rabefiraisana, J.-L.; Lazar, N.; De La Sierra-Gallay, I.L.; Ochsenbein, F.; Valerio-Lepiniec, M.; Urvoas, A.; Minard, P.; Mijakovic, I.; et al. Structural Analysis of the Hanks-Type Protein Kinase YabT From Bacillus subtilis Provides New Insights in its DNA-Dependent Activation. Front. Microbiol. 2019, 9, 3014. [Google Scholar] [CrossRef]
- Chao, J.D.; Wong, D.; Av-Gay, Y. Microbial Protein-tyrosine Kinases. J. Biol. Chem. 2014, 289, 9463–9472. [Google Scholar] [CrossRef] [Green Version]
- Grangeasse, C.; Terreux, R.; Nessler, S. Bacterial tyrosine-kinases: Structure–function analysis and therapeutic potential. Biochim. Biophys. Acta Proteins Proteom. 2010, 1804, 628–634. [Google Scholar] [CrossRef]
- Obadia, B.; Lacour, S.; Doublet, P.; Baubichon-Cortay, H.; Cozzone, A.J.; Grangeasse, C. Influence of Tyrosine-Kinase Wzc Activity on Colanic Acid Production in Escherichia coli K12 Cells. J. Mol. Biol. 2007, 367, 42–53. [Google Scholar] [CrossRef]
- Bechet, E.; Gruszczyk, J.; Terreux, R.; Gueguen-Chaignon, V.; Vigouroux, A.; Obadia, B.; Cozzone, A.J.; Nessler, S.; Grangeasse, C. Identification of structural and molecular determinants of the tyrosine-kinase Wzc and implications in capsular polysaccharide export. Mol. Microbiol. 2010, 77, 1315–1325. [Google Scholar] [CrossRef]
- Lee, D.C.; Zheng, J.; She, Y.-M.; Jia, Z. Structure of Escherichia coli tyrosine kinase Etk reveals a novel activation mechanism. EMBO J. 2008, 27, 1758–1766. [Google Scholar] [CrossRef] [Green Version]
- Gruszczyk, J.; Olivares-Illana, V.; Nourikyan, J.; Fleurie, A.; Béchet, E.; Gueguen-Chaignon, V.; Freton, C.; Aumont-Nicaise, M.; Moréra, S.; Grangeasse, C.; et al. Comparative Analysis of the Tyr-Kinases CapB1 and CapB2 Fused to Their Cognate Modulators CapA1 and CapA2 from Staphylococcus aureus. PLoS ONE 2013, 8, e75958. [Google Scholar] [CrossRef]
- Olivares-Illana, V.; Meyer, P.R.; Bechet, E.; Gueguen-Chaignon, V.; Soulat, D.; Lazereg-Riquier, S.; Mijakovic, I.; Deutscher, J.; Cozzone, A.J.; Laprévote, O.; et al. Structural Basis for the Regulation Mechanism of the Tyrosine Kinase CapB from Staphylococcus aureus. PLoS Biol. 2008, 6, e143. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: New York, NY, USA, 2005; pp. 571–607. [Google Scholar]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phasercrystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallogr. Sect. D Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazy, H.; Erez, E.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010, 38, W529–W533. [Google Scholar] [CrossRef] [Green Version]
- Landau, M.; Mayrose, I.; Rosenberg, Y.; Glaser, F.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2005: The projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005, 33, W299–W302. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.S. Global indicators of X-ray data quality. J. Appl. Crystallogr. 2001, 34, 130–135. [Google Scholar] [CrossRef]
- Karplus, P.A.; Diederichs, K. Linking Crystallographic Model and Data Quality. Science 2012, 336, 1030–1033. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.B.; Arendall, W.B., III; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunati, A.M.; Donella-Deana, A.; Ruzzene, M.; Marin, O.; Pinna, L.A. Site specificity of p72syk protein tyrosine kinase: Efficient phosphorylation of motifs recognized by Src homology 2 domains of the Src family. FEBS Lett. 1995, 367, 149–152. [Google Scholar] [CrossRef]
- Guskov, A.; Slotboom, D.J. Size exclusion chromatography with multi-angle laser light scattering (SEC-MALLS) to determine protein oligomeric states. In From Molecules to Living Organisms: An Interplay between Biology and Physics; Oxford University Press (OUP): Oxford, UK, 2016; pp. 169–183. [Google Scholar]
- Some, D.; Amartely, H.; Tsadok, A.; Lebendiker, M. Characterization of Proteins by Size-Exclusion Chromatography Coupled to Multi-Angle Light Scattering (SEC-MALS). J. Vis. Exp. 2019, e59615. [Google Scholar] [CrossRef] [Green Version]
- Spellberg, B. Antibiotic Resistance: Promoting Critically Needed Antibiotic Research and Development and Appropriate Use (“Stewardship”) of these Precious Drugs; Testimony of the Infectious Diseases Society of America (IDSA): Arlington, VA, USA, 2010. [Google Scholar]
- Ferreira, A.S.; Silva, I.N.; Oliveira, V.H.; Becker, J.D.; Givskov, M.; Ryan, R.P.; Fernandes, F.; Moreira, L.M. Comparative Transcriptomic Analysis of the Burkholderia cepacia Tyrosine Kinase bceF Mutant Reveals a Role in Tolerance to Stress, Biofilm Formation, and Virulence. Appl. Environ. Microbiol. 2013, 79, 3009–3020. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BceF | |
|---|---|
| PDB accession code | 6Z0P |
| Beamline | ESRF ID23-2 |
| Date | 25 June 2016 |
| Data collection | |
| Space group | P 1 21 1 |
| Cell dimensions | |
| a, b, c (Å) | 44.27 90.48 61.34 |
| α, β, γ (°) | 90.0 111.1 90.0 |
| Wavelength (Å) | 0.8729 |
| Resolution (Å) | 90.5-1.85 (2.2-1.85) |
| R-factor observed (%) | 9.5 (47.3) |
| aRmeas (%) | 11.2 (55.1) |
| I / sigma | 9.6 (2.7) |
| Total reflections | 142,307 (58,228) |
| Unique reflections | 38,309 (15,478) |
| Completeness (%) | 99.4 (99.5) |
| Multiplicity | 3.7 (3.8) |
| b CC1/2 (%) | 99.7 (80.6) |
| Refinement | |
| Resolution (Å) | 48.4-1.85 (1.90-1.85) |
| Completeness (%) | 99.3 (96.6) |
| c No. reflections | 36413 |
| dRwork (%) | 18.3 (25.6) |
| Rfree (%) | 20.2 (23.5) |
| Rfree value test set size (%) | 5 |
| No. atoms | 3780 |
| Protein | 1836 (Chain A; 271 residues) 1839 (Chain B; 271 residues) |
| Ligand/ion | 54 (ADP) |
| Water | 51 |
| B-factors | |
| Protein | 26.3 (Chain A) 24.8 (Chain B) |
| Ligand/ion | 20.6 (ADP) |
| Water | 24.8 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.005 |
| Bond angles (°) | 1.272 |
| Clash score * | 4.6 (97th percentile) |
| Molprobity score * | 1.24 (99th percentile) |
| Number of xtals used for scaling | 1 |
| ADP-Glo | Transcreener | |
|---|---|---|
| Vmax, μM min−1 | 0.1835 ± 0.0107 | 0.1634 ± 0.0198 |
| Km, μM | 123.06 ± 13.78 | 92.51 ± 22.58 |
| Kcat, min−1 | 0.0541 ± 0.0031 | 0.0481 ± 0.0058 |
| Kcat/Km, μM−1 min−1 | 0.0004 | 0.0005 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mayer, M.; Matiuhin, Y.; Nawatha, M.; Tabachnikov, O.; Fish, I.; Schutz, N.; Dvir, H.; Landau, M. Structural and Functional Insights into the Biofilm-Associated BceF Tyrosine Kinase Domain from Burkholderia cepacia. Biomolecules 2021, 11, 1196. https://doi.org/10.3390/biom11081196
Mayer M, Matiuhin Y, Nawatha M, Tabachnikov O, Fish I, Schutz N, Dvir H, Landau M. Structural and Functional Insights into the Biofilm-Associated BceF Tyrosine Kinase Domain from Burkholderia cepacia. Biomolecules. 2021; 11(8):1196. https://doi.org/10.3390/biom11081196
Chicago/Turabian StyleMayer, Michal, Yulia Matiuhin, Mickal Nawatha, Orly Tabachnikov, Inbar Fish, Nili Schutz, Hay Dvir, and Meytal Landau. 2021. "Structural and Functional Insights into the Biofilm-Associated BceF Tyrosine Kinase Domain from Burkholderia cepacia" Biomolecules 11, no. 8: 1196. https://doi.org/10.3390/biom11081196
APA StyleMayer, M., Matiuhin, Y., Nawatha, M., Tabachnikov, O., Fish, I., Schutz, N., Dvir, H., & Landau, M. (2021). Structural and Functional Insights into the Biofilm-Associated BceF Tyrosine Kinase Domain from Burkholderia cepacia. Biomolecules, 11(8), 1196. https://doi.org/10.3390/biom11081196