
FGF1 Fusions with the Fc Fragment of IgG1 for the Assembly of GFPpolygons-Mediated Multivalent Complexes Recognizing FGFRs

 , and
, and 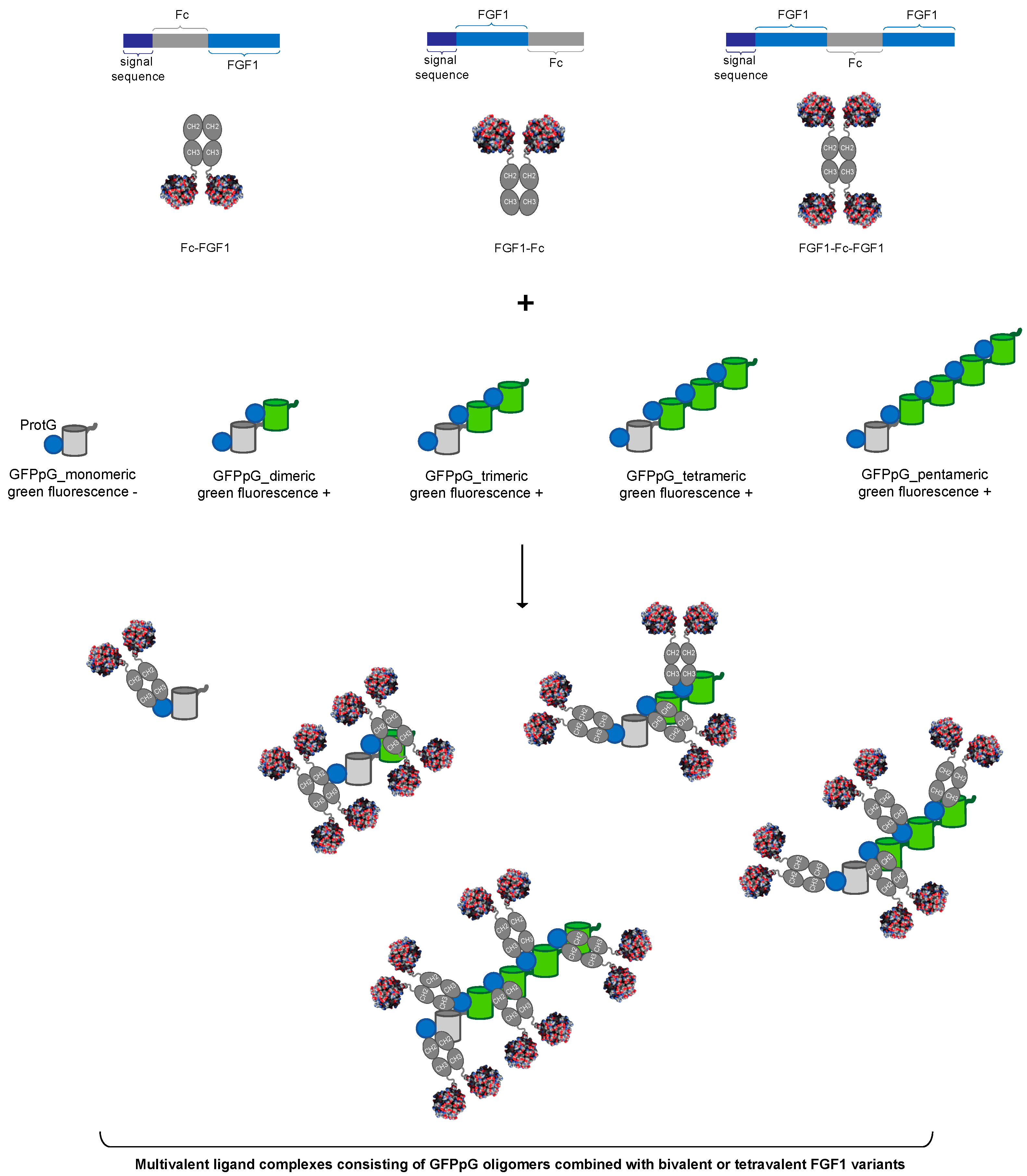{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. Cells
2.3. Recombinant Proteins
2.4. Activation of FGFR1 and Receptor-Downstream Signaling Cascades
2.5. Fluorescence Microscopy
2.6. Flow Cytometry
2.7. Blue Native-PAGE
2.8. BLI Measurements
3. Results
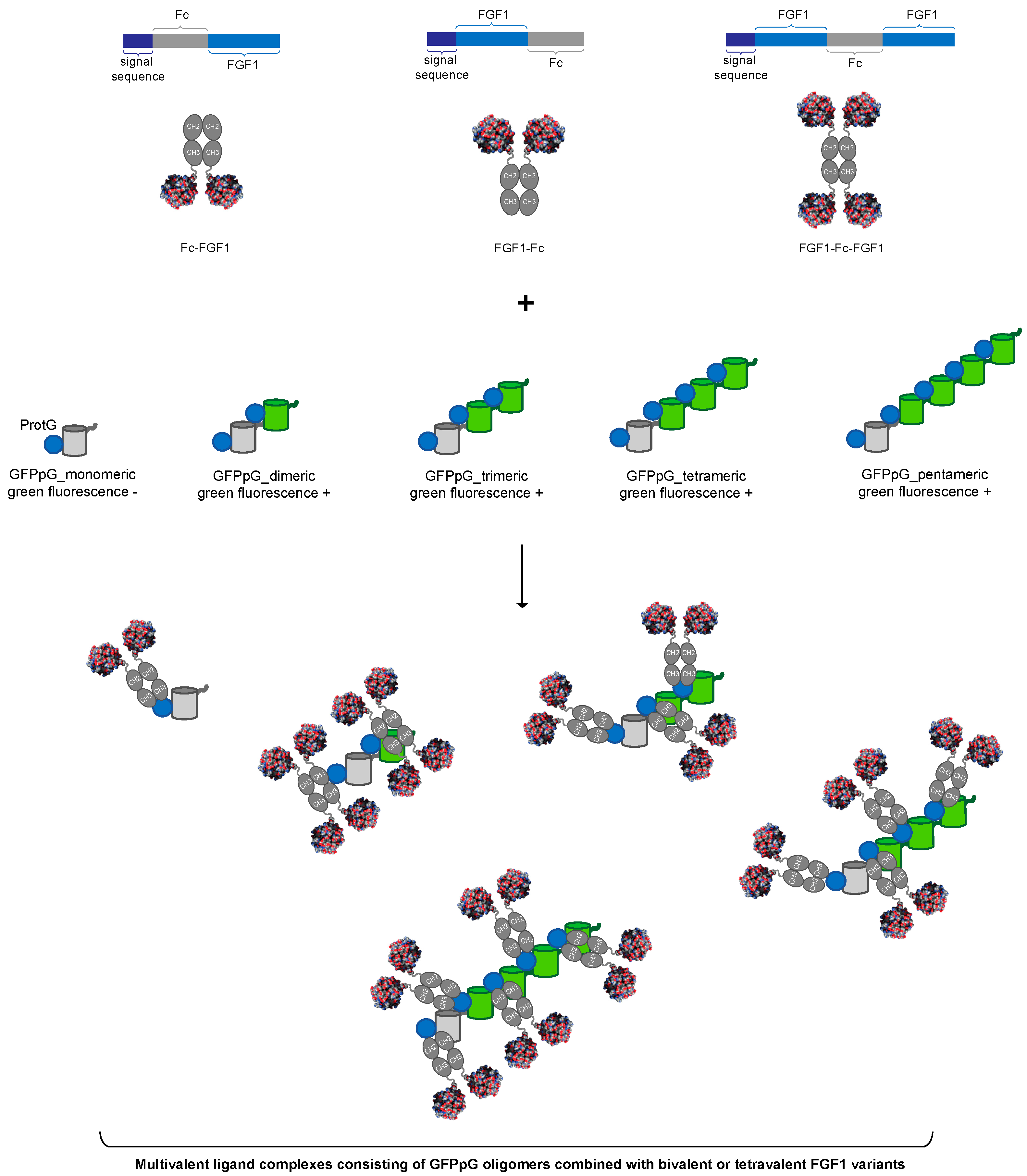
3.1. Strategy for Controlled Oligomerization of FGF1
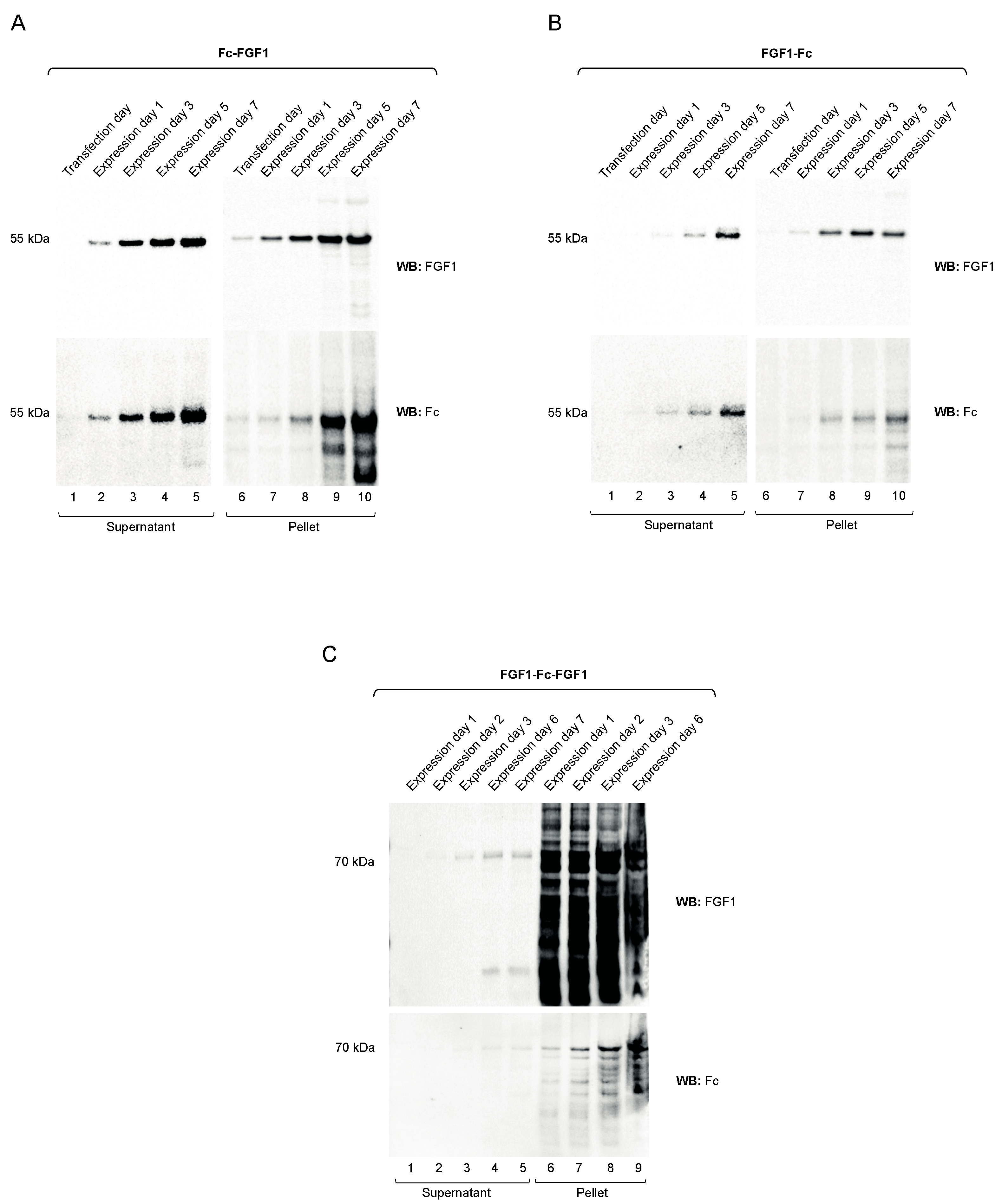
3.2. Preparation of FGF1 Fusions with the Fc
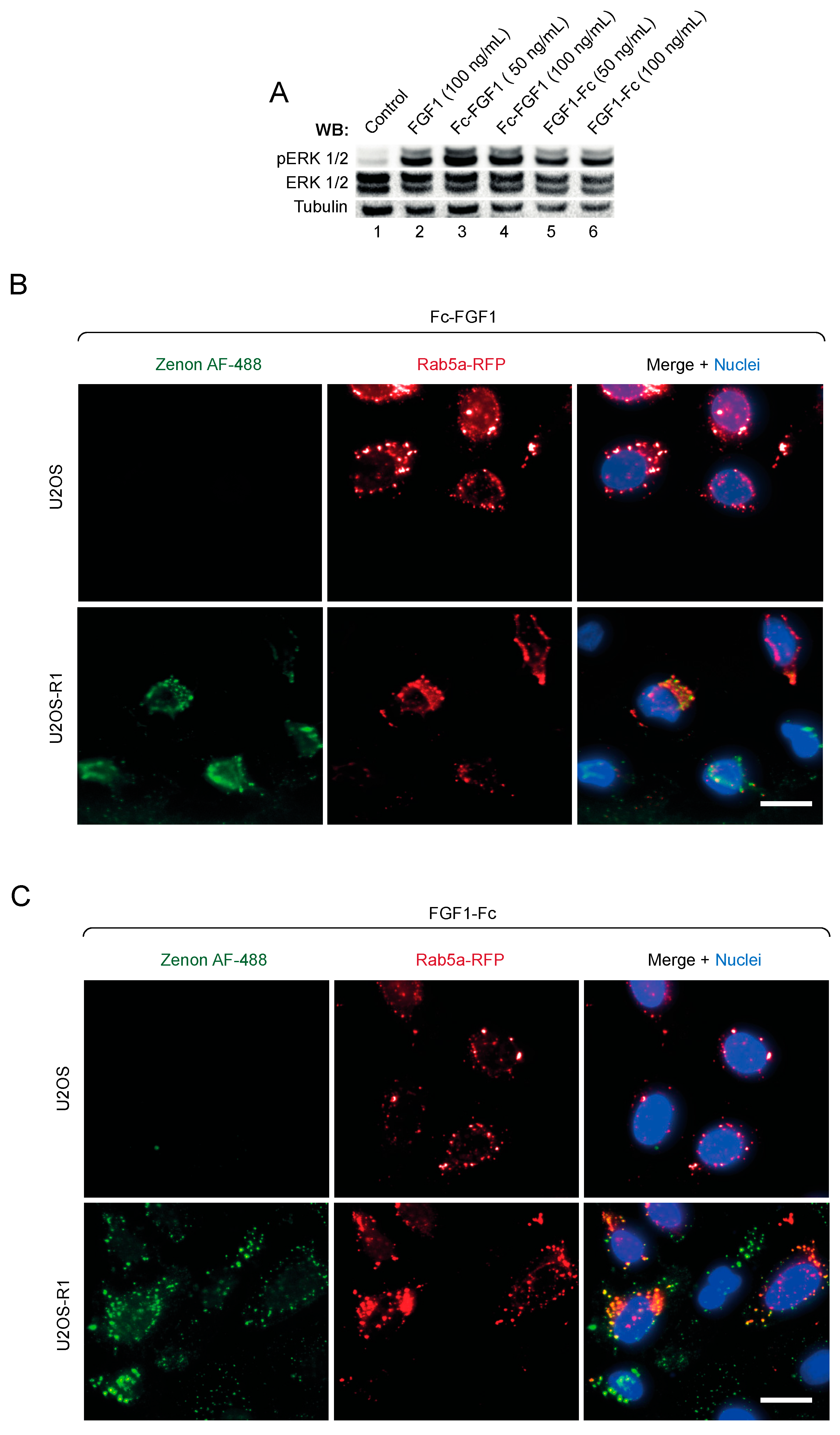
3.3. Biological Activity of FGF1 Fusions with the Fc Fragment
3.4. Assembly of Multivalent Complexes of Engineered FGF1 and GFPpolygons
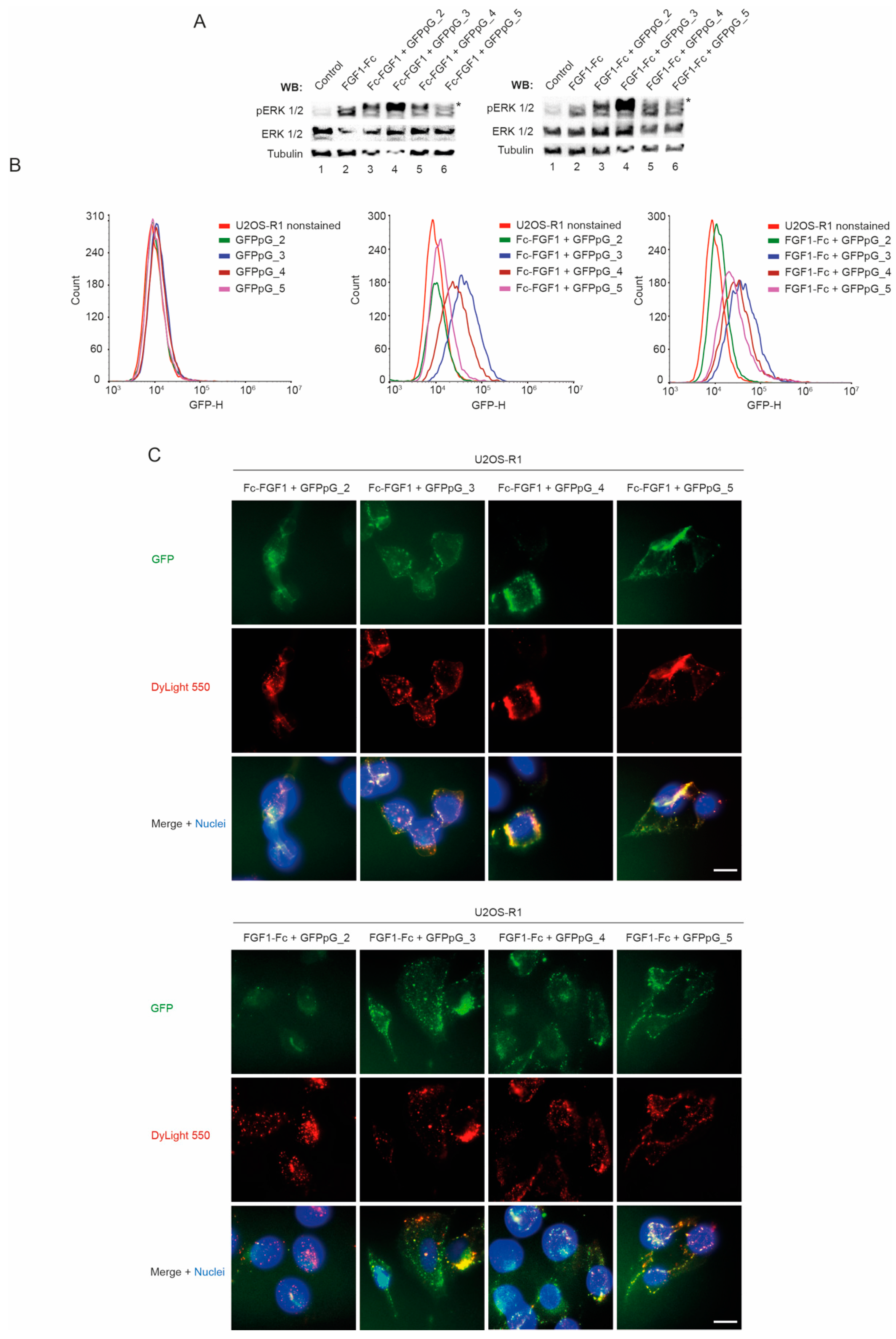
3.5. Biological Activity of Multivalent Complexes of Engineered FGF1 and GFPpolygons
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Farrell, B.; Breeze, A.L. Structure, activation and dysregulation of fibroblast growth factor receptor kinases: Perspectives for clinical targeting. Biochem. Soc. Trans. 2018, 46, 1753–1770. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Zhou, Z.; Chen, Z.; Xu, G.; Chen, Y. Fibroblast Growth Factor Receptors (FGFRs): Structures and Small Molecule Inhibitors. Cells 2019, 8, 614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, S.J.; Bruce, C.; Chioni, A.M.; Kocher, H.M.; Grose, R.P. The ins and outs of fibroblast growth factor receptor sig-nalling. Clin. Sci. 2014, 127, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Tiong, K.H.; Mah, L.Y.; Leong, C.-O. Functional roles of fibroblast growth factor receptors (FGFRs) signaling in human cancers. Apoptosis 2013, 18, 1447–1468. [Google Scholar] [CrossRef] [Green Version]
- Opaliński, Ł.; Sokolowska-Wedzina, A.; Szczepara, M.; Zakrzewska, M.; Otlewski, J. Antibody-induced dimerization of FGFR1 promotes receptor endocytosis independently of its kinase activity. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stauber, D.J.; DiGabriele, A.D.; Hendrickson, W.A. Structural interactions of fibroblast growth factor receptor with its ligands. Proc. Natl. Acad. Sci. USA 2000, 97, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Wesche, J.; Haglund, K.; Haugsten, E.M. Fibroblast growth factors and their receptors in cancer. Biochem. J. 2011, 437, 199–213. [Google Scholar] [CrossRef] [Green Version]
- Touat, M.; Ileana, E.; Postel-Vinay, S.; André, F.; Soria, J.-C. Targeting FGFR Signaling in Cancer. Clin. Cancer Res. 2015, 21, 2684–2694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinkle, A.; Mohammadi, M. A threshold model for receptor tyrosine kinase signaling specificity and cell fate determination. F1000Research 2018, 7, 872. [Google Scholar] [CrossRef] [PubMed]
- Latko, M.; Czyrek, A.; Porębska, N.; Kucińska, M.; Otlewski, J.; Zakrzewska, M.; Opaliński, Ł. Cross-Talk between Fibroblast Growth Factor Receptors and Other Cell Surface Proteins. Cells 2019, 8, 455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, N.; Ohta, H.; Konishi, M. Endocrine FGFs: Evolution, Physiology, Pathophysiology, and Pharmacotherapy. Front. Endocrinol. 2015, 6, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belov, A.A.; Mohammadi, M. Molecular Mechanisms of Fibroblast Growth Factor Signaling in Physiology and Pathology. Cold Spring Harb. Perspect. Biol. 2013, 5, a015958. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thisse, B.; Thisse, C. Functions and regulations of fibroblast growth factor signaling during embryonic development. Dev. Biol. 2005, 287, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Su, N.; Yang, J.; Tan, Q.; Huang, S.; Jin, M.; Ni, Z.; Zhang, B.; Zhang, D.; Luo, F.; et al. FGF/FGFR signaling in health and disease. Signal Transduct. Target. Ther. 2020, 5, 1–38. [Google Scholar] [CrossRef]
- Goh, L.K.; Sorkin, A. Endocytosis of Receptor Tyrosine Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a017459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarabipour, S.; Hristova, K. Mechanism of FGF receptor dimerization and activation. Nat. Commun. 2016, 7, 10262. [Google Scholar] [CrossRef]
- Sokolowska-Wedzina, A.; Chodaczek, G.; Chudzian, J.; Borek, A.; Zakrzewska, M.; Otlewski, J. High-Affinity Internalizing Human scFv-Fc Antibody for Targeting FGFR1-Overexpressing Lung Cancer. Mol. Cancer Res. 2017, 15, 1040–1050. [Google Scholar] [CrossRef] [Green Version]
- Opaliński, Ł.; Szymczyk, J.; Szczepara, M.; Kucińska, M.; Krowarsch, D.; Zakrzewska, M.; Otlewski, J. High Affinity Promotes Internalization of Engineered Antibodies Targeting FGFR1. Int. J. Mol. Sci. 2018, 19, 1435. [Google Scholar] [CrossRef] [Green Version]
- Pozniak, M.; Sokolowska-Wedzina, A.; Jastrzebski, K.; Szymczyk, J.; Porebska, N.; Krzyscik, M.; Zakrzewska, M.; Miaczynska, M.; Otlewski, J.; Opalinski, L. FGFR1 clustering with engineered tetravalent antibody improves the efficiency and modifies the mechanism of receptor internalization. Mol. Oncol. 2020, 14, 1998–2021. [Google Scholar] [CrossRef]
- Porebska, N.; Pozniak, M.; Krzyscik, M.A.; Knapik, A.; Czyrek, A.; Kucinska, M.; Jastrzebski, K.; Zakrzewska, M.; Otlewski, J.; Opalinski, L. Dissecting biological activities of fibroblast growth factor receptors by the coiled-coil-mediated oligomerization of FGF1. Int. J. Biol. Macromol. 2021, 180, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Poźniak, M.; Porębska, N.; Krzyścik, M.A.; Sokołowska-Wędzina, A.; Jastrzębski, K.; Sochacka, M.; Szymczyk, J.; Zakrzewska, M.; Otlewski, J.; Opaliński, Ł. The cytotoxic conjugate of highly internalizing tetravalent antibody for targeting FGFR1-overproducing cancer cells. Mol. Med. 2021, 27, 1–13. [Google Scholar] [CrossRef]
- Kim, Y.E.; Kim, Y.-N.; Kim, J.A.; Kim, H.M.; Jung, Y. Green fluorescent protein nanopolygons as monodisperse supramolecular assemblies of functional proteins with defined valency. Nat. Commun. 2015, 6, 7134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haugsten, E.M.; Sørensen, V.; Brech, A.; Olsnes, S.; Wesche, J. Different intracellular trafficking of FGF1 endocytosed by the four homologous FGF receptors. J. Cell Sci. 2005, 118, 3869–3881. [Google Scholar] [CrossRef] [Green Version]
- Auciello, G.; Cunningham, D.; Tatar, T.; Heath, J.K.; Rappoport, J.Z. Regulation of fibroblast growth factor receptor signalling and trafficking by Src and Eps8. J. Cell Sci. 2013, 126, 613–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H. Endocytosis and membrane receptor internalization implication of F-BAR protein Carom. Front. Biosci. 2017, 22, 1439–1457. [Google Scholar] [CrossRef] [Green Version]
- Volinsky, N.; Kholodenko, B.N. Complexity of Receptor Tyrosine Kinase Signal Processing. Cold Spring Harb. Perspect. Biol. 2013, 5, a009043. [Google Scholar] [CrossRef] [Green Version]
- Bergeron, J.J.; Di Guglielmo, G.M.; Dahan, S.; Dominguez, M.; Posner, B.I. Spatial and Temporal Regulation of Receptor Tyrosine Kinase Activation and Intracellular Signal Transduction. Annu. Rev. Biochem. 2016, 85, 573–597. [Google Scholar] [CrossRef]
- Dikov, M.M.; Reich, M.B.; Dworkin, L.; Thomas, J.W.; Miller, G.G. A Functional Fibroblast Growth Factor-1 Immunoglobulin Fusion Protein. J. Biol. Chem. 1998, 273, 15811–15817. [Google Scholar] [CrossRef] [Green Version]
- Kucińska, M.; Porębska, N.; Lampart, A.; Latko, M.; Knapik, A.; Zakrzewska, M.; Otlewski, J.; Opaliński, Ł. Differential regulation of fibroblast growth factor receptor 1 trafficking and function by extracellular galectins. Cell Commun. Signal. 2019, 17, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porębska, N.; Poźniak, M.; Matynia, A.; Żukowska, D.; Zakrzewska, M.; Otlewski, J.; Opaliński, Ł. Galectins as modulators of receptor tyrosine kinases signaling in health and disease. Cytokine Growth Factor Rev. 2021, 60, 89–106. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poźniak, M.; Zarzycka, W.; Porębska, N.; Knapik, A.; Marczakiewicz-Perera, P.; Zakrzewska, M.; Otlewski, J.; Opaliński, Ł. FGF1 Fusions with the Fc Fragment of IgG1 for the Assembly of GFPpolygons-Mediated Multivalent Complexes Recognizing FGFRs. Biomolecules 2021, 11, 1088. https://doi.org/10.3390/biom11081088
Poźniak M, Zarzycka W, Porębska N, Knapik A, Marczakiewicz-Perera P, Zakrzewska M, Otlewski J, Opaliński Ł. FGF1 Fusions with the Fc Fragment of IgG1 for the Assembly of GFPpolygons-Mediated Multivalent Complexes Recognizing FGFRs. Biomolecules. 2021; 11(8):1088. https://doi.org/10.3390/biom11081088
Chicago/Turabian StylePoźniak, Marta, Weronika Zarzycka, Natalia Porębska, Agata Knapik, Paulina Marczakiewicz-Perera, Malgorzata Zakrzewska, Jacek Otlewski, and Łukasz Opaliński. 2021. "FGF1 Fusions with the Fc Fragment of IgG1 for the Assembly of GFPpolygons-Mediated Multivalent Complexes Recognizing FGFRs" Biomolecules 11, no. 8: 1088. https://doi.org/10.3390/biom11081088
APA StylePoźniak, M., Zarzycka, W., Porębska, N., Knapik, A., Marczakiewicz-Perera, P., Zakrzewska, M., Otlewski, J., & Opaliński, Ł. (2021). FGF1 Fusions with the Fc Fragment of IgG1 for the Assembly of GFPpolygons-Mediated Multivalent Complexes Recognizing FGFRs. Biomolecules, 11(8), 1088. https://doi.org/10.3390/biom11081088