
Epigenetic Alterations in Pancreatic Cancer Metastasis


Abstract
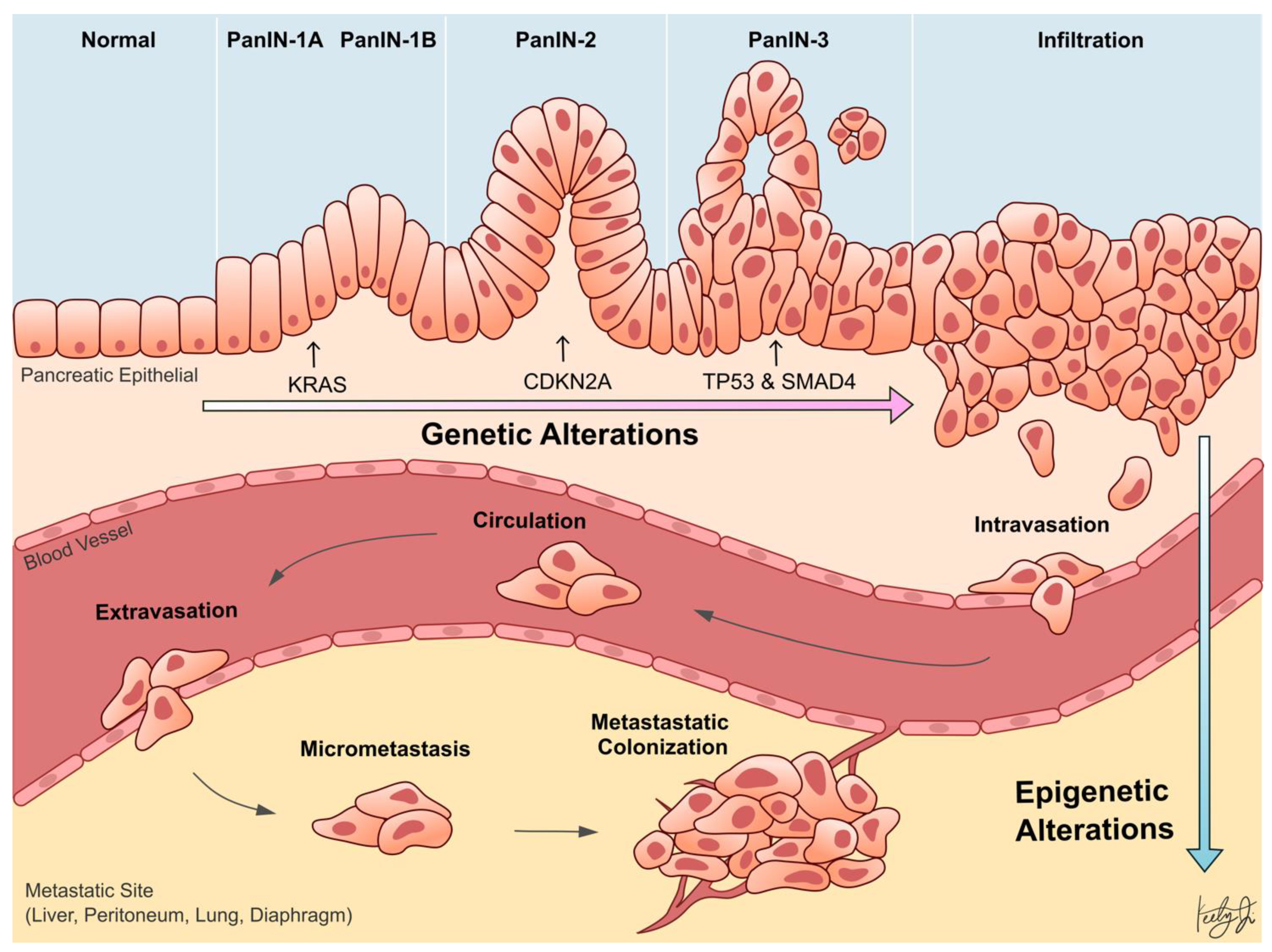
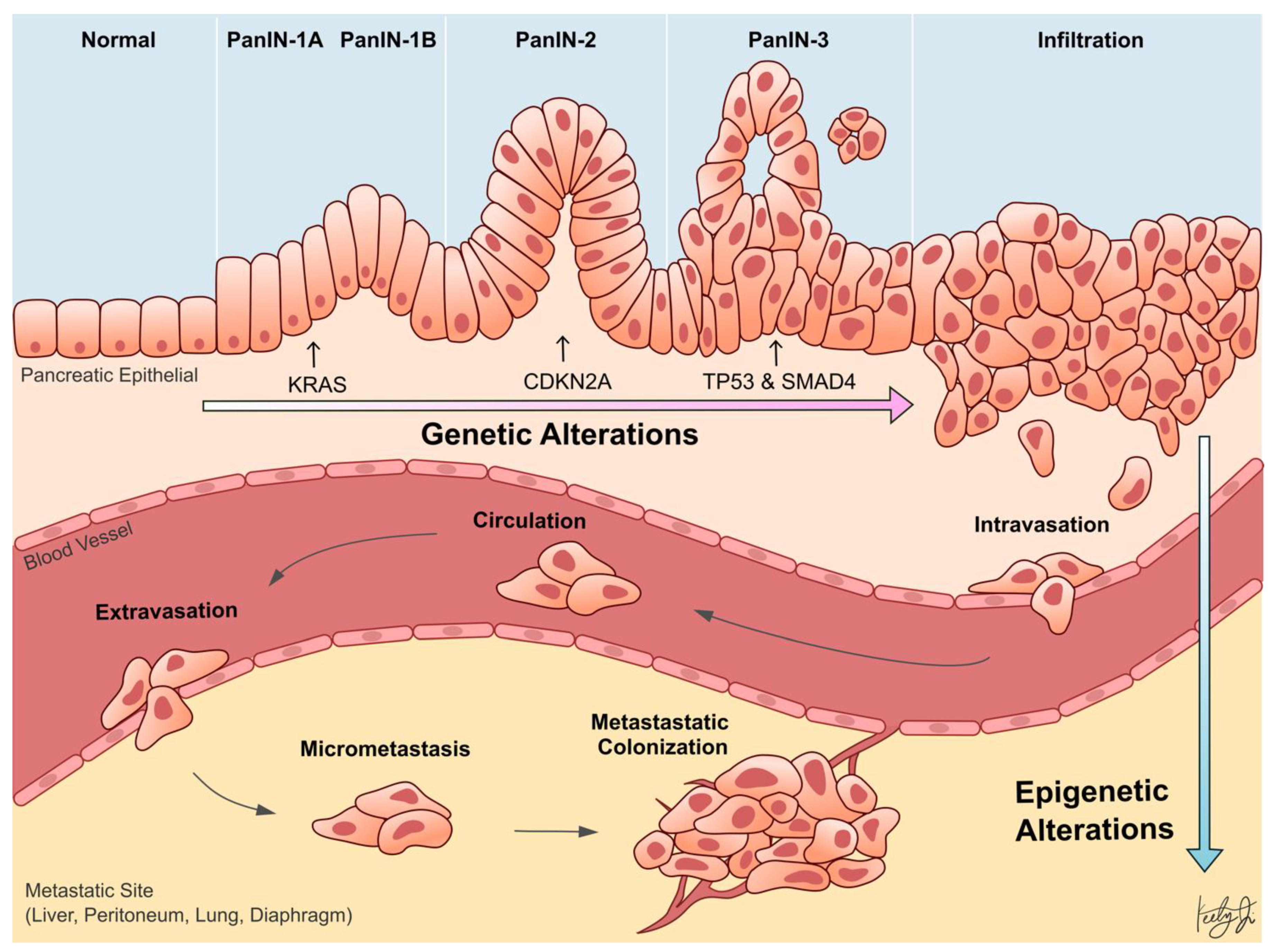
:1. Introduction
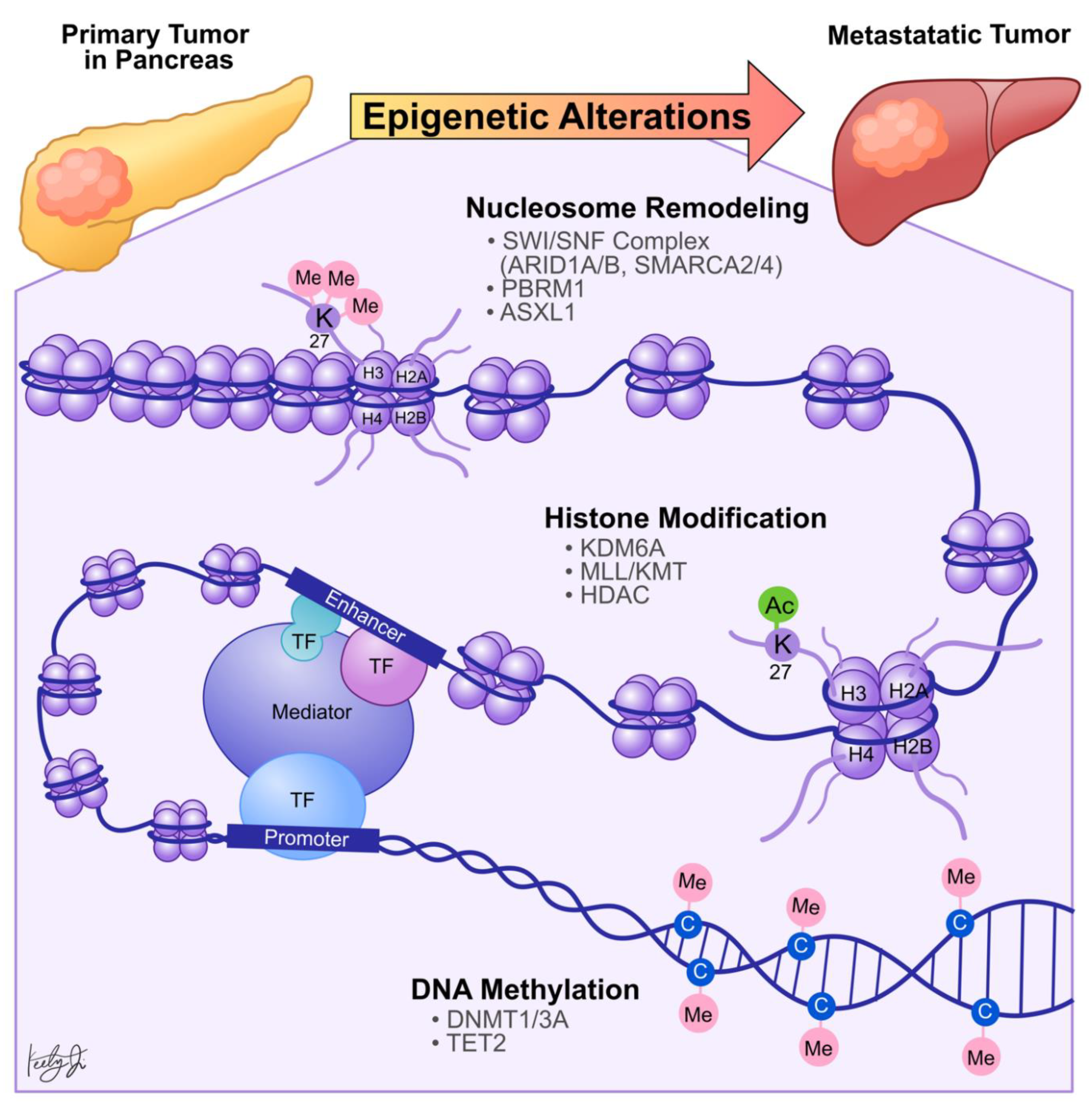
2. Genetic Alterations in Epigenetic Regulators
3. Chromatin Accessibility and Metastasis
4. Transcription Factor-Mediated Histone Modification and Metastasis
5. Transcription Factor-Mediated Enhancer Regulation in Aggressive PDA Molecular Subtype Differentiation
6. DNA Methylation and Metastasis
7. Therapeutic Implications of Epigenetic Regulators
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA. Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Dey, P.; Yao, W.; Kimmelman, A.C.; Draetta, G.F.; Maitra, A.; Depinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016, 30, 355–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef] [Green Version]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Baylin, S.B.; Jones, P.A. Epigenetic determinants of cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [Green Version]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome-biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef]
- Ting, A.H.; McGarvey, K.M.; Baylin, S.B. The cancer epigenome—Components and functional correlates. Genes Dev. 2006, 20, 3215–3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, N.J.; Norris, A.L.; Petersen, G.M.; Bondy, M.L.; Brand, R.; Gallinger, S.; Kurtz, R.C.; Olson, S.H.; Rustgi, A.K.; Schwartz, A.G.; et al. Whole genome sequencing defines the genetic heterogeneity of familial pancreatic cancer. Cancer Discov. 2016, 6, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Mann, K.M.; Ward, J.M.; Yew, C.C.K.; Kovochich, A.; Dawson, D.W.; Black, M.A.; Brett, B.T.; Sheetz, T.E.; Dupuy, A.J.; Chang, D.K.; et al. Sleeping Beauty mutagenesis reveals cooperating mutations and pathways in pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2012, 109, 5934–5941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Mancera, P.A.; Rust, A.G.; Van Der Weyden, L.; Kristiansen, G.; Li, A.; Sarver, A.L.; Silverstein, K.A.T.; Grützmann, R.; Aust, D.; Rümmele, P.; et al. The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature 2012, 486, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Dhara, S.; Chhangawala, S.; Chintalapudi, H.; Askan, G.; Aveson, V.; Massa, A.L.; Zhang, L.; Torres, D.; Makohon-Moore, A.P.; Lecomte, N.; et al. Pancreatic cancer prognosis is predicted by an ATAC-array technology for assessing chromatin accessibility. Nat. Commun. 2021, 12, 3044. [Google Scholar] [CrossRef] [PubMed]
- Roe, J.S.; Hwang, C.I.; Somerville, T.D.D.; Milazzo, J.P.; Lee, E.J.; Da Silva, B.; Maiorino, L.; Tiriac, H.; Young, C.M.; Miyabayashi, K.; et al. Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell 2017, 170, 875–888.e20. [Google Scholar] [CrossRef] [Green Version]
- Somerville, T.D.D.; Xu, Y.; Miyabayashi, K.; Tiriac, H.; Cleary, C.R.; Maia-Silva, D.; Milazzo, J.P.; Tuveson, D.A.; Vakoc, C.R. TP63-Mediated Enhancer Reprogramming Drives the Squamous Subtype of Pancreatic Ductal Adenocarcinoma. Cell Rep. 2018, 25, 1741–1755.e7. [Google Scholar] [CrossRef] [Green Version]
- Sato, N.; Parker, A.R.; Fukushima, N.; Miyagi, Y.; Iacobuzio-Donahue, C.A.; Eshleman, J.R.; Goggins, M. Epigenetic inactivation of TFPI-2 as a common mechanism associated with growth and invasion of pancreatic ductal adenocarcinoma. Oncogene 2005, 24, 850–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, N.; Fukushima, N.; Chang, R.; Matsubayashi, H.; Goggins, M. Differential and epigenetic gene expression profiling identifies frequent disruption of the RELN pathway in pancreatic cancers. Gastroenterology 2006, 130, 548–565. [Google Scholar] [CrossRef]
- Nones, K.; Waddell, N.; Song, S.; Patch, A.M.; Miller, D.; Johns, A.; Wu, J.; Kassahn, K.S.; Wood, D.; Bailey, P.; et al. Genome-wide DNA methylation patterns in pancreatic ductal adenocarcinoma reveal epigenetic deregulation of SLIT-ROBO, ITGA2 and MET signaling. Int. J. Cancer 2014, 135, 1110–1118. [Google Scholar] [CrossRef] [Green Version]
- Campbell, P.J.; Yachida, S.; Mudie, L.J.; Stephens, P.J.; Pleasance, E.D.; Stebbings, L.A.; Morsberger, L.A.; Latimer, C.; McLaren, S.; Lin, M.L.; et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 2010, 467, 1109–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Lemire, M.; Zhang, A.; Chan-Seng-Yue, M.; Wilson, G.; Grant, R.C.; Merico, D.; Lungu, I.; et al. Integration of Genomic and Transcriptional Features in Pancreatic Cancer Reveals Increased Cell Cycle Progression in Metastases. Cancer Cell 2019, 35, 267–282.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makohon-Moore, A.P.; Zhang, M.; Reiter, J.G.; Bozic, I.; Allen, B.; Kundu, D.; Chatterjee, K.; Wong, F.; Jiao, Y.; Kohutek, Z.A.; et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat. Genet. 2017, 49, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Priestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; de Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haidari, S.; van Hoeck, A.; Onstenk, W.; et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019, 575, 210–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Embuscado, E.E.; Laheru, D.; Ricci, F.; Yun, K.J.; Witzel, S.D.B.; Seigel, A.; Flickinger, K.; Hidalgo, M.; Bova, G.S.; Iacobuzio-Donahue, C.A. Immortalizing the complexity of cancer metastasis: Genetic features of lethal metastatic pancreatic cancer obtained from rapid autopsy. Cancer Biol. Ther. 2005, 4, 548–554. [Google Scholar] [CrossRef] [Green Version]
- McGinty, R.K.; Tan, S. Nucleosome structure and function. Chem. Rev. 2015, 115, 2255–2273. [Google Scholar] [CrossRef] [Green Version]
- Tamaru, H. Confining euchromatin/heterochromatin territory: Jumonji crosses the line. Genes Dev. 2010, 24, 1465–1478. [Google Scholar] [CrossRef] [Green Version]
- Mayran, A.; Drouin, J. Pioneer transcription factors shape the epigenetic landscape. J. Biol. Chem. 2018, 293, 13795–13804. [Google Scholar] [CrossRef] [Green Version]
- Maston, G.A.; Evans, S.K.; Green, M.R. Transcriptional regulatory elements in the human genome. Annu. Rev. Genomics Hum. Genet. 2006, 7, 29–59. [Google Scholar] [CrossRef] [Green Version]
- Shlyueva, D.; Stampfel, G.; Stark, A. Transcriptional enhancers: From properties to genome-wide predictions. Nat. Rev. Genet. 2014, 15, 272–286. [Google Scholar] [CrossRef]
- Denny, S.K.; Yang, D.; Chuang, C.H.; Brady, J.J.; Lim, J.S.S.; Grüner, B.M.; Chiou, S.H.; Schep, A.N.; Baral, J.; Hamard, C.; et al. Nfib Promotes Metastasis through a Widespread Increase in Chromatin Accessibility. Cell 2016, 166, 328–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, 324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.M.; Wong, C.C.L.; Ng, Y.L.; Au, S.L.K.; Ko, F.C.F.; Ng, I.O.L. Transcriptional repressive H3K9 and H3K27 methylations contribute to DNMT1-mediated DNA methylation recovery. PLoS ONE 2011, 6, e16702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, O.G.; Li, X.; Saunders, T.; Tryggvadottir, R.; Mentch, S.J.; Warmoes, M.O.; Word, A.E.; Carrer, A.; Salz, T.H.; Natsume, S.; et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet. 2017, 49, 367–376. [Google Scholar] [CrossRef]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [Green Version]
- Gao, N.; LeLay, J.; Vatamaniuk, M.Z.; Rieck, S.; Friedman, J.R.; Kaestner, K.H. Dynamic regulation of Pdx1 enhancers by Foxa1 and Foxa2 is essential for pancreas development. Genes Dev. 2008, 22, 3435–3448. [Google Scholar] [CrossRef] [Green Version]
- Vigil, D.; Martin, T.D.; Williams, F.; Jen Yeh, J.; Campbell, S.L.; Der, C.J. Aberrant overexpression of the Rgl2 Ral small GTPase-specific guanine nucleotide exchange factor promotes pancreatic cancer growth through Ral-dependent and Ral-independent mechanisms. J. Biol. Chem. 2010, 285, 34729–34740. [Google Scholar] [CrossRef] [Green Version]
- Larsen, H.L.; Grapin-Botton, A. The molecular and morphogenetic basis of pancreas organogenesis. Semin. Cell Dev. Biol. 2017, 66, 51–68. [Google Scholar] [CrossRef]
- Kim, M.P.; Li, X.; Deng, J.; Zhang, Y.; Dai, B.; Allton, K.L.; Hughes, T.G.; Siangco, C.; Augustine, J.J.; Kang, Y.; et al. Oncogenic KRAS recruits an expansive transcriptional network through mutant p53 to drive pancreatic cancer metastasis. Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Andricovich, J.; Perkail, S.; Kai, Y.; Casasanta, N.; Peng, W.; Tzatsos, A. Loss of KDM6A Activates Super-Enhancers to Induce Gender-Specific Squamous-like Pancreatic Cancer and Confers Sensitivity to BET Inhibitors. Cancer Cell 2018, 33, 512–526.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavery, W.J.; Barski, A.; Wiley, S.; Schorry, E.K.; Lindsley, A.W. KMT2C/D COMPASS complex-associated diseases [KCDCOM-ADs]: An emerging class of congenital regulopathies. Clin. Epigenet. 2020, 12, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Piunti, A.; Shilatifard, A. Epigenetic balance of gene expression by polycomb and compass families. Science 2016, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, E.; Zhang, Y. DNA methylation in mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a019133. [Google Scholar] [CrossRef]
- Wu, S.C.; Zhang, Y. Active DNA demethylation: Many roads lead to Rome. Nat. Rev. Mol. Cell Biol. 2010, 11, 607–620. [Google Scholar] [CrossRef] [Green Version]
- Valinluck, V.; Sowers, L.C. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007, 67, 946–950. [Google Scholar] [CrossRef] [Green Version]
- Brenet, F.; Moh, M.; Funk, P.; Feierstein, E.; Viale, A.J.; Socci, N.D.; Scandura, J.M. DNA Methylation of the First Exon Is Tightly Linked to Transcriptional Silencing. PLoS ONE 2011, 6, e14524. [Google Scholar] [CrossRef]
- Angeloni, A.; Bogdanovic, O. Enhancer DNA methylation: Implications for gene regulation. Essays Biochem. 2019, 63, 707–715. [Google Scholar]
- Vincent, A.; Omura, N.; Hong, S.M.; Jaffe, A.; Eshleman, J.; Goggins, M. Genome-wide analysis of promoter methylation associated with gene expression profile in pancreatic adenocarcinoma. Clin. Cancer Res. 2011, 17, 4341–4354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ball, M.P.; Li, J.B.; Gao, Y.; Lee, J.H.; Leproust, E.M.; Park, I.H.; Xie, B.; Daley, G.Q.; Church, G.M. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 2009, 27, 361–368. [Google Scholar] [CrossRef] [Green Version]
- Aran, D.; Toperoff, G.; Rosenberg, M.; Hellman, A. Replication timing-related and gene body-specific methylation of active human genes. Hum. Mol. Genet. 2011, 20, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Paz, M.F.; Fraga, M.F.; Avila, S.; Guo, M.; Pollan, M.; Herman, J.G.; Esteller, M. A systematic profile of DNA methylation in human cancer cell lines. Cancer Res. 2003, 63, 1114–1121. [Google Scholar]
- Saghafinia, S.; Mina, M.; Riggi, N.; Hanahan, D.; Ciriello, G. Pan-Cancer Landscape of Aberrant DNA Methylation across Human Tumors. Cell Rep. 2018, 25, 1066–1080.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salhab, A.; Nordström, K.; Gasparoni, G.; Kattler, K.; Ebert, P.; Ramirez, F.; Arrigoni, L.; Müller, F.; Polansky, J.K.; Cadenas, C.; et al. A comprehensive analysis of 195 DNA methylomes reveals shared and cell-specific features of partially methylated domains. Genome Biol. 2018, 19, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Thompson, M.J.; Rubbi, L.; Dawson, D.W.; Donahue, T.R.; Pellegrini, M. Pancreatic Cancer Patient Survival Correlates with DNA Methylation of Pancreas Development Genes. PLoS ONE 2015, 10, e0128814. [Google Scholar] [CrossRef] [Green Version]
- Mishra, N.K.; Southekal, S.; Guda, C. Survival analysis of multi-omics data identifies potential prognostic markers of pancreatic ductal adenocarcinoma. Front. Genet. 2019, 10, 624. [Google Scholar] [CrossRef] [Green Version]
- Mishra, N.K.; Guda, C. Genome-wide DNA methylation analysis reveals molecular subtypes of pancreatic cancer. Oncotarget 2017, 8, 28990–29012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.; Singh, A.P.; Gupta, D. Unsupervised subtyping and methylation landscape of pancreatic ductal adenocarcinoma. Heliyon 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; DePinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006, 20, 1218–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koizumi, M.; Doi, R.; Toyoda, E.; Masui, T.; Tulachan, S.S.; Kawaguchi, Y.; Fujimoto, K.; Gittes, G.K.; Imamura, M. Increased PDX-1 expression is associated with outcome in patients with pancreatic cancer. Surgery 2003, 134, 260–266. [Google Scholar] [CrossRef]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Espinet, E.; Gu, Z.; Imbusch, C.D.; Giese, N.A.; Buscher, M.; Safavi, M.; Weisenburger, S.; Klein, C.; Vogel, V.; Falcone, M.; et al. Aggressive PDACs show hypomethylation of repetitive elements and the execution of an intrinsic IFN program linked to a ductal cell-of-origin. Cancer Discov. 2020, 11, 638–659. [Google Scholar] [CrossRef]
- Tan, A.C.; Jimeno, A.; Lin, S.H.; Wheelhouse, J.; Chan, F.; Solomon, A.; Rajeshkumar, N.V.; Rubio-Viqueira, B.; Hidalgo, M. Characterizing DNA methylation patterns in pancreatic cancer genome. Mol. Oncol. 2009, 3, 425–438. [Google Scholar] [CrossRef] [Green Version]
- Kelly, T.K.; De Carvalho, D.D.; Jones, P.A. Epigenetic modifications as therapeutic targets. Nat. Biotechnol. 2010, 28, 1069–1078. [Google Scholar] [CrossRef] [Green Version]
- Hessmann, E.; Johnsen, S.A.; Siveke, J.T.; Ellenrieder, V. Epigenetic treatment of pancreatic cancer: Is there a therapeutic perspective on the horizon? Gut 2017, 66, 168–179. [Google Scholar] [CrossRef]
- Brancaccio, M.; Natale, F.; Falco, G.; Angrisano, T. Cell-Free DNA Methylation: The New Frontiers of Pancreatic Cancer Biomarkers’ Discovery. Genes 2019, 11, 14. [Google Scholar] [CrossRef] [Green Version]
- Shinjo, K.; Hara, K.; Nagae, G.; Umeda, T.; Katsushima, K.; Suzuki, M.; Murofushi, Y.; Umezu, Y.; Takeuchi, I.; Takahashi, S.; et al. A novel sensitive detection method for DNA methylation in circulating free DNA of pancreatic cancer. PLoS ONE 2020, 15, 1–18. [Google Scholar] [CrossRef]
- Singh, N.; Rashid, S.; Rashid, S.; Dash, N.R.; Gupta, S.; Saraya, A. Clinical significance of promoter methylation status of tumor suppressor genes in circulating DNA of pancreatic cancer patients. J. Cancer Res. Clin. Oncol. 2020, 146, 897–907. [Google Scholar] [CrossRef]
- Nishizawa, N.; Harada, H.; Kumamoto, Y.; Kaizu, T.; Katoh, H.; Tajima, H.; Ushiku, H.; Yokoi, K.; Igarashi, K.; Fujiyama, Y.; et al. Diagnostic potential of hypermethylation of the cysteine dioxygenase 1 gene (CDO 1) promoter DNA in pancreatic cancer. Cancer Sci. 2019, 110, 2846–2855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manuyakorn, A.; Paulus, R.; Farrell, J.; Dawson, N.A.; Tze, S.; Cheung-Lau, G.; Hines, O.J.; Reber, H.; Seligson, D.B.; Horvath, S.; et al. Cellular histone modification patterns predict prognosis and treatment response in resectable pancreatic adenocarcinoma: Results from RTOG 9704. J. Clin. Oncol. 2010, 28, 1358–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Category | Gene | Molecular Function | Molecular Phenotype in PDA | Functional Phenotype in PDA | Reference |
|---|---|---|---|---|---|
| Germline PTV Mutation in Epigenetic Regulators | TET2 | Dioxygenase of 5-methylcytosine, involved in demethylation of cytosines | loss of function in encoded protein | ↓ patient survival | [12] |
| DNMT3a | DNA methyltransferase, involved in de novo DNA methylation | loss of function in encoded protein | ↓ patient survival | [12] | |
| ASXL1 | Polycomb group protein, involved in gene transcriptional regulation and chromatin architecture maintenance | loss of function in encoded protein | ↑ proliferation, ↓ patient survival | [12] | |
| Somatic Mutation in Epigenetic Regulators | ARID1A | Chromatin remodeler, involved in chromatin remodeling and gene transcriptional regulation | loss of function in encoded protein | ↑ progression, ↓ survival | [6,7,8,13,14] |
| KDM6A | Lysine-specific histone demethylase, involved in promoter and enhancer activities | loss of function in encoded protein | ↑ squamous identity, ↓ survival | [6,7,13,14] | |
| Chromatin Accessibility | ZKSCAN1 | Transcription factor, involved in proliferation and differentiation | ↑ TF binding via open chromatin | ↑ metastasis | [15] |
| HNF1B | Transcription factor, involved in beta cell development in the pancreas | ↓ TF binding via closed chromatin | ↑ metastasis | [15] | |
| Transcription Factor-Mediated Histone Modification | FOXA1 | Transcription factor, involved in cell differentiation and chromatin remodeling | ↑ enhancer activation (H3K27ac) | ↑ cell growth, ↑ invasion, ↑ progression | [16] |
| TP63 | Transcription factor, involved in cell proliferation, differentiation, and apoptosis | ↑ enhancer activation (H3K27ac) | ↑ squamous identity | [17] | |
| DNA Methylation | TFPI-2 | Serine proteinase inhibitor, involved in negative regulation of pro-metastasis extracellular matrix degradation | ↓ expression via hypermethylation | ↑ progression, ↑ proliferation, ↑ migration | [18] |
| RELN | Extracellular matrix serine protease, involved in neuronal migration | ↓ expression via hypermethylation | ↓ patient survival, ↑ migration, ↑ invasion, ↑ colony formation | [19] | |
| MET | Receptor tyrosine kinase, involved in cell survival, migration, and invasion | ↑ expression via hypomethylation | ↓ patient survival | [20] | |
| ITGA2 | Integrin, involved in adhesion of cells to the extracellular matrix | ↑ expression via hypomethylation | ↓ patient survival | [20] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.S.; Xu, J.; Ji, K.Y.; Hwang, C.-I. Epigenetic Alterations in Pancreatic Cancer Metastasis. Biomolecules 2021, 11, 1082. https://doi.org/10.3390/biom11081082
Wang SS, Xu J, Ji KY, Hwang C-I. Epigenetic Alterations in Pancreatic Cancer Metastasis. Biomolecules. 2021; 11(8):1082. https://doi.org/10.3390/biom11081082
Chicago/Turabian StyleWang, Sarah S., Jihao Xu, Keely Y. Ji, and Chang-Il Hwang. 2021. "Epigenetic Alterations in Pancreatic Cancer Metastasis" Biomolecules 11, no. 8: 1082. https://doi.org/10.3390/biom11081082
APA StyleWang, S. S., Xu, J., Ji, K. Y., & Hwang, C.-I. (2021). Epigenetic Alterations in Pancreatic Cancer Metastasis. Biomolecules, 11(8), 1082. https://doi.org/10.3390/biom11081082