
Polycationic Monomeric and Homodimeric Asymmetric Monomethine Cyanine Dyes with Hydroxypropyl Functionality—Strong Affinity Nucleic Acids Binders

 ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis
2.1.1. NMR Spectroscopy
2.1.2. Synthesis of Dyes 4a–4e
2.2. Study of DNA/RNA Interactions
2.2.1. General Procedures
2.2.2. UV/Vis, CD and Fluorescence Titrations
2.2.3. Thermal Melting Experiments
2.3. Evaluation of the Antiproliferative Effect
2.3.1. Cell Lines and Culturing
2.3.2. Proliferation Assays
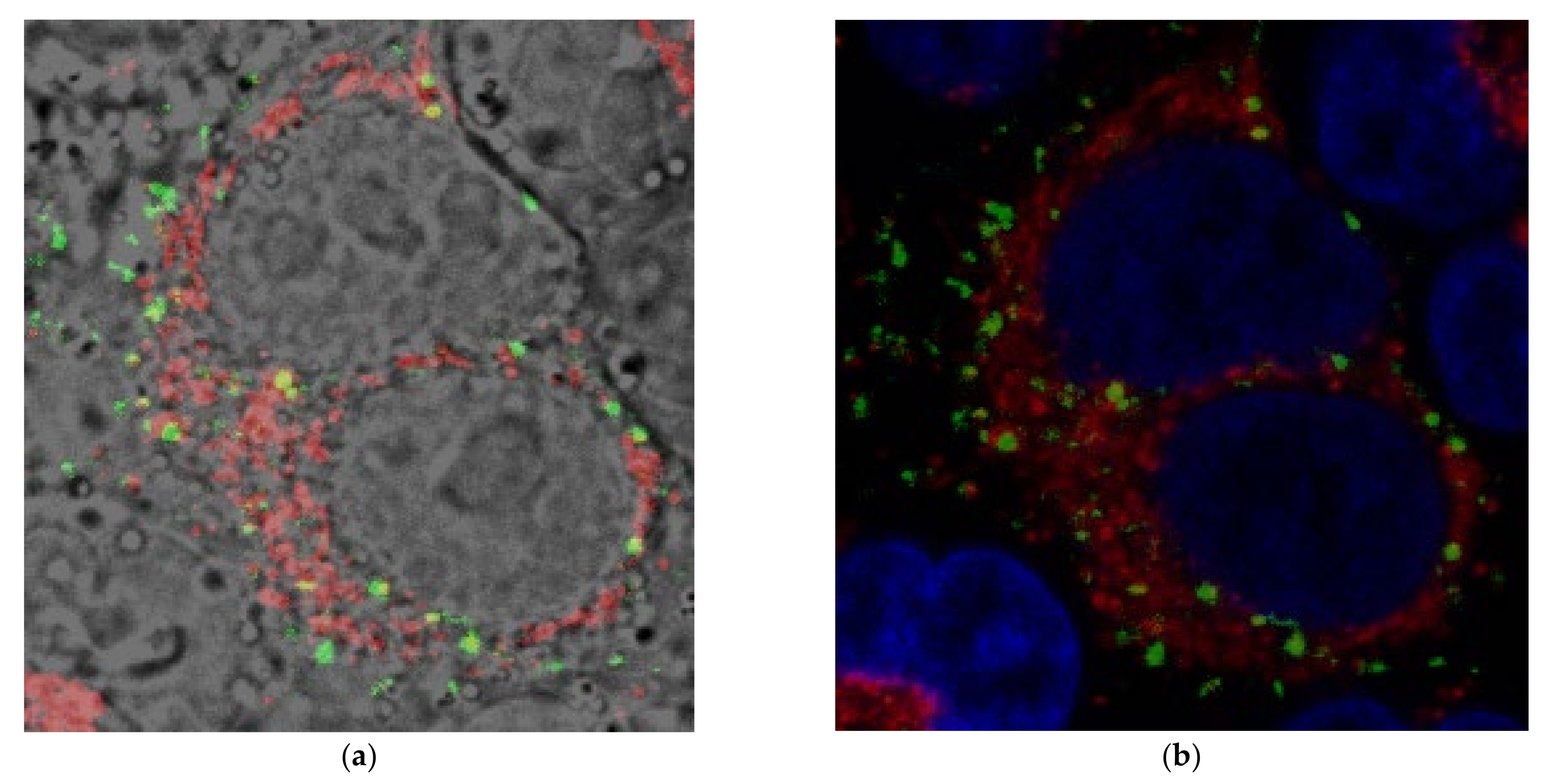
2.3.3. Confocal Microscopy
3. Results and Discussion
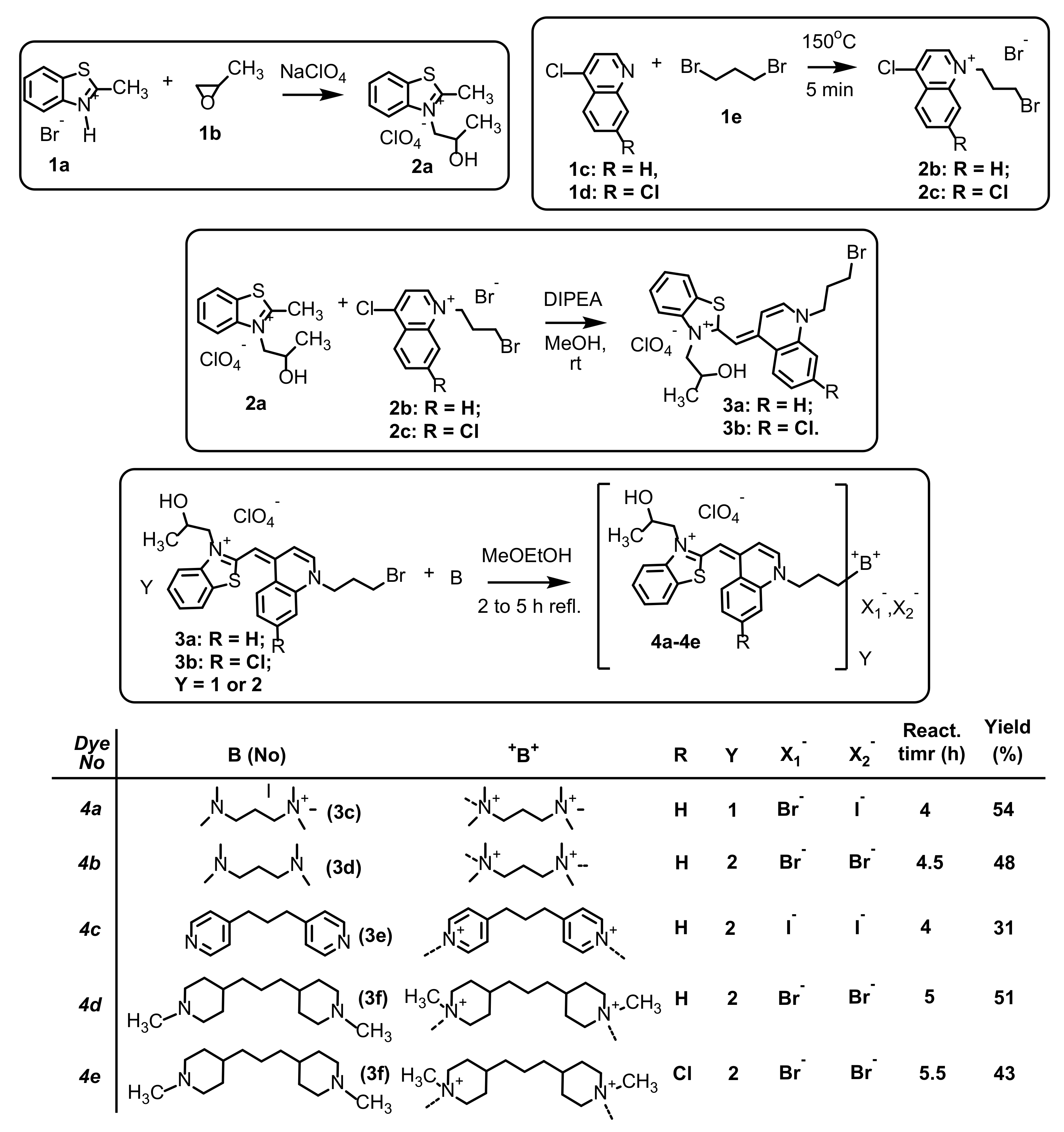
3.1. Synthesis
3.2. Characterization of the Photophysical Properties of Dyes 4a–4e in the Aqueous Medium
3.3. Interactions of 4a–4e with ds-Polynucleotides in an Aqueous Medium
3.3.1. Thermal Melting Studies
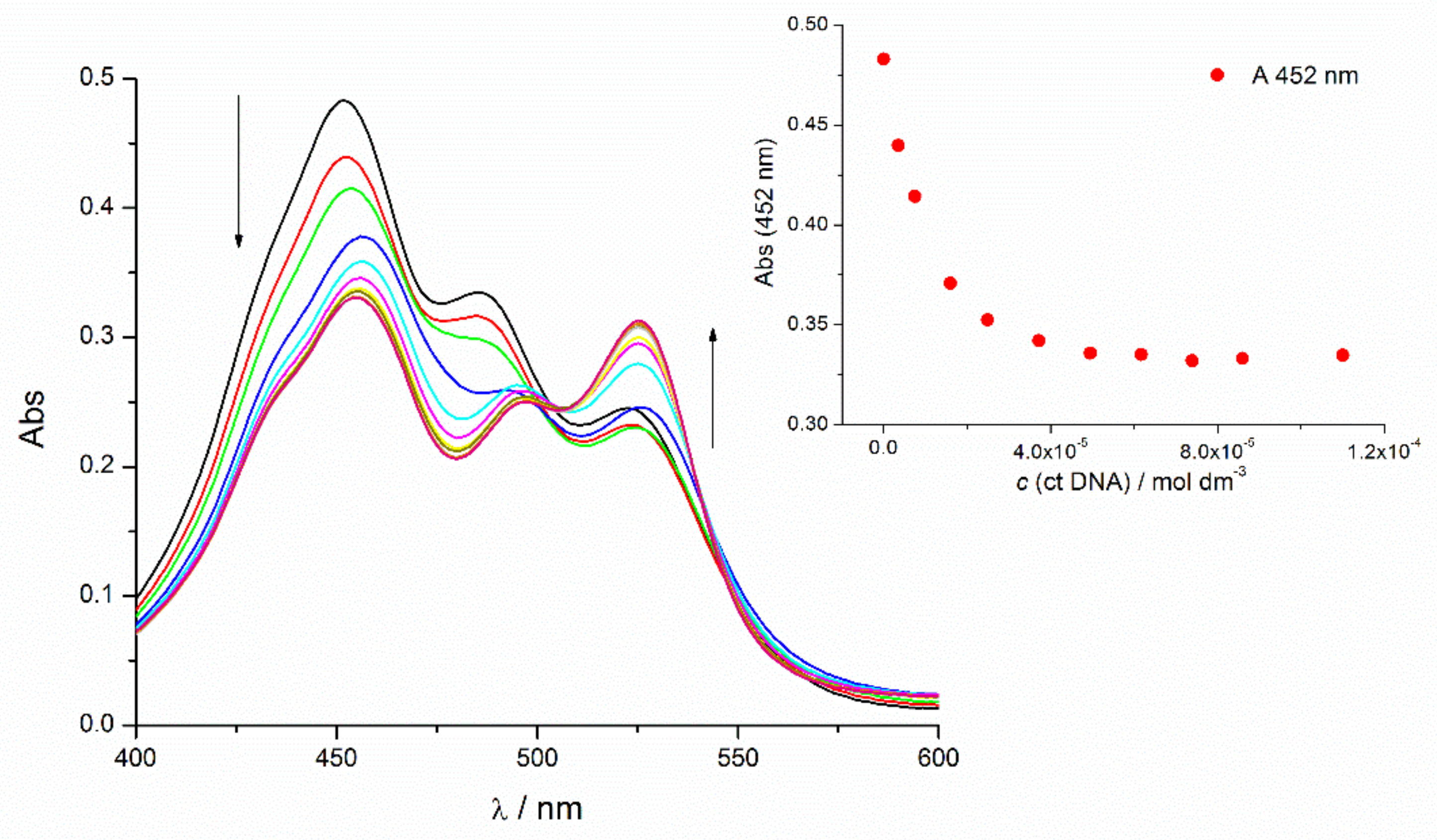
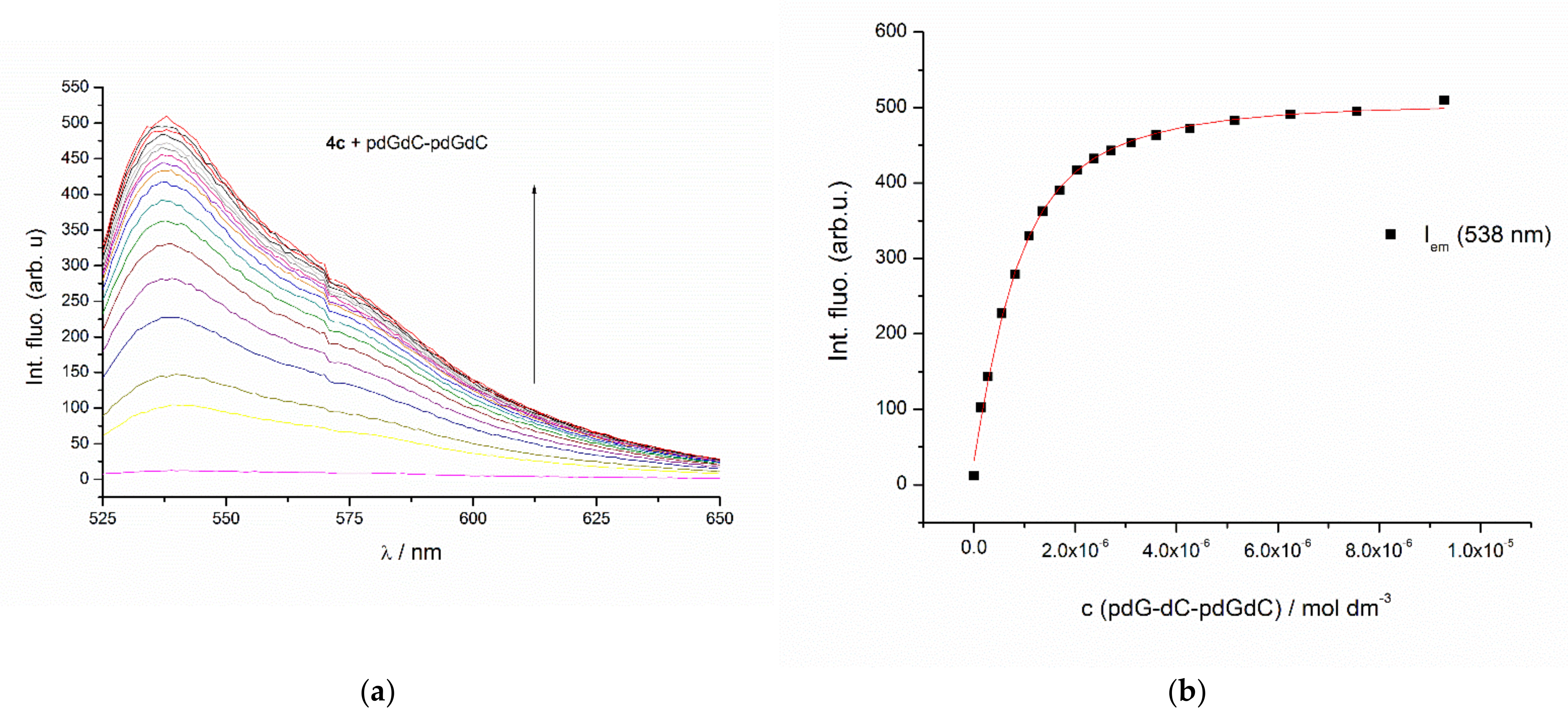
3.3.2. Spectrophotometric Titrations
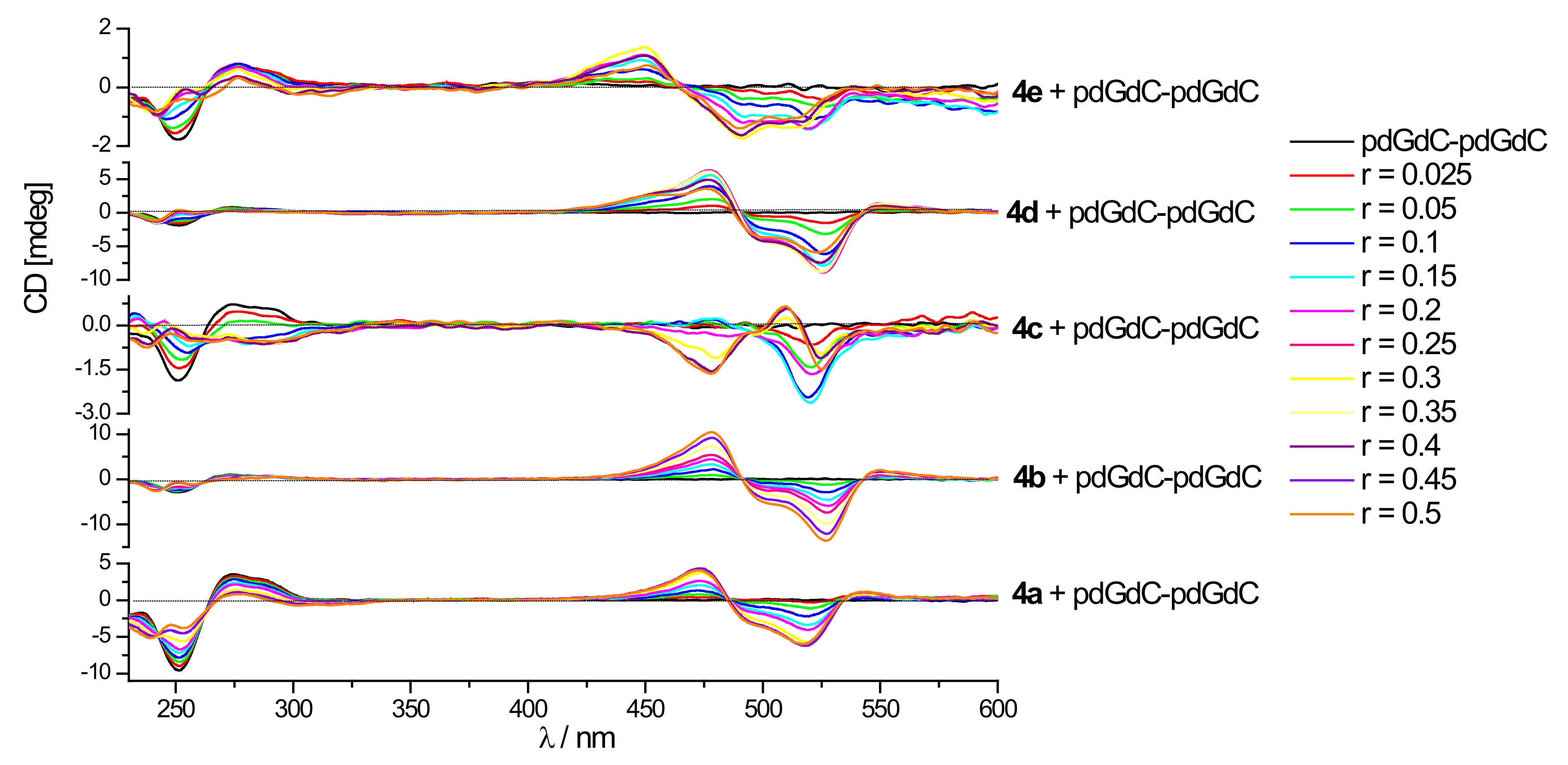
3.3.3. Circular Dichroism (CD) Experiments
3.3.4. Discussion of the Spectroscopic Results
3.4. Antiproliferative Effect of Compounds In Vitro
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chastain, M.; Tinoco, I. Structural Elements in RNA. In Progress in Nucleic Acid Research and Molecular Biology; Cohn, W.E., Moldave, K., Eds.; Academic Press: Cambridge, MA, USA, 1991; Volume 41, pp. 131–177. [Google Scholar]
- Saenger, W. Intercalation. In Principles of Nucleic Acid Structure; Saenger, W., Ed.; Springer: New York, NY, USA, 1984; pp. 350–367. [Google Scholar]
- Chiang, S.-Y.; Welch, J.; Rauscher, F.J.; Beerman, T.A. Effects of Minor Groove Binding Drugs on the Interaction of TATA Box Binding Protein and TFIIA with DNA. Biochemistry 1994, 33, 7033–7040. [Google Scholar] [CrossRef] [PubMed]
- Silverman, R.B.; Holladay, M.W. Chapter 6—DNA-Interactive Agents. In The Organic Chemistry of Drug Design and Drug Action, 3rd ed.; Silverman, R.B., Holladay, M.W., Eds.; Academic Press: Boston, MA, USA, 2014; pp. 275–331. [Google Scholar]
- Demeunynck, M.; Bailly, C.; Wilson, W.D. (Eds.) Small Molecule DNA and RNA Binders: From Synthesis to Nucleic Acid Complexes; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- Piantanida, I.; Palm, B.S.; Čudić, P.; Žinić, M.; Schneider, H.J. Interactions of acyclic and cyclic bis-phenanthridinium derivatives with ss- and ds-polynucleotides. Tetrahedron 2004, 60, 6225–6231. [Google Scholar] [CrossRef]
- Šmidlehner, T.; Badovinac, M.; Piantanida, I. Pyrene–cyanine conjugates as multipurpose fluorescent probes for non-covalent recognition of ds-DNA, RNA and proteins. New J. Chem. 2018, 42, 6655–6663. [Google Scholar] [CrossRef]
- Armitage, B.A. Cyanine Dye–DNA Interactions: Intercalation, Groove Binding, and Aggregation. In DNA Binders and Related Subjects; Waring, M.J., Chaires, J.B., Eds.; Springer: Berlin/Heidelberg, Germany, 2005; pp. 55–76. [Google Scholar]
- Rye, H.S.; Glazer, A.N. Interaction of dimeric intercalating dyes with single-stranded DNA. Nucleic Acids Res. 1995, 23, 1215–1222. [Google Scholar] [CrossRef] [Green Version]
- Glavaš-Obrovac, L.; Piantanida, I.; Marczi, S.; Mašić, L.; Timcheva, I.I.; Deligeorgiev, T.G. Minor structural differences of monomethine cyanine derivatives yield strong variation in their interactions with DNA, RNA as well as on their in vitro antiproliferative activity. Bioorg. Med. Chem. 2009, 17, 4747–4755. [Google Scholar] [CrossRef]
- Tumir, L.M.; Crnolatac, I.; Deligeorgiev, T.; Vasilev, A.; Kaloyanova, S.; Branilović, M.G.; Tomić, S.; Piantanida, I. Kinetic Differentiation between Homo- and Alternating AT DNA by Sterically Restricted Phosphonium Dyes. Chem. A Eur. J. 2012, 18, 3859–3864. [Google Scholar] [CrossRef]
- Crnolatac, I.; Tumir, L.M.; Lesev, N.Y.; Vasilev, A.A.; Deligeorgiev, T.G.; Mišković, K.; Glavaš-Obrovac, L.; Vugrek, O.; Piantanida, I. Probing the Structural Properties of DNA/RNA Grooves with Sterically Restricted Phosphonium Dyes: Screening of Dye Cytotoxicity and Uptake. ChemMedChem 2013, 8, 1093–1103. [Google Scholar] [CrossRef]
- Rožman, A.; Crnolatac, I.; Deligeorgiev, T.; Piantanida, I. Strong impact of chloro substituent on TOTO and YOYO ds-DNA/RNA sensing. J. Lumin. 2019, 205, 87–96. [Google Scholar] [CrossRef]
- Vasilev, A.; Lesev, N.; Dimitrova, S.; Nedelcheva-Veleva, M.; Stoynov, S.; Angelova, S. Bright fluorescent dsDNA probes: Novel polycationic asymmetric monomethine cyanine dyes based on thiazolopyridine-quinolinium chromophore. Coloration Technol. 2015, 131, 94–103. [Google Scholar] [CrossRef]
- Deligeorgiev, T.; Gadjev, N.; Vasilev, A.; Drexhage, K.H.; Yarmoluk, S.M. Synthesis of novel monomeric and homodimeric cyanine dyes with thioacetyl substituents for nucleic acid detection. Dye. Pigment. 2007, 72, 28–32. [Google Scholar] [CrossRef]
- Deligeorgiev, T.; Vasilev, A.; Drexhage, K.-H. Synthesis of novel monomeric cyanine dyes containing 2-hydroxypropyl and 3-chloro-2-hydroxypropyl substituents—Noncovalent labels for nucleic acids. Dye. Pigment. 2007, 73, 69–75. [Google Scholar] [CrossRef]
- Vassilev, A.; Dikova, I.; Deligeorgiev, T.; Drexhage, K.H. Quaternization of N-Heterocycles with Propylene Oxide and Epichlorohydrin. Synth. Commun. 2004, 34, 2539–2547. [Google Scholar] [CrossRef]
- Chaires, J.B.; Dattagupta, N.; Crothers, D.M. Studies on interaction of anthracycline antibiotics and deoxyribonucleic acid: Equilibrium binding studies on interaction of daunomycin with deoxyribonucleic acid. Biochemistry 1982, 21, 3933–3940. [Google Scholar] [CrossRef]
- Chalikian, T.V.; Völker, J.; Plum, G.E.; Breslauer, K.J. A more unified picture for the thermodynamics of nucleic acid duplex melting: A characterization by calorimetric and volumetric techniques. Proc. Natl. Acad. Sci. USA 1999, 96, 7853–7858. [Google Scholar] [CrossRef] [Green Version]
- Wheelhouse, R.T.; Chaires, J.B. Drug binding to DNA x RNA hybrid structures. Methods Mol. Biol. 2010, 613, 55–70. [Google Scholar]
- Ragazzon, P.; Chaires, J.B. Use of competition dialysis in the discovery of G-quadruplex selective ligands. Methods 2007, 43, 313–323. [Google Scholar] [CrossRef] [Green Version]
- Scatchard, G. The attractions of proteins for small molecules and ions. Ann. N. Y. Acad. Sci. 1949, 51, 660–672. [Google Scholar] [CrossRef]
- McGhee, J.D.; von Hippel, P.H. Theoretical aspects of DNA-protein interactions: Co-operative and non-co-operative binding of large ligands to a one-dimensional homogeneous lattice. J. Mol. Biol. 1974, 86, 469–489. [Google Scholar] [CrossRef]
- Boyd, M.R.; Paull, K.D. Some practical considerations and applications of the national cancer institute in vitro anticancer drug discovery screen. Drug Dev. Res. 1995, 34, 91–109. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Nygren, J.; Svanvik, N.; Kubista, M. The interactions between the fluorescent dye thiazole orange and DNA. Biopolymers 1998, 46, 39–51. [Google Scholar] [CrossRef]
- Nygren, J.; Andrade, J.M.; Kubista, M. Characterization of a Single Sample by Combining Thermodynamic and Spectroscopic Information in Spectral Analysis. Anal. Chem. 1996, 68, 1706–1710. [Google Scholar] [CrossRef]
- Herz, A.H. Dye-dye interactions of cyanines in solution and at agbr surfaces. Photogr. Sci. Eng. 1974, 18, 323–335. [Google Scholar]
- Joseph, M.J.; Taylor, J.C.; McGown, L.B.; Pitner, J.B.; Linn, C.P. Spectroscopic studies of YO and YOYO fluorescent dyes in a thrombin-binding DNA ligand. Biospectroscopy 1996, 2, 173–183. [Google Scholar] [CrossRef]
- Mergny, J.L.; Lacroix, L. Analysis of thermal melting curves. Oligonucleotides 2003, 13, 515–537. [Google Scholar] [CrossRef]
- Crothers, D.M. Calculation of binding isotherms for heterogeneous polymers. Biopolymers 1968, 6, 575–584. [Google Scholar] [CrossRef]
- Wakelin, L.P.G. Polyfunctional DNA intercalating agents. Med. Res. Rev. 1986, 6, 275–340. [Google Scholar] [CrossRef]
- Mao, F.; Leung, W.-Y.; Xin, X. Characterization of EvaGreen and the implication of its physicochemical properties for qPCR applications. BMC Biotechnol. 2007, 7, 76. [Google Scholar]
- Eriksson, M.; Nordén, B. Linear and circular dichroism of drug-nucleic acid complexes. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2001; Volume 340, pp. 68–98. [Google Scholar]
- Berova, N.; Nakanishi, K.; Woody, R. Circular Dichroism Principles and Applications, 2nd ed.; Wiley-VCH: New York NY, USA, 2000. [Google Scholar]
- Rodger, A.; Norden, B. Circular Dichroism and Linear Dichroism, Chapter 2; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Garbett, N.C.; Ragazzon, P.A.; Chaires, J.B. Circular dichroism to determine binding mode and affinity of ligand–DNA interactions. Nat. Protoc. 2007, 2, 3166–3172. [Google Scholar] [CrossRef] [PubMed]
- Šmidlehner, T.; Piantanida, I.; Pescitelli, G. Polarization spectroscopy methods in the determination of interactions of small molecules with nucleic acids—tutorial. Beilstein J. Org. Chem. 2018, 14, 84–105. [Google Scholar] [CrossRef] [PubMed]
- Garoff, R.A.; Litzinger, E.A.; Connor, R.E.; Fishman, I.; Armitage, B.A. Helical Aggregation of Cyanine Dyes on DNA Templates: Effect of Dye Structure on Formation of Homo- and Heteroaggregates. Langmuir 2002, 18, 6330–6337. [Google Scholar] [CrossRef]
- Tanaka, T.; Ishitobi, M.; Aoyama, T.; Matsumoto, S. Highly Oriented J-Aggregates of Nitroazo Dye and Its Surface-Induced Chromism. Langmuir 2016, 32, 4710–4718. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yang, C.-X.; Chen, L.-G.; Yan, X.-P. Dual-stimuli responsive and reversibly activatable theranostic nanoprobe for precision tumor-targeting and fluorescence-guided photothermal therapy. Nat. Commun. 2017, 8, 14998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummen, G.P.C. Fluorescent Probes and Fluorescence (Microscopy) Techniques—Illuminating Biological and Biomedical Research. Molecules 2012, 17, 14067–14090. [Google Scholar] [CrossRef] [Green Version]
- Trinquet, E.; Mathis, G. Fluorescence technologies for the investigation of chemical libraries. Mol. Biosyst. 2006, 2, 380–387. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ΔTm/°C | ||||
|---|---|---|---|---|
| Compound | r | ct-DNA | Poly dAdT–Poly dAdT | Poly A–Poly U |
| 4a | 0.1 0.2 0.3 | >20 c | 5.5 28.6 31.6 | 19.2 24.0 26.0 |
| 4b | 0.1 0.2 0.3 | 2.2 5.3 8.6 | 2.7 8.9 12.4 | 1.3 3.0 3.4 |
| 4c | 0.05 0.1 0.2 0.3 | 3.95 12.3 >15 c >15 c | 2.1 >20 c >20 c d | 9.1 10.6 −19.1 −16.8 |
| 4d | 0.05 0.1 0.2 0.3 | 1.0 2.3 4.5 6.4 | 1.9 5.7 10.7 11.9 | 0.5 0 0 0 |
| 4e | 0.05 0.1 0.2 0.3 | 0.7 2.7 5.2d | 1.5 4.2 7.1 10.0 | 0 0.5 0.6 0 |
| log Ks | Δ Icalc b | ||
|---|---|---|---|
| 4a λexc 510 nm | ct–DNA | 6.57 | 377 |
| poly dA–poly dT | 6.41 | 712 | |
| poly dA-dT–poly dA-dT | 6.65 | 358 | |
| poly rA–poly rU | 6.11 | 1573 | |
| poly dG–poly dC | 6.79 | 561 | |
| poly dG-dC–poly dG-dC | 6.98 | 895 | |
| poly rG–poly rC | 5.23 | 799 | |
| 4b λexc 478 nm | ct–DNA | 6.93 | 157 |
| poly dA–poly dT | 6.19 | 44 | |
| poly dA-dT–poly dA-dT | 7.23 | 75 | |
| poly rA–poly rU | 7.15 | 158 | |
| poly dG–poly dC | 7.55 | 114 | |
| poly dG-dC–poly dG-dC | 7.64 | 65 | |
| poly rG–poly rC | 4.94 | 618 | |
| 4c λexc 488 nm | ct–DNA | 6.62 | 386 |
| poly dA–poly dT | 6.23 | 151 | |
| poly dA-dT–poly dA-dT | 6.68 | 100 | |
| poly rA–poly rU | 6.22 | 255 | |
| poly dG–poly dC | 6.78 | 298 | |
| poly dG-dC–poly dG-dC | 7.04 | 485 | |
| poly rG–poly rC | 4.76 | 63 | |
| 4d λexc 478 nm | ct–DNA | 6.87 | 340 |
| poly dA–poly dT | 5.89 | 143 | |
| poly dA-dT–poly dA-dT | 6.98 | 117 | |
| poly rA–poly rU | 6.14 | 78 | |
| poly dG–poly dC | 6.63 | 120 | |
| poly dG-dC–poly dG-dC | 7.20 | 285 | |
| poly rG–poly rC | 4.79 | 572 | |
| 4e λexc 452 nm | ct–DNA | 5.94 | 216 |
| poly dA–poly dT | 5.91 | 329 | |
| poly dA-dT–poly dA-dT | 6.05 | 439 | |
| poly rA–poly rU | 6.14 | 35 | |
| poly dG–poly dC | 7.41 | 155 | |
| poly dG-dC–poly dG-dC | 7.40 | 285 | |
| poly rG–poly rC | 4.95 | 218 |
| GI50 a (μM) | |||
|---|---|---|---|
| Compound | Cell Lines | ||
| HCT 116 | MCF-7 | H460 | |
| 4a | 6 ± 3 | 5 ± 4 | 10 ± 7 |
| 4b | 4.5 ± 0.7 | 2 ± 0.6 | 7 ± 2 |
| 4c | 2.2 ± 0.6 | 3.2 ± 1.9 | 1.9 ± 1.0 |
| 4d | 2.9 ± 0.1 | 3.5 ± 1.2 | 6.1 ± 4.4 |
| 4e | 3.3 ± 0.2 | 9.7 ± 2.9 | 4.6 ± 3.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikulin, I.; Ljubić, I.; Piantanida, I.; Vasilev, A.; Mondeshki, M.; Kandinska, M.; Uzelac, L.; Martin-Kleiner, I.; Kralj, M.; Tumir, L.-M. Polycationic Monomeric and Homodimeric Asymmetric Monomethine Cyanine Dyes with Hydroxypropyl Functionality—Strong Affinity Nucleic Acids Binders. Biomolecules 2021, 11, 1075. https://doi.org/10.3390/biom11081075
Mikulin I, Ljubić I, Piantanida I, Vasilev A, Mondeshki M, Kandinska M, Uzelac L, Martin-Kleiner I, Kralj M, Tumir L-M. Polycationic Monomeric and Homodimeric Asymmetric Monomethine Cyanine Dyes with Hydroxypropyl Functionality—Strong Affinity Nucleic Acids Binders. Biomolecules. 2021; 11(8):1075. https://doi.org/10.3390/biom11081075
Chicago/Turabian StyleMikulin, Ivana, Ivana Ljubić, Ivo Piantanida, Aleksey Vasilev, Mihail Mondeshki, Meglena Kandinska, Lidija Uzelac, Irena Martin-Kleiner, Marijeta Kralj, and Lidija-Marija Tumir. 2021. "Polycationic Monomeric and Homodimeric Asymmetric Monomethine Cyanine Dyes with Hydroxypropyl Functionality—Strong Affinity Nucleic Acids Binders" Biomolecules 11, no. 8: 1075. https://doi.org/10.3390/biom11081075
APA StyleMikulin, I., Ljubić, I., Piantanida, I., Vasilev, A., Mondeshki, M., Kandinska, M., Uzelac, L., Martin-Kleiner, I., Kralj, M., & Tumir, L.-M. (2021). Polycationic Monomeric and Homodimeric Asymmetric Monomethine Cyanine Dyes with Hydroxypropyl Functionality—Strong Affinity Nucleic Acids Binders. Biomolecules, 11(8), 1075. https://doi.org/10.3390/biom11081075