
Gatekeepers of the Gut: The Roles of Proteasomes at the Gastrointestinal Barrier

{kind=link}
{kind=link}
{kind=link}
Abstract
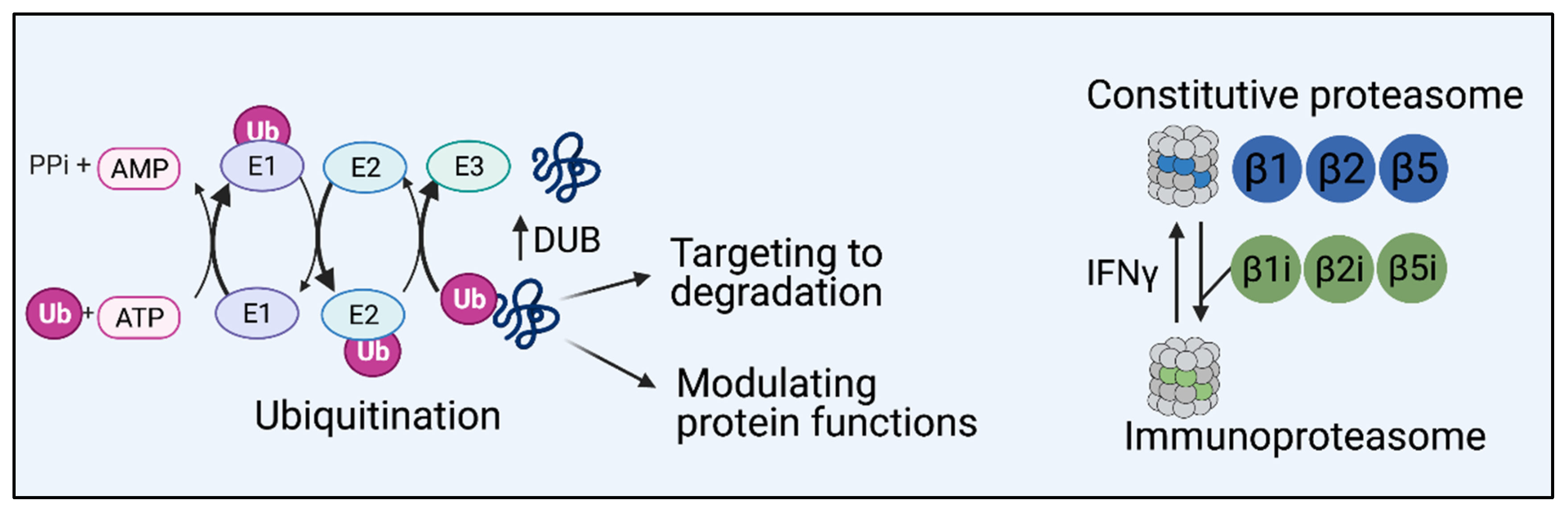
1. Introduction
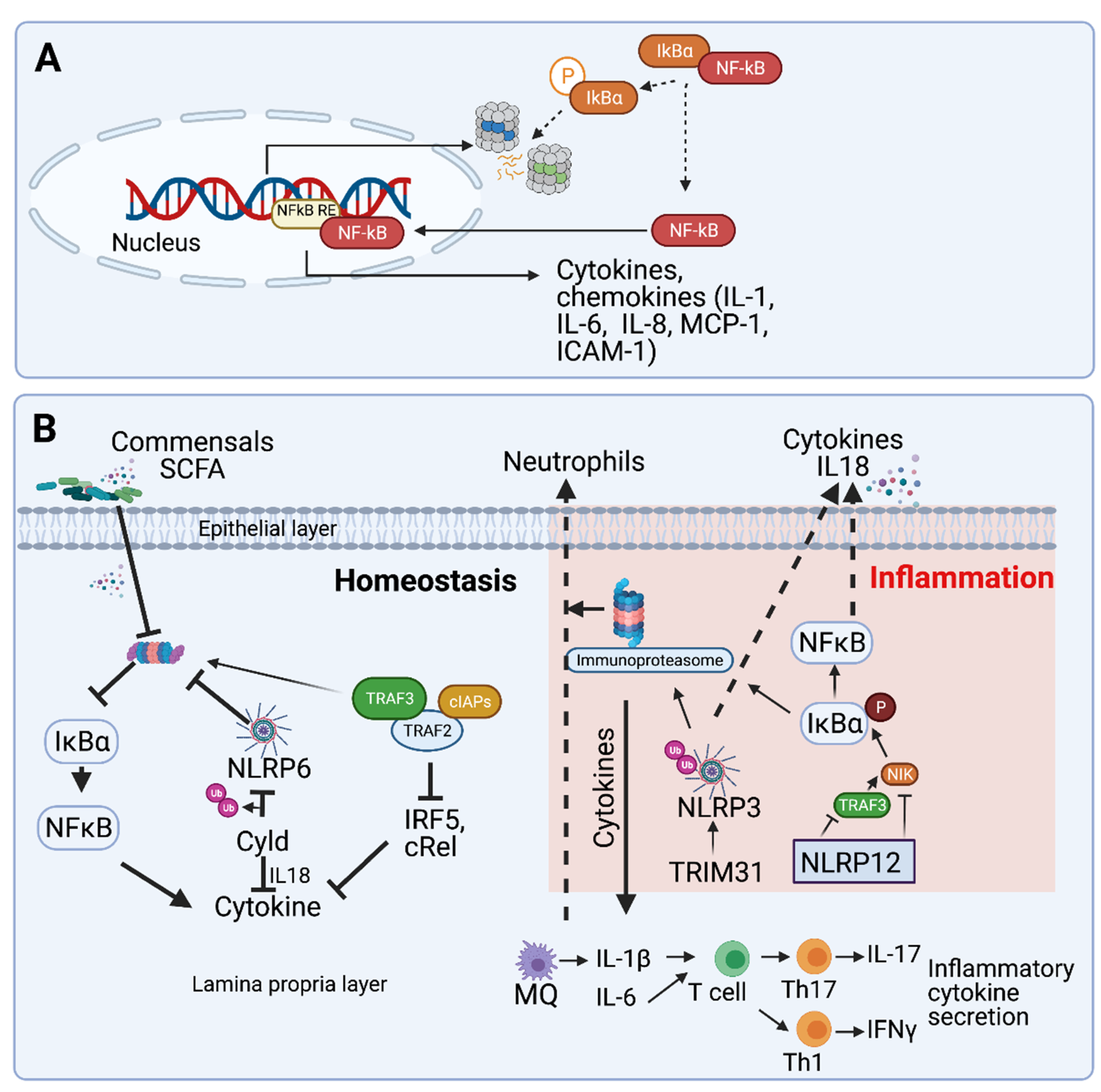
2. UPS-Dependent Gastrointestinal Inflammation
2.1. The Exploitation of Proteasomal Degradation by the Gut Microbiota and Enteric Pathogens
2.2. Proteasomes in Inflammatory Bowel Disease
2.3. Ubiquitin E3 Ligases in Gut Inflammation
2.3.1. Inflammasome Activation
2.3.2. TLR Activation
2.4. Proteasome Activity at the Immune Compartment of the Gut Barrier
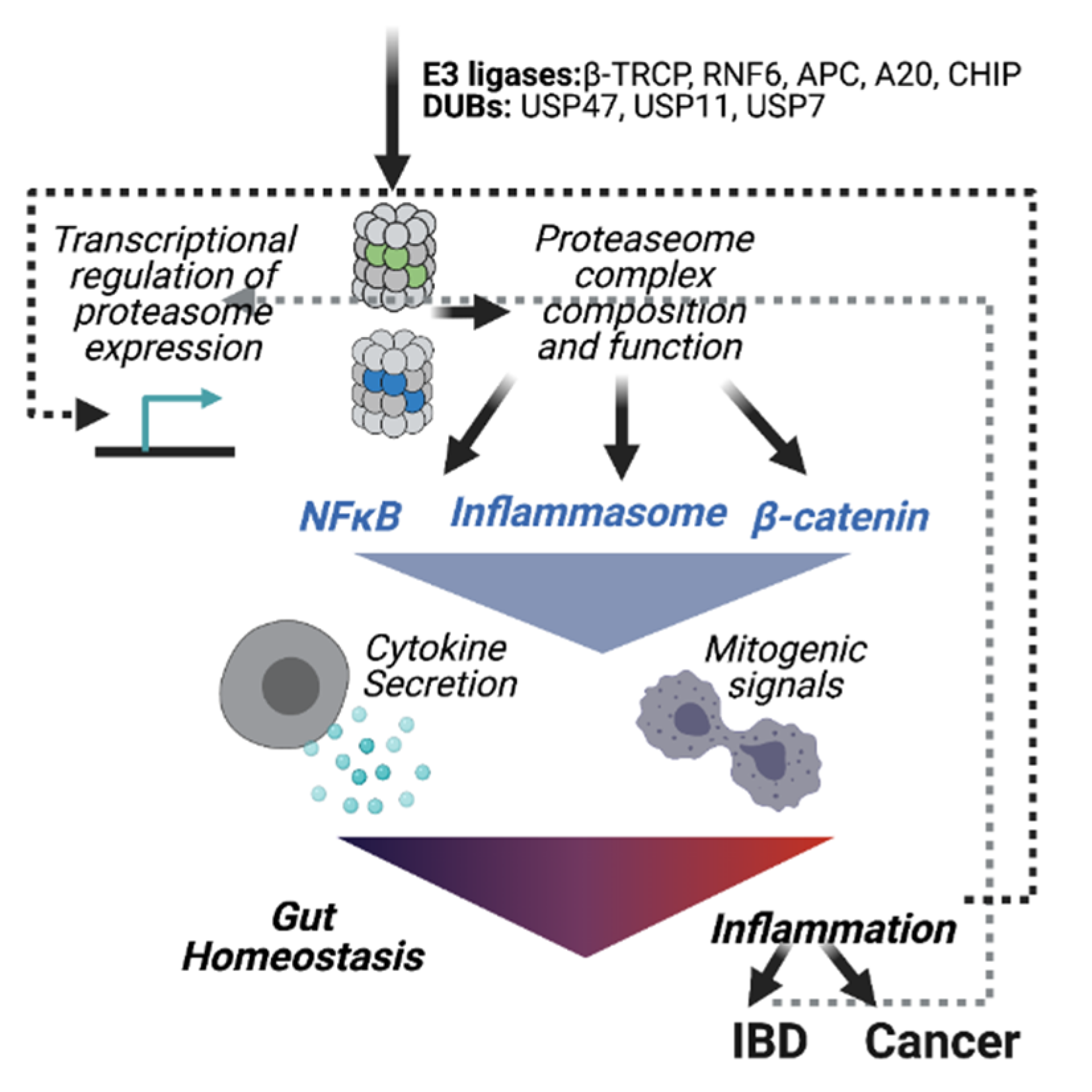
3. The UPS in Colorectal Cancer Development
Enzymes of the Ubiquitination Machinery in Colon Cancer
4. Concluding Remarks and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lechuga, S.; Ivanov, A.I. Disruption of the epithelial barrier during intestinal inflammation: Quest for new molecules and mechanisms. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1183–1194. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef] [PubMed]
- Tuganbaev, T.; Mor, U.; Bashiardes, S.; Liwinski, T.; Nobs, S.P.; Leshem, A.; Dori-Bachash, M.; Thaiss, C.A.; Pinker, E.Y.; Ratiner, K.; et al. Diet Diurnally Regulates Small Intestinal Microbiome-Epithelial-Immune Homeostasis and Enteritis. Cell 2020, 182, 1441–1459.e21. [Google Scholar] [CrossRef]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Vancamelbeke, M.; Vermeire, S. The intestinal barrier: A fundamental role in health and disease. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 821–834. [Google Scholar] [CrossRef]
- Swiatczak, B.; Cohen, I.R. Gut feelings of safety: Tolerance to the microbiota mediated by innate immune receptors. Microbiol. Immunol. 2015, 59, 573–585. [Google Scholar] [CrossRef]
- Mowat, A.M. To respond or not to respond—A personal perspective of intestinal tolerance. Nat. Rev. Immunol. 2018, 18, 405–415. [Google Scholar] [CrossRef]
- Meizlish, M.L.; Franklin, R.A.; Zhou, X.; Medzhitov, R. Tissue Homeostasis and Inflammation. Annu. Rev. Immunol. 2021, 39, 557–581. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Nowarski, R.; Thaiss, C.A.; Hu, B.; Jin, C.; Flavell, R.A. Inflammation-induced cancer: Crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 2013, 13, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Shalapour, S.; Karin, M. Pas de Deux: Control of Anti-tumor Immunity by Cancer-Associated Inflammation. Immunity 2019, 51, 15–26. [Google Scholar] [CrossRef]
- Niec, R.E.; Rudensky, A.Y.; Fuchs, E. Inflammatory adaptation in barrier tissues. Cell 2021, 184, 3361–3375. [Google Scholar] [CrossRef]
- Zmora, N.; Levy, M.; Pevsner-Fischer, M.; Elinav, E. Inflammasomes and intestinal inflammation. Mucosal Immunol. 2017. [Google Scholar] [CrossRef]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef]
- Pasparakis, M. Regulation of tissue homeostasis by NF-κB signalling: Implications for inflammatory diseases. Nat. Rev. Immunol. 2009, 9, 778–788. [Google Scholar] [CrossRef]
- Kumar, A.; Wu, H.; Collier-Hyams, L.S.; Hansen, J.M.; Li, T.; Yamoah, K.; Pan, Z.Q.; Jones, D.P.; Neish, A.S. Commensal bacteria modulate cullin-dependent signaling via generation of reactive oxygen species. EMBO J. 2007, 26, 4457–4466. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Torres, R.J.; Chamaillard, M. The Ubiquitin Code of NODs Signaling Pathways in Health and Disease. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Lee, K.H.; Biswas, A.; Liu, Y.J.; Kobayashi, K.S. Proteasomal degradation of Nod2 protein mediates tolerance to bacterial cell wall components. J. Biol. Chem. 2012, 287, 39800–39811. [Google Scholar] [CrossRef] [PubMed]
- Vlantis, K.; Polykratis, A.; Welz, P.S.; Van Loo, G.; Pasparakis, M.; Wullaert, A. TLR-independent anti-inflammatory function of intestinal epithelial TRAF6 signalling prevents DSS-induced colitis in mice. Gut 2016, 65, 935–943. [Google Scholar] [CrossRef]
- Kathania, M.; Tsakem, E.L.; Theiss, A.L.; Venuprasad, K. Gut Microbiota Contributes to Spontaneous Colitis in E3 Ligase Itch-Deficient Mice. J. Immunol. 2020, 204, 2277–2284. [Google Scholar] [CrossRef]
- Cleynen, I.; Vazeille, E.; Artieda, M.; Verspaget, H.W.; Szczypiorska, M.; Bringer, M.-A.; Lakatos, P.L.; Seibold, F.; Parnell, K.; Weersma, R.K.; et al. Genetic and microbial factors modulating the ubiquitin proteasome system in inflammatory bowel disease. Gut 2014, 63, 1265–1274. [Google Scholar] [CrossRef]
- Kanarek, N.; Ben-Neriah, Y. Regulation of NF-κB by ubiquitination and degradation of the IκBs. Immunol. Rev. 2012, 246, 77–94. [Google Scholar] [CrossRef]
- Kravtsova-Ivantsiv, Y.; Ciechanover, A. The ubiquitin-proteasome system and activation of NF-κB: Involvement of the ubiquitin ligase KPC1 in p105 processing and tumor suppression. Mol. Cell. Oncol. 2015, 2, e1054552. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Livneh, I.; Cohen-Kaplan, V.; Cohen-Rosenzweig, C.; Avni, N.; Ciechanover, A. The life cycle of the 26S proteasome: From birth, through regulation and function, and onto its death. Cell Res. 2016, 26, 869–885. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.L.; Ciechanover, A. Targeting proteins for destruction by the ubiquitin system: Implications for human pathobiology. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 73–96. [Google Scholar] [CrossRef]
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef]
- Bassermann, F.; Eichner, R.; Pagano, M. The ubiquitin proteasome system—Implications for cell cycle control and the targeted treatment of cancer. Biochim. Biophys. Acta 2014, 1843, 150–162. [Google Scholar] [CrossRef]
- Calise, J.; Powell, S.R. The ubiquitin proteasome system and myocardial ischemia. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H337–H349. [Google Scholar] [CrossRef] [PubMed]
- Durairaj, G.; Kaiser, P. The 26S proteasome and initiation of gene transcription. Biomolecules 2014, 4, 827–847. [Google Scholar] [CrossRef]
- Gupta, I.; Singh, K.; Varshney, N.K.; Khan, S. Delineating Crosstalk Mechanisms of the Ubiquitin Proteasome System That Regulate Apoptosis. Front. Cell Dev. Biol. 2018, 6, 11. [Google Scholar] [CrossRef]
- Goetzke, C.C.; Ebstein, F.; Kallinich, T. Role of Proteasomes in Inflammation. J. Clin. Med. 2021, 10, 1783. [Google Scholar] [CrossRef] [PubMed]
- Sitaraman, S.; Na, C.-L.; Yang, L.; Filuta, A.; Bridges, J.P.; Weaver, T.E. Proteasome dysfunction in alveolar type 2 epithelial cells is associated with acute respiratory distress syndrome. Sci. Rep. 2019, 9, 12509. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Gramm, C.; Rothstein, L.; Clark, K.; Stein, R.; Dick, L.; Hwang, D.; Goldberg, A.L. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 1994, 78, 761–771. [Google Scholar] [CrossRef]
- Rock, K.L.; York, I.A.; Saric, T.; Goldberg, A.L. Protein degradation and the generation of MHC class I-presented peptides. Adv. Immunol. 2002, 80, 1–70. [Google Scholar]
- Ferrington, D.A.; Gregerson, D.S. Immunoproteasomes: Structure, function, and antigen presentation. Prog. Mol. Biol. Transl. Sci. 2012, 109, 75–112. [Google Scholar] [CrossRef]
- Ciechanover, A.; Schwartz, A.L. The ubiquitin-proteasome pathway: The complexity and myriad functions of proteins death. Proc. Natl. Acad. Sci. USA 1998, 95, 2727–2730. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef]
- Besche, H.C.; Peth, A.; Goldberg, A.L. Getting to first base in proteasome assembly. Cell 2009, 138, 25–28. [Google Scholar] [CrossRef]
- Bard, J.A.M.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef]
- Groll, M.; Huber, R. Substrate access and processing by the 20S proteasome core particle. Int. J. Biochem. Cell Biol. 2003, 35, 606–616. [Google Scholar] [CrossRef]
- Huber, E.M.; Basler, M.; Schwab, R.; Heinemeyer, W.; Kirk, C.J.; Groettrup, M.; Groll, M. Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell 2012, 148, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Kniepert, A.; Groettrup, M. The unique functions of tissue-specific proteasomes. Trends Biochem. Sci. 2014, 39, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, E.Z.; Che, J.W.; York, I.; Escobar, H.; Reyes-Vargas, E.; Delgado, J.C.; Welsh, R.M.; Karow, M.L.; Murphy, A.J.; Valenzuela, D.M.; et al. Mice completely lacking immunoproteasomes show major changes in antigen presentation. Nat. Immunol. 2012, 13, 129–135. [Google Scholar] [CrossRef]
- Winter, M.B.; La Greca, F.; Arastu-Kapur, S.; Caiazza, F.; Cimermancic, P.; Buchholz, T.J.; Anderl, J.L.; Ravalin, M.; Bohn, M.F.; Sali, A.; et al. Immunoproteasome functions explained by divergence in cleavage specificity and regulation. Elife 2017, 6. [Google Scholar] [CrossRef]
- Ebstein, F.; Kloetzel, P.M.; Krüger, E.; Seifert, U. Emerging roles of immunoproteasomes beyond MHC class i antigen processing. Cell. Mol. Life Sci. 2012, 69, 2543–2558. [Google Scholar] [CrossRef]
- Murata, S.; Takahama, Y.; Kasahara, M.; Tanaka, K. The immunoproteasome and thymoproteasome: Functions, evolution and human disease. Nat. Immunol. 2018, 19, 923–931. [Google Scholar] [CrossRef]
- Tien, M.-T.; Girardin, S.E.; Regnault, B.; Le Bourhis, L.; Dillies, M.-A.; Coppée, J.-Y.; Bourdet-Sicard, R.; Sansonetti, P.J.; Pédron, T. Anti-Inflammatory Effect of Lactobacillus casei on Shigella -Infected Human Intestinal Epithelial Cells. J. Immunol. 2006, 176, 1228–1237. [Google Scholar] [CrossRef]
- Mukherjee, S.; Kumar, R.; Tsakem Lenou, E.; Basrur, V.; Kontoyiannis, D.L.; Ioakeimidis, F.; Mosialos, G.; Theiss, A.L.; Flavell, R.A.; Venuprasad, K. Deubiquitination of NLRP6 inflammasome by Cyld critically regulates intestinal inflammation. Nat. Immunol. 2020, 21, 626–635. [Google Scholar] [CrossRef]
- Jin, J.; Xiao, Y.; Hu, H.; Zou, Q.; Li, Y.; Gao, Y.; Ge, W.; Cheng, X.; Sun, S.-C. Proinflammatory TLR signalling is regulated by a TRAF2-dependent proteolysis mechanism in macrophages. Nat. Commun. 2015, 6, 5930. [Google Scholar] [CrossRef]
- Petrof, E.O.; Kojima, K.; Ropeleski, M.J.; Musch, M.W.; Tao, Y.; De Simone, C.; Chang, E.B. Probiotics inhibit nuclear factor-κB and induce heat shock proteins in colonic epithelial cells through proteasome inhibition. Gastroenterology 2004, 127, 1474–1487. [Google Scholar] [CrossRef]
- Neish, A.S. Prokaryotic Regulation of Epithelial Responses by Inhibition of Ikappa B-alpha Ubiquitination. Science 2000, 289, 1560–1563. [Google Scholar] [CrossRef]
- Sun, S.-C. CYLD: A tumor suppressor deubiquitinase regulating NF-kappaB activation and diverse biological processes. Cell Death Differ. 2010, 17, 25–34. [Google Scholar] [CrossRef]
- Zhang, L.; Wei, N.; Cui, Y.; Hong, Z.; Liu, X.; Wang, Q.; Li, S.; Liu, H.; Yu, H.; Cai, Y.; et al. The deubiquitinase CYLD is a specific checkpoint of the STING antiviral signaling pathway. PLoS Pathog. 2018, 14. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Khateb, A.; Li, Y.; Tinoco, R.; Zhang, T.; Bar-Yoseph, H.; Tam, M.A.; Chowers, Y.; Sabo, E.; Gerassy-Vainberg, S.; et al. Regulation of S100A8 Stability by RNF5 in Intestinal Epithelial Cells Determines Intestinal Inflammation and Severity of Colitis. Cell Rep. 2018, 24, 3296–3311.e6. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhou, L.; Ji, L.; Chen, F.; Fortmann, K.; Zhang, K.; Liu, Q.; Li, K.; Wang, W.; Wang, H.; et al. The REGγ-proteasome forms a regulatory circuit with IκBɛ and NFκB in experimental colitis. Nat. Commun. 2016, 7, 10761. [Google Scholar] [CrossRef]
- Song, H.; Liu, B.; Huai, W.; Yu, Z.; Wang, W.; Zhao, J.; Han, L.; Jiang, G.; Zhang, L.; Gao, C.; et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat. Commun. 2016, 7, 13727. [Google Scholar] [CrossRef]
- Visekruna, A.; Joeris, T.; Seidel, D.; Kroesen, A.; Loddenkemper, C.; Zeitz, M.; Kaufmann, S.H.E.; Schmidt-Ullrich, R.; Steinhoff, U. Proteasome-mediated degradation of IkappaBalpha and processing of p105 in Crohn disease and ulcerative colitis. J. Clin. Investig. 2006, 116, 3195–3203. [Google Scholar] [CrossRef]
- Inoue, S.; Nakase, H.; Matsuura, M.; Mikami, S.; Ueno, S.; Uza, N.; Chiba, T. The effect of proteasome inhibitor MG132 on experimental inflammatory bowel disease. Clin. Exp. Immunol. 2009, 156, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Griffin, T.A.; Nandi, D.; Cruz, M.; Fehling, H.J.; Van Kaer, L.; Monaco, J.J.; Colbert, R.A. Immunoproteasome Assembly: Cooperative Incorporation of Interferon γ (IFN-γ)–inducible Subunits. J. Exp. Med. 1998, 187, 97–104. [Google Scholar] [CrossRef]
- Visekruna, A.; Joeris, T.; Schmidt, N.; Lawrenz, M.; Ritz, J.-P.; Buhr, H.J.; Steinhoff, U. Comparative expression analysis and characterization of 20S proteasomes in human intestinal tissues: The proteasome pattern as diagnostic tool for IBD patients. Inflamm. Bowel Dis. 2009, 15, 526–533. [Google Scholar] [CrossRef]
- Fitzpatrick, L.R.; Small, J.S.; Poritz, L.S.; McKenna, K.J.; Koltun, W.A. Enhanced intestinal expression of the proteasome subunit low molecular mass polypeptide 2 in patients with inflammatory bowel disease. Dis. Colon Rectum 2007, 50, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Jang, E.R.; Lee, N.-R.; Han, S.; Wu, Y.; Sharma, L.K.; Carmony, K.C.; Marks, J.; Lee, D.-M.; Ban, J.-O.; Wehenkel, M.; et al. Revisiting the role of the immunoproteasome in the activation of the canonical NF-κB pathway. Mol. Biosyst. 2012, 8, 2295–2302. [Google Scholar] [CrossRef]
- Collett, A.; Higgs, N.B.; Gironella, M.; Zeef, L.A.H.; Hayes, A.; Salmo, E.; Haboubi, N.; Iovanna, J.L.; Carlson, G.L.; Warhurst, G. Early molecular and functional changes in colonic epithelium that precede increased gut permeability during colitis development in mdr1a(-/-) mice. Inflamm. Bowel Dis. 2008, 14, 620–631. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, L.R.; Khare, V.; Small, J.S.; Koltun, W.A. Dextran sulfate sodium-induced colitis is associated with enhanced low molecular mass polypeptide 2 (LMP2) expression and is attenuated in LMP2 knockout mice. Dig. Dis. Sci. 2006, 51, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Basler, M.; Dajee, M.; Moll, C.; Groettrup, M.; Kirk, C.J. Prevention of experimental colitis by a selective inhibitor of the immunoproteasome. J. Immunol. 2010, 185, 634–641. [Google Scholar] [CrossRef]
- Schmidt, N.; Gonzalez, E.; Visekruna, A.; Kühl, A.A.; Loddenkemper, C.; Mollenkopf, H.; Kaufmann, S.H.E.; Steinhoff, U.; Joeris, T. Targeting the proteasome: Partial inhibition of the proteasome by bortezomib or deletion of the immunosubunit LMP7 attenuates experimental colitis. Gut 2010, 59, 896–906. [Google Scholar] [CrossRef]
- Coëffier, M.; Gloro, R.; Boukhettala, N.; Aziz, M.; Lecleire, S.; Vandaele, N.; Antonietti, M.; Savoye, G.; Bôle-Feysot, C.; Déchelotte, P.; et al. Increased proteasome-mediated degradation of occludin in irritable bowel syndrome. Am. J. Gastroenterol. 2010, 105, 1181–1188. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Zaki, M.H.; Boyd, K.L.; Vogel, P.; Kastan, M.B.; Lamkanfi, M.; Kanneganti, T.-D. The NLRP3 Inflammasome Protects against Loss of Epithelial Integrity and Mortality during Experimental Colitis. Immunity 2010, 32, 379–391. [Google Scholar] [CrossRef]
- Chung, I.-C.; Yuan, S.-N.; OuYang, C.-N.; Lin, H.-C.; Huang, K.-Y.; Chen, Y.-J.; Chung, A.-K.; Chu, C.-L.; Ojcius, D.M.; Chang, Y.-S.; et al. Src-family kinase-Cbl axis negatively regulates NLRP3 inflammasome activation. Cell Death Dis. 2018, 9, 1109. [Google Scholar] [CrossRef] [PubMed]
- Leaphart, C.L.; Cavallo, J.; Gribar, S.C.; Cetin, S.; Li, J.; Branca, M.F.; Dubowski, T.D.; Sodhi, C.P.; Hackam, D.J. A Critical Role for TLR4 in the Pathogenesis of Necrotizing Enterocolitis by Modulating Intestinal Injury and Repair. J. Immunol. 2007, 179, 4808–4820. [Google Scholar] [CrossRef] [PubMed]
- Afrazi, A.; Sodhi, C.P.; Good, M.; Jia, H.; Siggers, R.; Yazji, I.; Ma, C.; Neal, M.D.; Prindle, T.; Grant, Z.S.; et al. Intracellular heat shock protein-70 negatively regulates TLR4 signaling in the newborn intestinal epithelium. J. Immunol. 2012, 188, 4543–4557. [Google Scholar] [CrossRef]
- De Souza, H.S.P.; West, G.A.; Rebert, N.; de la Motte, C.; Drazba, J.; Fiocchi, C. Increased levels of survivin, via association with heat shock protein 90, in mucosal T cells from patients with Crohn’s disease. Gastroenterology 2012, 143, 1017–1026.e9. [Google Scholar] [CrossRef]
- Qiao, Y.Q.; Shen, J.; Gu, Y.; Tong, J.L.; Xu, X.T.; Huang, M.L.; Ran, Z.H. Gene expression of tumor necrosis factor receptor associated-factor (TRAF)-1 and TRAF-2 in inflammatory bowel disease. J. Dig. Dis. 2013, 14, 244–250. [Google Scholar] [CrossRef]
- Todoric, J.; Karin, M. The Fire within: Cell-Autonomous Mechanisms in Inflammation-Driven Cancer. Cancer Cell 2019, 35, 714–720. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Hibino, S.; Kawazoe, T.; Kasahara, H.; Itoh, S.; Ishimoto, T.; Sakata-Yanagimoto, M.; Taniguchi, K. Inflammation-Induced Tumorigenesis and Metastasis. Int. J. Mol. Sci. 2021, 22, 5421. [Google Scholar] [CrossRef]
- Zhang, H.; Ramakrishnan, S.K.; Triner, D.; Centofanti, B.; Maitra, D.; Győrffy, B.; Sebolt-Leopold, J.S.; Dame, M.K.; Varani, J.; Brenner, D.E.; et al. Tumor-selective proteotoxicity of verteporfin inhibits colon cancer progression independently of YAP1. Sci. Signal. 2015, 8, ra98. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, K.; Jin, Y.; Sheng, X. Endoplasmic reticulum proteostasis control and gastric cancer. Cancer Lett. 2019, 449, 263–271. [Google Scholar] [CrossRef]
- Sekine, H.; Okazaki, K.; Kato, K.; Alam, M.M.; Shima, H.; Katsuoka, F.; Tsujita, T.; Suzuki, N.; Kobayashi, A.; Igarashi, K.; et al. O -GlcNAcylation Signal Mediates Proteasome Inhibitor Resistance in Cancer Cells by Stabilizing NRF1. Mol. Cell. Biol. 2018, 38. [Google Scholar] [CrossRef]
- Arlt, A.; Bauer, I.; Schafmayer, C.; Tepel, J.; Müerköster, S.S.; Brosch, M.; Röder, C.; Kalthoff, H.; Hampe, J.; Moyer, M.P.; et al. Increased proteasome subunit protein expression and proteasome activity in colon cancer relate to an enhanced activation of nuclear factor E2-related factor 2 (Nrf2). Oncogene 2009, 28, 3983–3996. [Google Scholar] [CrossRef]
- Osburn, W.O.; Kensler, T.W. Nrf2 signaling: An adaptive response pathway for protection against environmental toxic insults. Mutat. Res. 2008, 659, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Waku, T.; Nakamura, N.; Koji, M.; Watanabe, H.; Katoh, H.; Tatsumi, C.; Tamura, N.; Hatanaka, A.; Hirose, S.; Katayama, H.; et al. NRF3-POMP-20S Proteasome Assembly Axis Promotes Cancer Development via Ubiquitin-Independent Proteolysis of p53 and Retinoblastoma Protein. Mol. Cell. Biol. 2020, 40, e00597-19. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.; Minis, A.; Lalazar, G.; Rodriguez, J.; Steller, H. PSMD5 inactivation promotes 26S proteasome assembly during colorectal tumor progression. Cancer Res. 2018, 78, 3458–3468. [Google Scholar] [CrossRef]
- Kimura, H.; Caturegli, P.; Takahashi, M.; Suzuki, K. New Insights into the Function of the Immunoproteasome in Immune and Nonimmune Cells. J. Immunol. Res. 2015, 2015, 541984. [Google Scholar] [CrossRef] [PubMed]
- Krüger, E.; Kloetzel, P.-M. Immunoproteasomes at the interface of innate and adaptive immune responses: Two faces of one enzyme. Curr. Opin. Immunol. 2012, 24, 77–83. [Google Scholar] [CrossRef]
- Koerner, J.; Brunner, T.; Groettrup, M. Inhibition and deficiency of the immunoproteasome subunit LMP7 suppress the development and progression of colorectal carcinoma in mice. Oncotarget 2017, 8, 50873–50888. [Google Scholar] [CrossRef]
- Vachharajani, N.; Joeris, T.; Luu, M.; Hartmann, S.; Pautz, S.; Jenike, E.; Pantazis, G.; Prinz, I.; Hofer, M.J.; Steinhoff, U.; et al. Prevention of colitis-associated cancer by selective targeting of immunoproteasome subunit LMP7. Oncotarget 2017, 8, 50447–50459. [Google Scholar] [CrossRef]
- Fellerhoff, B.; Gu, S.; Laumbacher, B.; Nerlich, A.G.; Weiss, E.H.; Glas, J.; Kopp, R.; Johnson, J.P.; Wank, R. The LMP7-K Allele of the Immunoproteasome Exhibits Reduced Transcript Stability and Predicts High Risk of Colon Cancer. Cancer Res. 2011, 71, 7145–7154. [Google Scholar] [CrossRef]
- Allen, I.C.; Wilson, J.E.; Schneider, M.; Lich, J.D.; Roberts, R.A.; Arthur, J.C.; Woodford, R.-M.T.; Davis, B.K.; Uronis, J.M.; Herfarth, H.H.; et al. NLRP12 Suppresses Colon Inflammation and Tumorigenesis through the Negative Regulation of Noncanonical NF-κB Signaling. Immunity 2012, 36, 742–754. [Google Scholar] [CrossRef]
- Hernández, A.R.; Klein, A.M.; Kirschner, M.W. Kinetic Responses of β-Catenin Specify the Sites of Wnt Control. Science 2012, 338, 1337–1340. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, W.; Evans, P.M.; Chen, X.; He, X.; Liu, C. Adenomatous Polyposis Coli (APC) Differentially Regulates β-Catenin Phosphorylation and Ubiquitination in Colon Cancer Cells. J. Biol. Chem. 2006, 281, 17751–17757. [Google Scholar] [CrossRef] [PubMed]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, Y.; Wong, C.C.; Zhang, J.; Dong, Y.; Li, X.; Kang, W.; Chan, F.K.L.; Sung, J.J.Y.; Yu, J. RNF6 Promotes Colorectal Cancer by Activating the Wnt/β-Catenin Pathway via Ubiquitination of TLE3. Cancer Res. 2018, 78, 1958–1971. [Google Scholar] [CrossRef] [PubMed]
- Abed, M.; Abed, M.; Heuberger, J.; Novak, R.; Zohar, Y.; Beltran Lopez, A.P.; Trausch-Azar, J.S.; Ilagan, M.X.G.; Benhamou, D.; Dittmar, G.; et al. RNF4-Dependent Oncogene Activation by Protein Stabilization. Cell Rep. 2016, 16, 3388–3400. [Google Scholar] [CrossRef]
- Glaeser, K.; Urban, M.; Fenech, E.; Voloshanenko, O.; Kranz, D.; Lari, F.; Christianson, J.C.; Boutros, M. ERAD-dependent control of the Wnt secretory factor Evi. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Xiao, Y.; Huang, Q.; Wu, Z.; Chen, W. Roles of protein ubiquitination in inflammatory bowel disease. Immunobiology 2020, 225, 152026. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ren, F.; Wang, Y.; Feng, Y.; Wang, D.; Jia, B.; Qiu, Y.; Wang, S.; Yu, J.; Sung, J.J.Y.; et al. CHIP/Stub1 functions as a tumor suppressor and represses NF-κB-mediated signaling in colorectal cancer. Carcinogenesis 2014, 35, 983–991. [Google Scholar] [CrossRef]
- Barbash, O.; Lee, E.K.; Diehl, J.A. Phosphorylation-dependent regulation of SCF Fbx4 dimerization and activity involves a novel component, 14-3-3. Oncogene 2011, 30, 1995–2002. [Google Scholar] [CrossRef][Green Version]
- Rape, M.; Kirschner, M.W. Autonomous regulation of the anaphase-promoting complex couples mitosis to S-phase entry. Nature 2004, 432, 588–595. [Google Scholar] [CrossRef]
- Yamano, H. APC/C: Current understanding and future perspectives. F1000Research 2019, 8, 725. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.Y.; Kwon, H.Y.; Park, K.H.; Kim, D.S. Anaphase-Promoting Complex 7 is a Prognostic Factor in Human Colorectal Cancer. Ann. Coloproctol. 2017, 33, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-Z.; Song, Y.; Zhang, H.-H.; Jin, B.-X.; Liu, Y.; Liu, W.-B.; Zhang, X.-D.; Du, R.-L. UbcH10 overexpression increases carcinogenesis and blocks ALLN susceptibility in colorectal cancer. Sci. Rep. 2014, 4, 6910. [Google Scholar] [CrossRef] [PubMed]
- Hillert, E.-K.; Brnjic, S.; Zhang, X.; Mazurkiewicz, M.; Saei, A.A.; Mofers, A.; Selvaraju, K.; Zubarev, R.; Linder, S.; D’Arcy, P. Proteasome inhibitor b-AP15 induces enhanced proteotoxicity by inhibiting cytoprotective aggresome formation. Cancer Lett. 2019, 448, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.-J.; Park, S.-A.; Lee, S.-Y.; Cha, Y.N.; Surh, Y.-J. Hypoxia induces epithelial-mesenchymal transition in colorectal cancer cells through ubiquitin-specific protease 47-mediated stabilization of Snail: A potential role of Sox9. Sci. Rep. 2017, 7, 15918. [Google Scholar] [CrossRef]
- Sun, H.; Ou, B.; Zhao, S.; Liu, X.; Song, L.; Liu, X.; Wang, R.; Peng, Z. USP11 promotes growth and metastasis of colorectal cancer via PPP1CA-mediated activation of ERK/MAPK signaling pathway. EBioMedicine 2019, 48, 236–247. [Google Scholar] [CrossRef]
- Zhou, Z.; Yao, X.; Li, S.; Xiong, Y.; Dong, X.; Zhao, Y.; Jiang, J.; Zhang, Q. Deubiquitination of Ci/Gli by Usp7/HAUSP Regulates Hedgehog Signaling. Dev. Cell 2015, 34, 58–72. [Google Scholar] [CrossRef]
- An, T.; Gong, Y.; Li, X.; Kong, L.; Ma, P.; Gong, L.; Zhu, H.; Yu, C.; Liu, J.; Zhou, H.; et al. USP7 inhibitor P5091 inhibits Wnt signaling and colorectal tumor growth. Biochem. Pharmacol. 2017, 131, 29–39. [Google Scholar] [CrossRef]
- Li, X.; Kong, L.; Yang, Q.; Duan, A.; Ju, X.; Cai, B.; Chen, L.; An, T.; Li, Y. Parthenolide inhibits ubiquitin-specific peptidase 7 (USP7), Wnt signaling, and colorectal cancer cell growth. J. Biol. Chem. 2020, 295, 3576–3589. [Google Scholar] [CrossRef]
- Wolf-Levy, H.; Javitt, A.; Eisenberg-Lerner, A.; Kacen, A.; Ulman, A.; Sheban, D.; Dassa, B.; Fishbain-Yoskovitz, V.; Carmona-Rivera, C.; Kramer, M.P.; et al. Revealing the cellular degradome by mass spectrometry analysis of proteasome-cleaved peptides. Nat. Biotechnol. 2018, 36, 1110–1116. [Google Scholar] [CrossRef]
- Javitt, A.; Merbl, Y. Global views of proteasome-mediated degradation by mass spectrometry. Expert Rev. Proteomics 2019, 16, 711–716. [Google Scholar] [CrossRef]
- Verma, R.; Mohl, D.; Deshaies, R.J. Harnessing the Power of Proteolysis for Targeted Protein Inactivation. Mol. Cell 2020, 77, 446–460. [Google Scholar] [CrossRef] [PubMed]
- Burslem, G.M.; Crews, C.M. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020, 181, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Jevtić, P.; Haakonsen, D.L.; Rapé, M. An E3 ligase guide to the galaxy of small-molecule-induced protein degradation. Cell Chem. Biol. 2021. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohapatra, G.; Eisenberg-Lerner, A.; Merbl, Y. Gatekeepers of the Gut: The Roles of Proteasomes at the Gastrointestinal Barrier. Biomolecules 2021, 11, 989. https://doi.org/10.3390/biom11070989
Mohapatra G, Eisenberg-Lerner A, Merbl Y. Gatekeepers of the Gut: The Roles of Proteasomes at the Gastrointestinal Barrier. Biomolecules. 2021; 11(7):989. https://doi.org/10.3390/biom11070989
Chicago/Turabian StyleMohapatra, Gayatree, Avital Eisenberg-Lerner, and Yifat Merbl. 2021. "Gatekeepers of the Gut: The Roles of Proteasomes at the Gastrointestinal Barrier" Biomolecules 11, no. 7: 989. https://doi.org/10.3390/biom11070989
APA StyleMohapatra, G., Eisenberg-Lerner, A., & Merbl, Y. (2021). Gatekeepers of the Gut: The Roles of Proteasomes at the Gastrointestinal Barrier. Biomolecules, 11(7), 989. https://doi.org/10.3390/biom11070989