
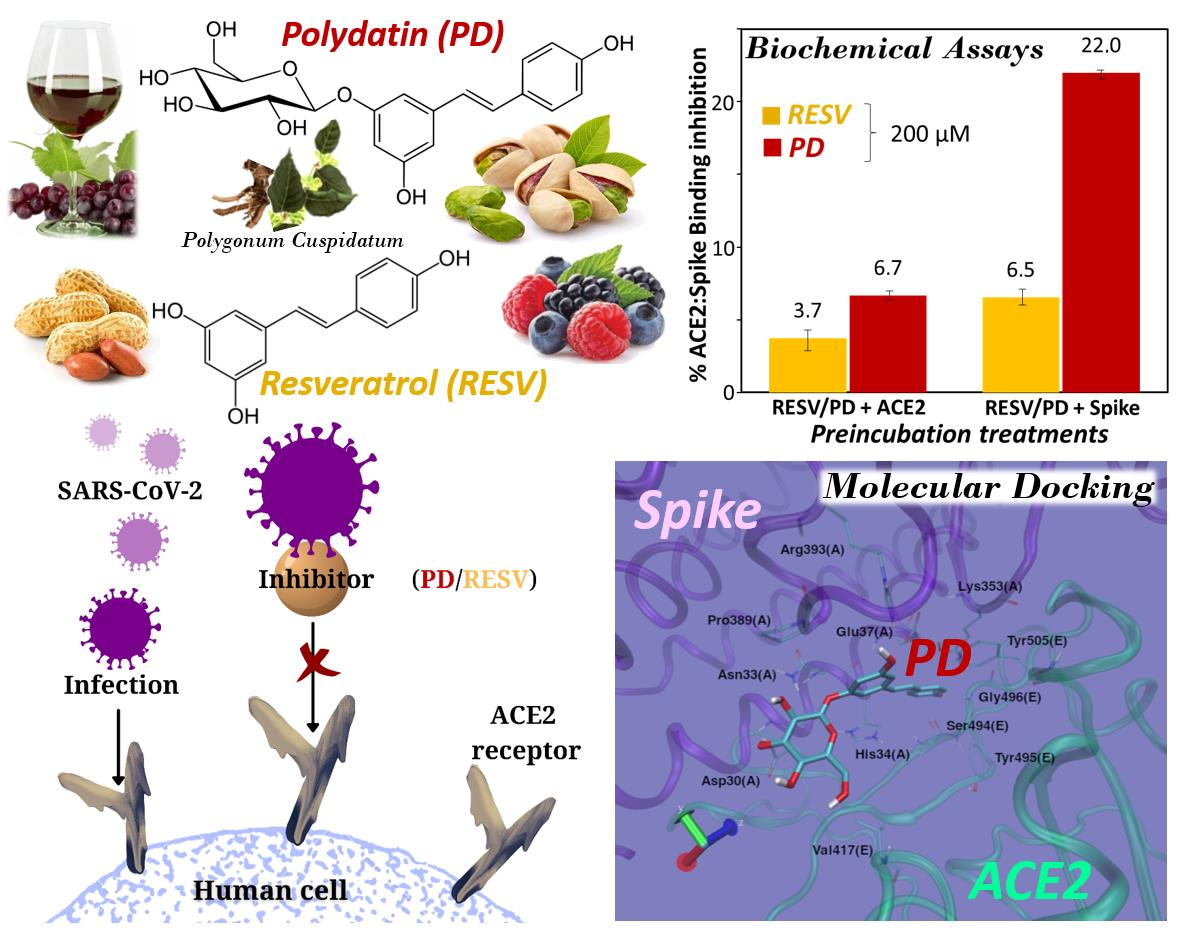
Interference of Polydatin/Resveratrol in the ACE2:Spike Recognition during COVID-19 Infection. A Focus on Their Potential Mechanism of Action through Computational and Biochemical Assays

 ,
,  , , ,
, , ,  ,
,  , ,
, ,  and
and 
Abstract
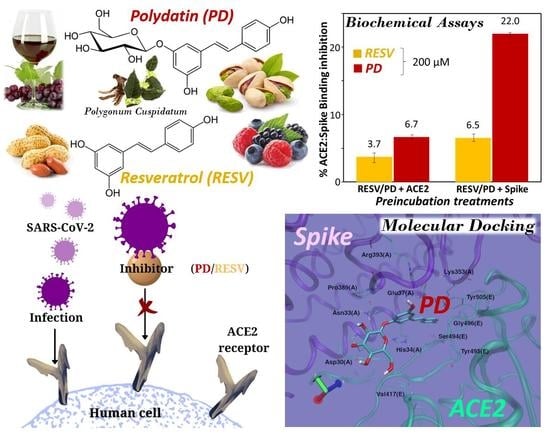
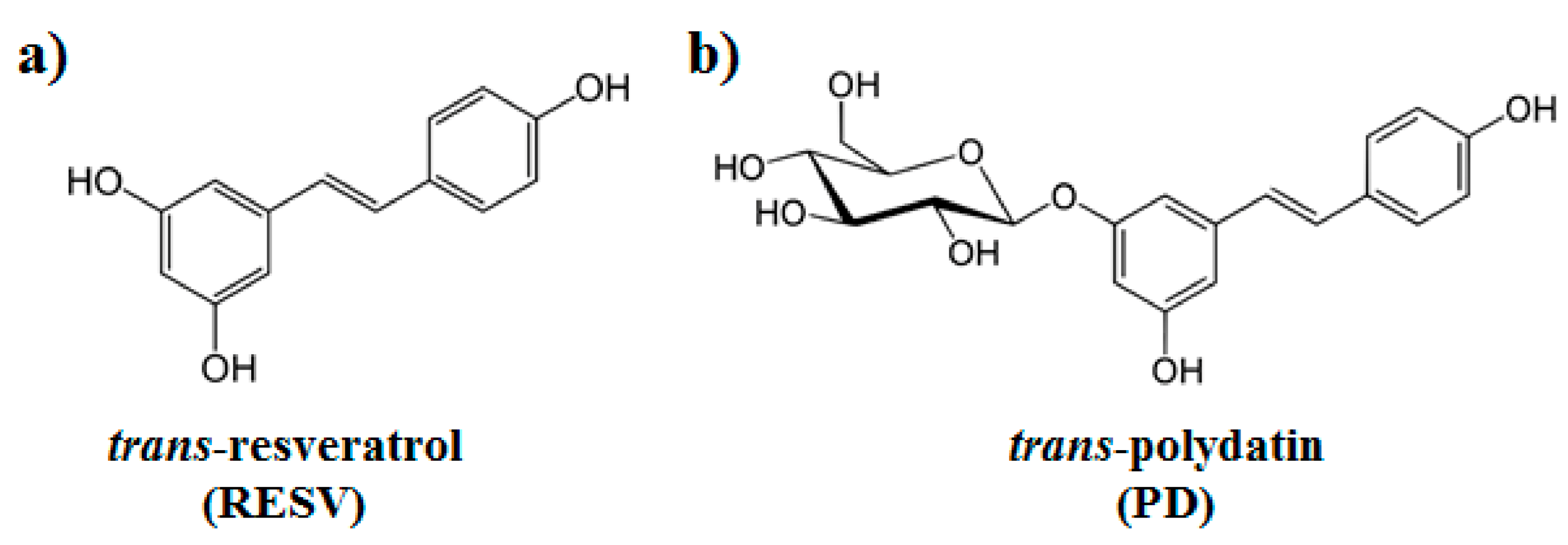
:
1. Introduction
2. Materials and Methods
2.1. Computational Details
2.2. Biochemical Assays
2.2.1. Instrumentations
2.2.2. Chemicals
2.2.3. Proteins and Inhibitors Solutions Preparation
2.2.4. Spike-ACE2 Binding Assay
3. Results and Discussion
3.1. Molecular Docking Simulations
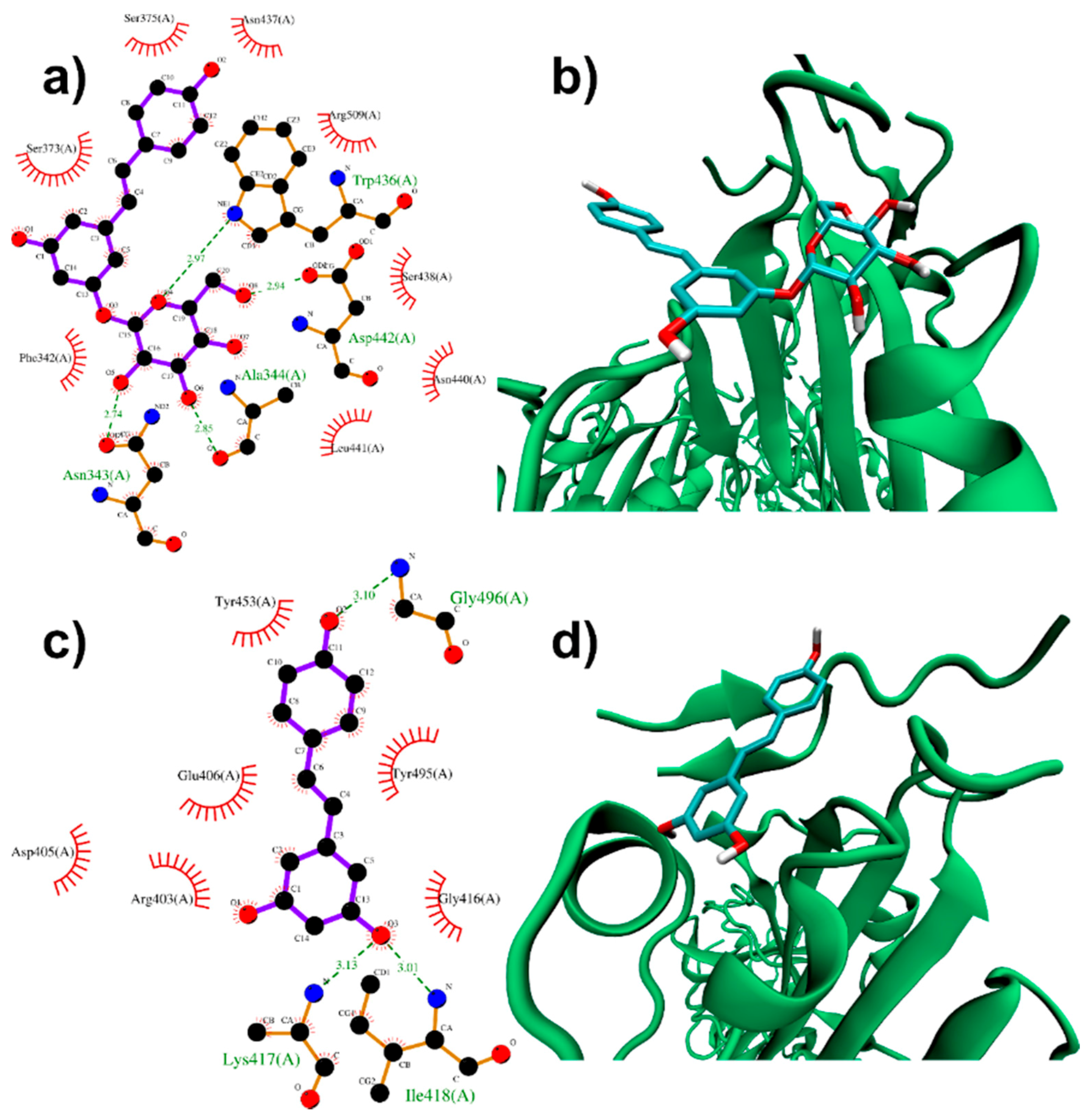
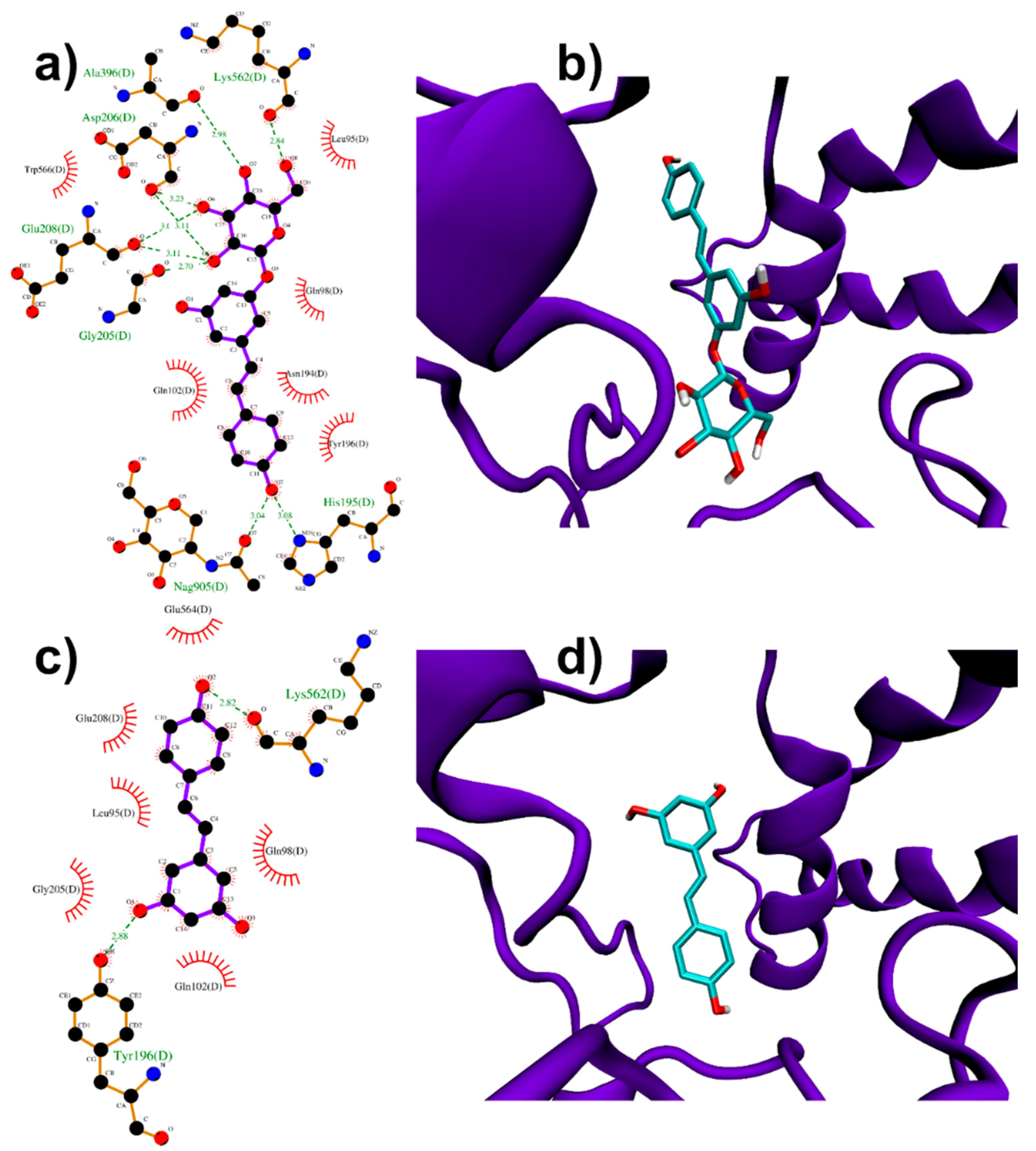
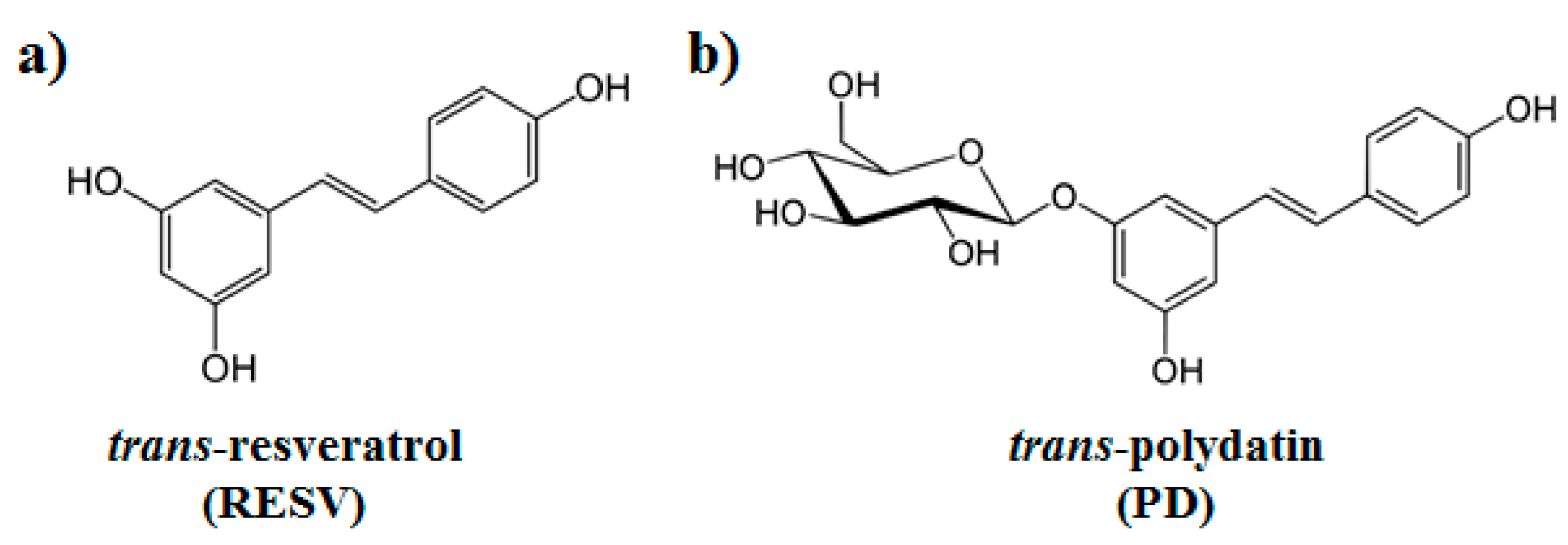
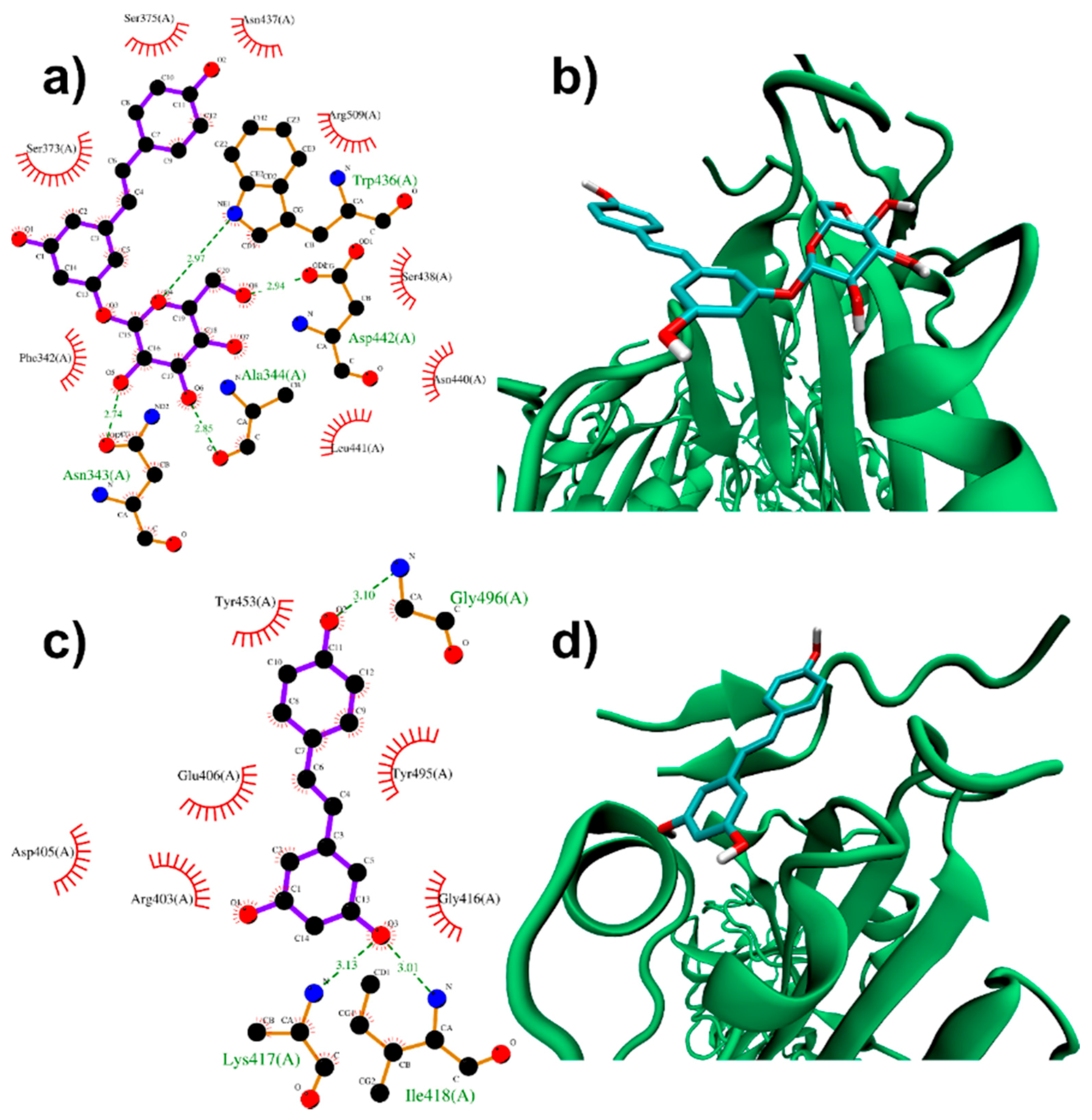
3.1.1. Binding to SARS-CoV-2 Spike Protein
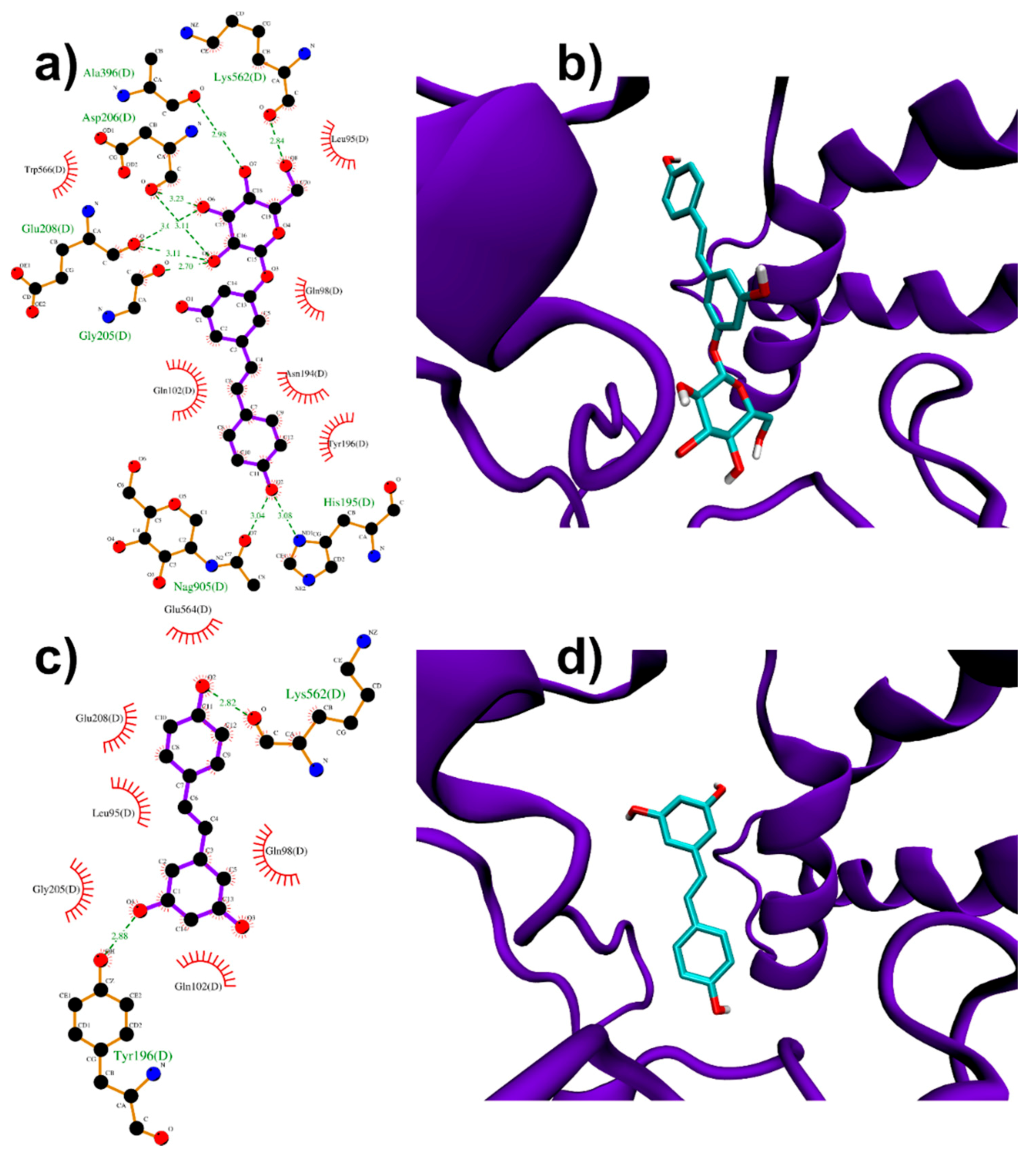
3.1.2. Binding to ACE2 Receptor
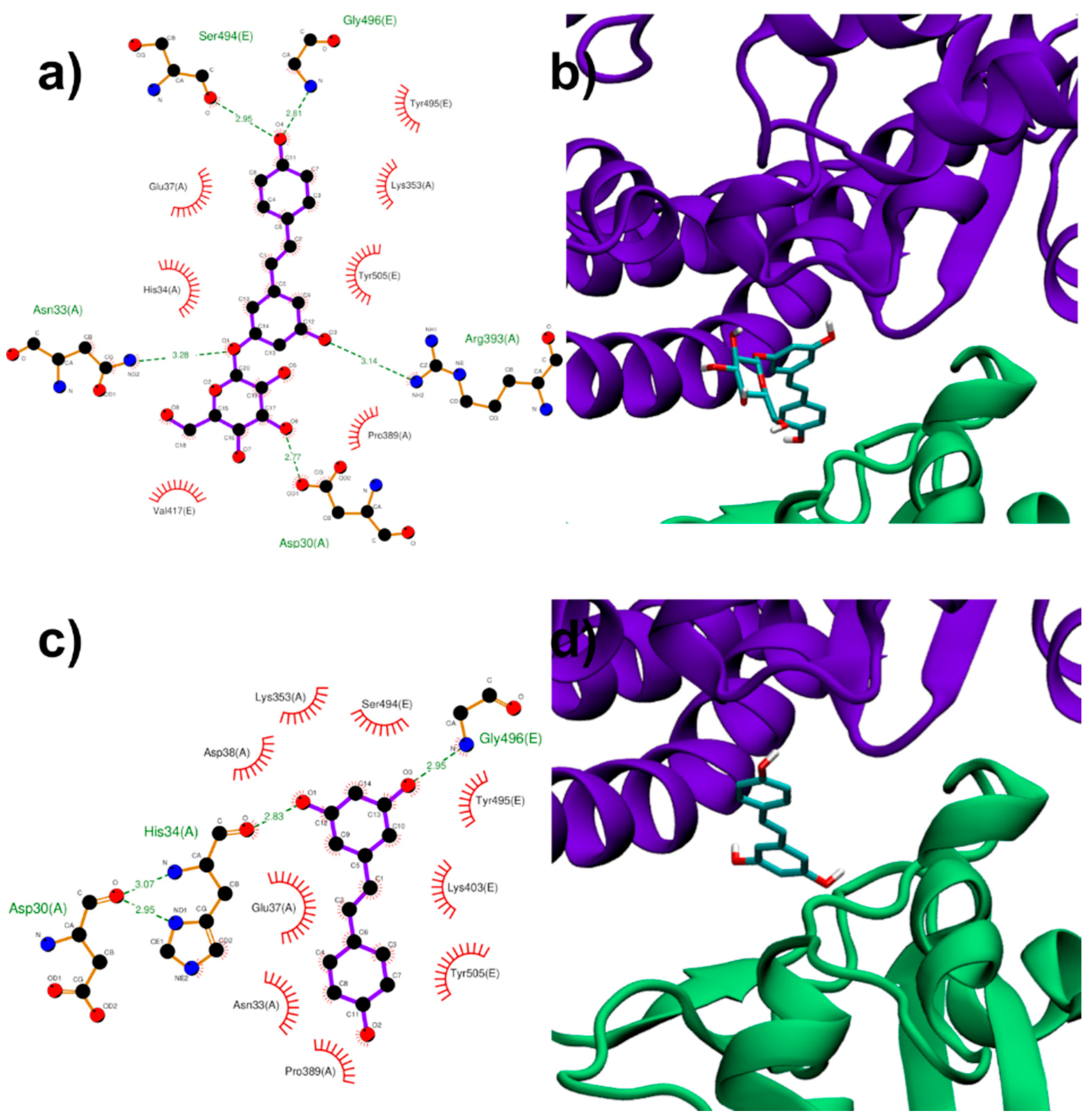
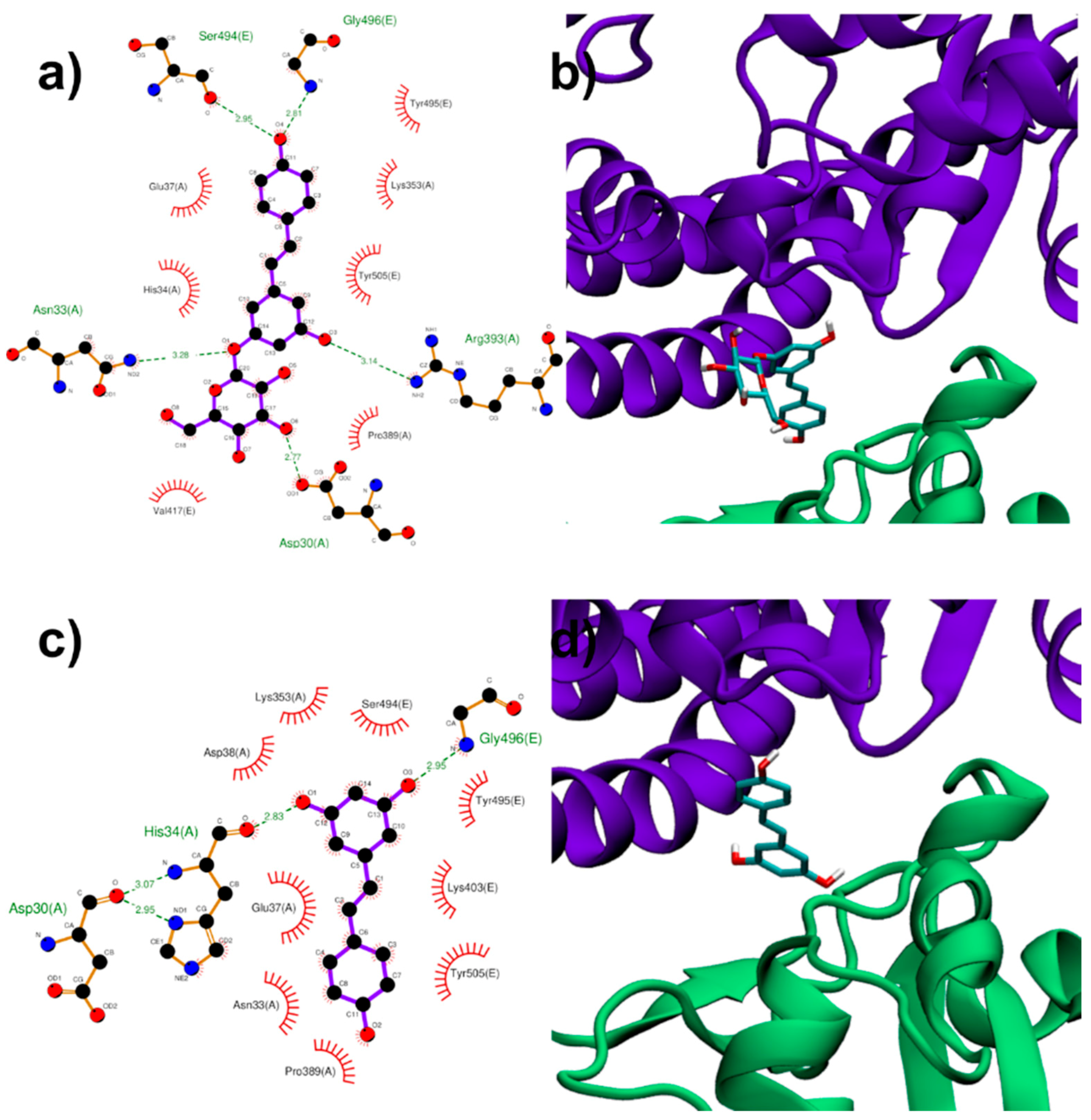
3.1.3. Binding to ACE2:Spike Complex
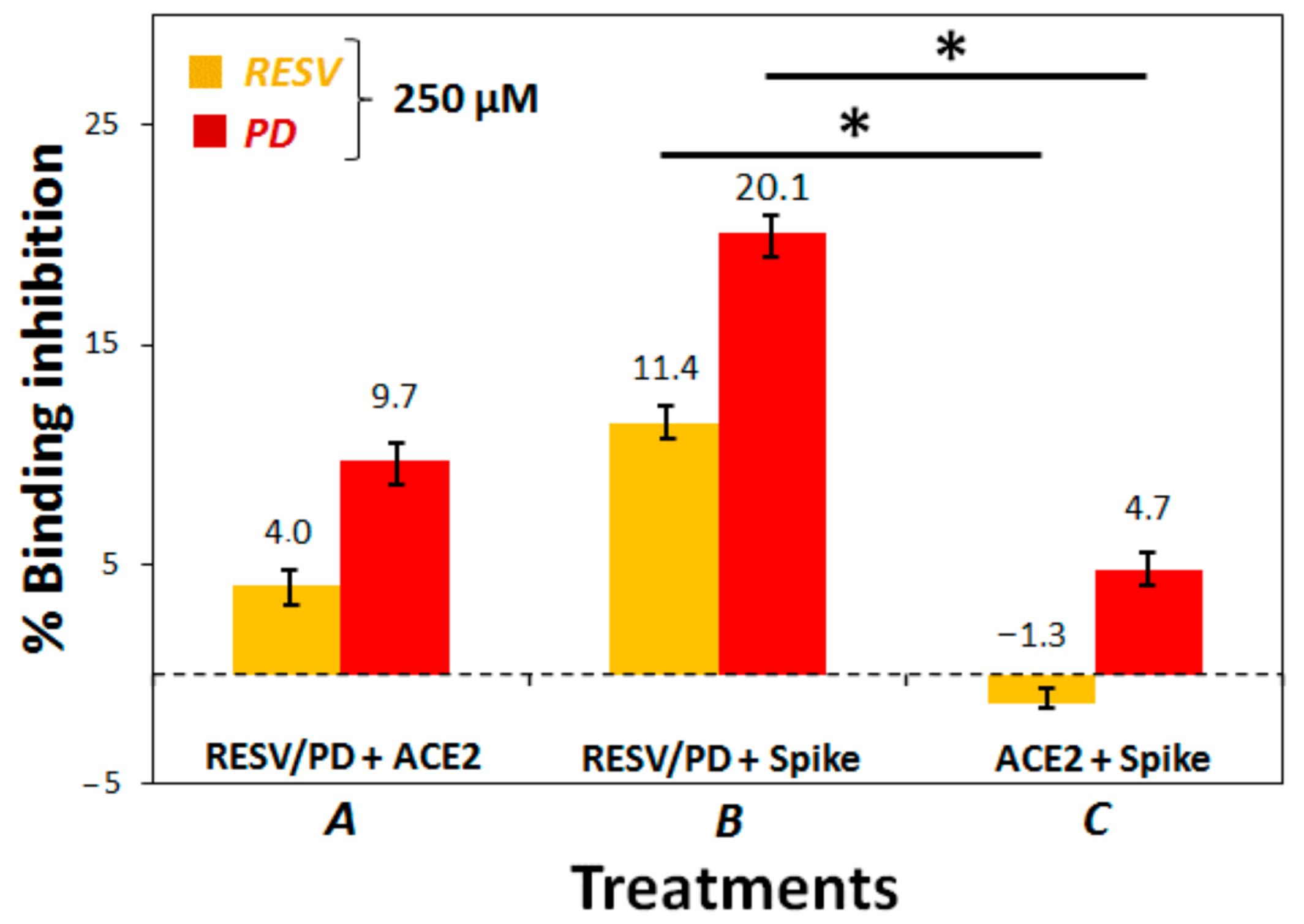
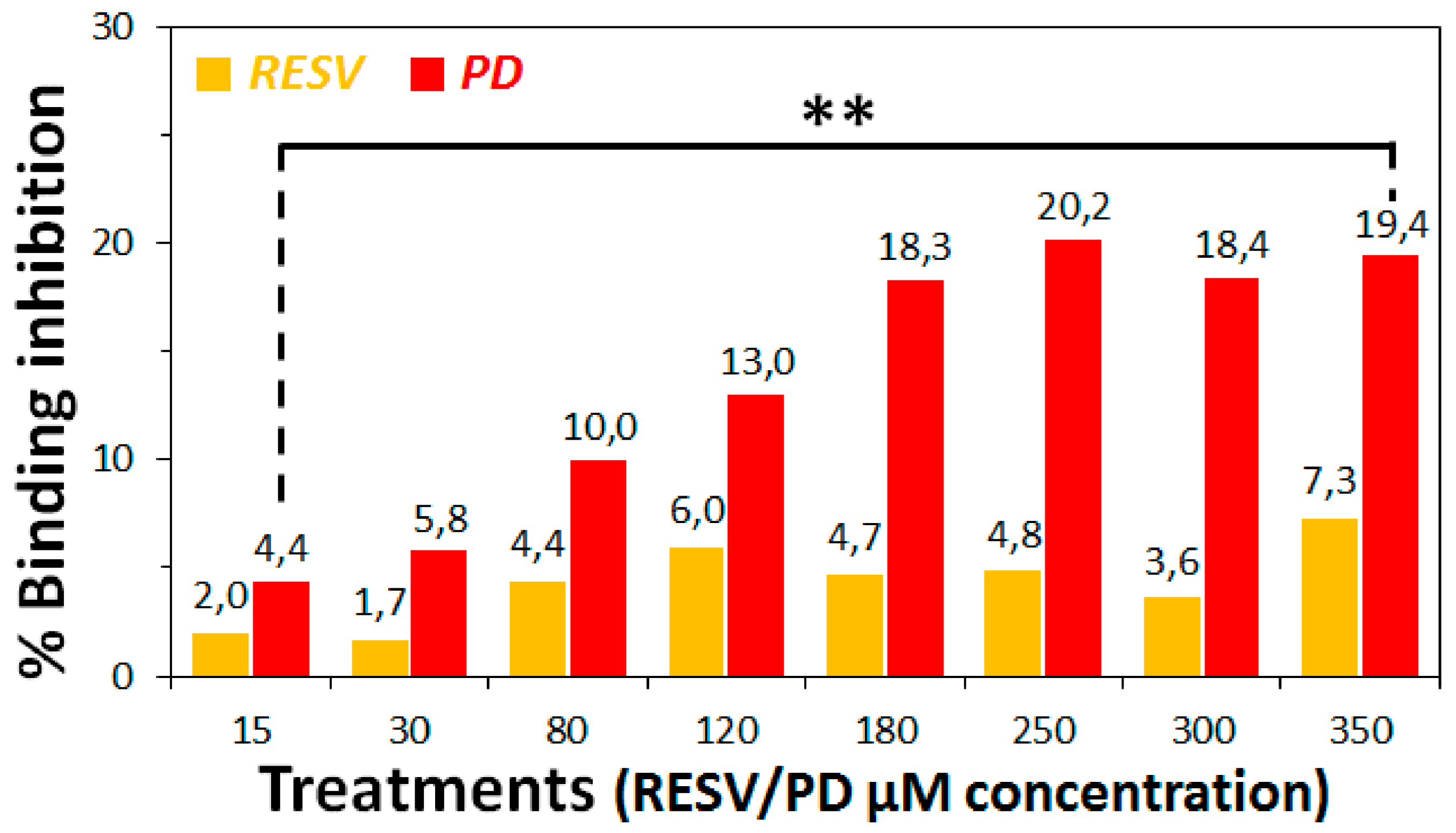
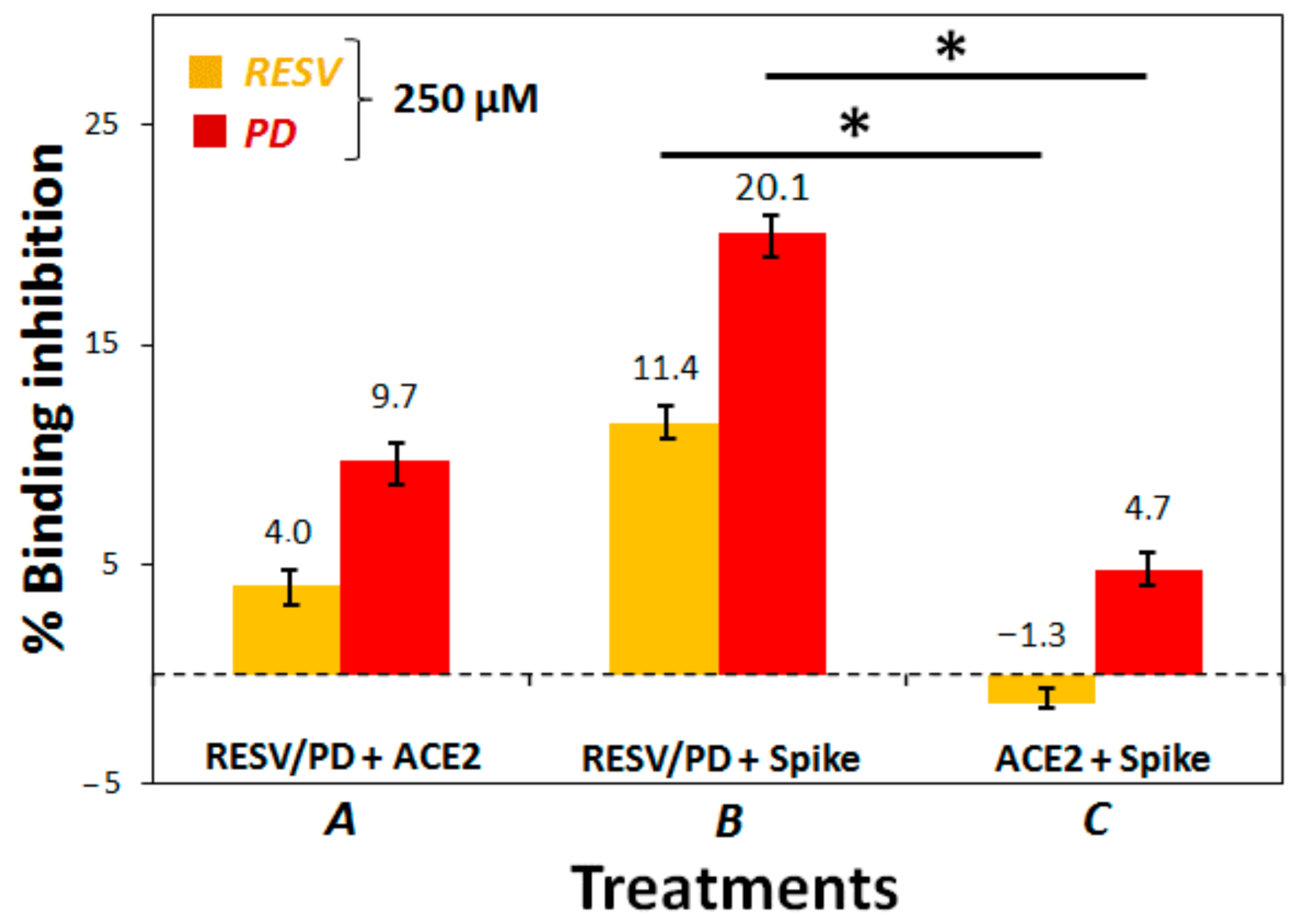
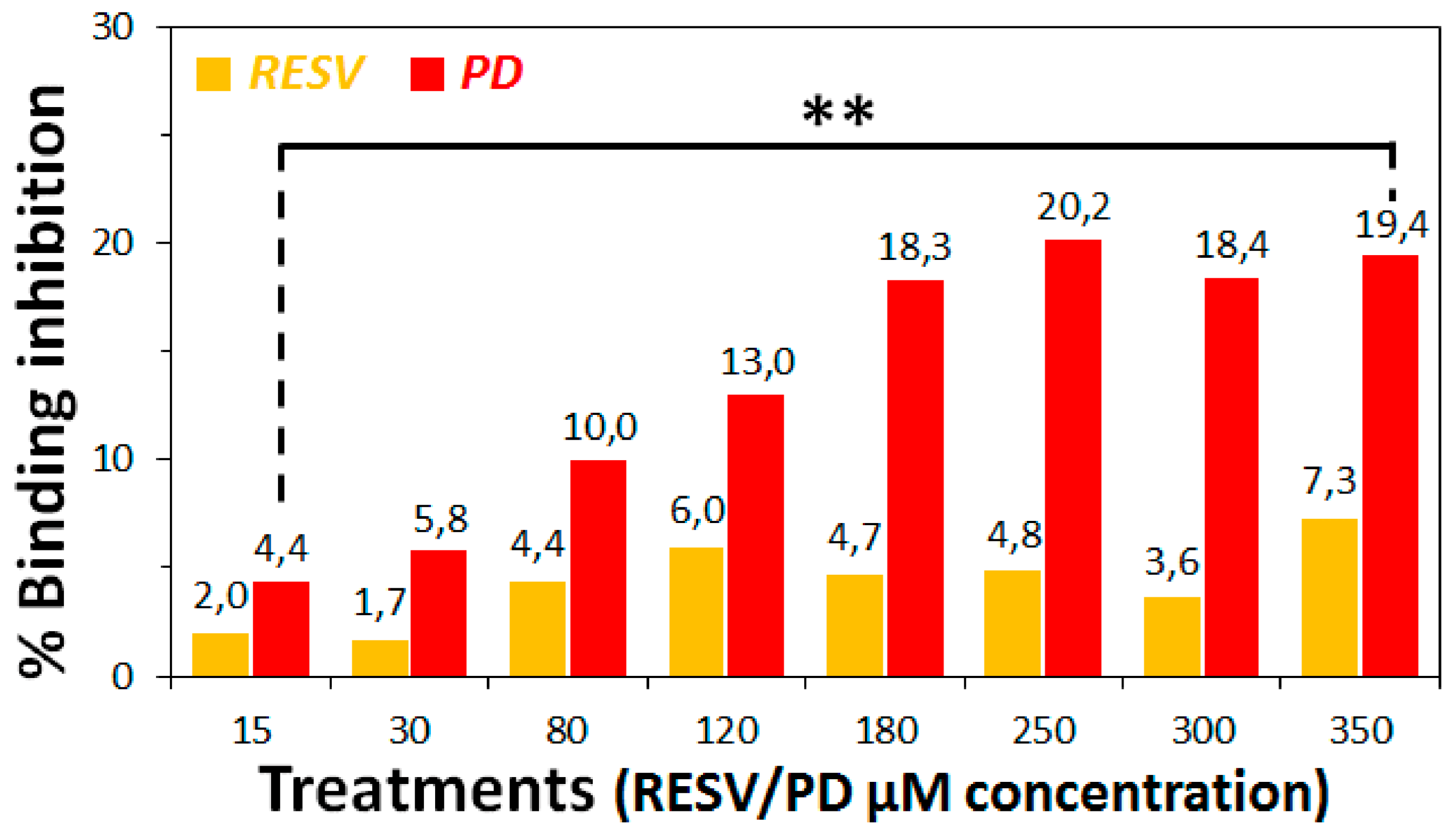
3.2. Preliminary ACE2:Spike Binding Inhibition Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. In Coronaviruses: Methods and Protocols; Springer: New York, NY, USA, 2015; pp. 1–23. [Google Scholar]
- Masters, P.S. The Molecular Biology of Coronaviruses. Adv. Virus Res. 2006, 65, 193–292. [Google Scholar]
- Corman, V.M.; Lienau, J.; Witzenrath, M. Coronaviruses as the Cause of Respiratory Infections. Internist 2019, 60, 1136–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Siu, Y.L.; Teoh, K.T.; Lo, J.; Chan, C.M.; Kien, F.; Escriou, N.; Tsao, S.W.; Nicholls, J.M.; Altmeyer, R.; Peiris, J.S.M.; et al. The M, E, and N Structural Proteins of the Severe Acute Respiratory Syndrome Coronavirus Are Required for Efficient Assembly, Trafficking, and Release of Virus-Like Particles. J. Virol. 2008, 82, 11318–11330. [Google Scholar] [CrossRef] [Green Version]
- Belouzard, S.; Millet, J.K.; Licitra, B.N.; Whittaker, G.R. Mechanisms of Coronavirus Cell Entry Mediated by the Viral Spike Protein. Viruses 2012, 4, 1011–1033. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell Entry Mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural Basis of Receptor Recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef] [Green Version]
- Bourgonje, A.R.; Abdulle, A.E.; Timens, W.; Hillebrands, J.L.; Navis, G.J.; Gordijn, S.J.; Bolling, M.C.; Dijkstra, G.; Voors, A.A.; Osterhaus, A.D.M.E.; et al. Angiotensin-Converting Enzyme 2 (ACE2), SARS-CoV-2 and the Pathophysiology of Coronavirus Disease 2019 (COVID-19). J. Pathol. 2020, 251, 228–248. [Google Scholar] [CrossRef]
- Lukassen, S.; Chua, R.L.; Trefzer, T.; Kahn, N.C.; Schneider, M.A.; Muley, T.; Winter, H.; Meister, M.; Veith, C.; Boots, A.W.; et al. SARS-CoV-2 Receptor ACE 2 and TMPRSS 2 Are Primarily Expressed in Bronchial Transient Secretory Cells. EMBO J. 2020, 39, e105114. [Google Scholar] [CrossRef]
- Magrone, T.; Magrone, M.; Jirillo, E. Focus on Receptors for Coronaviruses with Special Reference to Angiotensin- Converting Enzyme 2 as a Potential Drug Target—A Perspective. Endocr. Metab. Immune Disord. Drug Targets 2020, 20, 807–811. [Google Scholar] [CrossRef]
- Chi, X.; Yan, R.; Zhang, J.; Zhang, G.; Zhang, Y.; Hao, M.; Zhang, Z.; Fan, P.; Dong, Y.; Yang, Y.; et al. A Neutralizing Human Antibody Binds to the N-Terminal Domain of the Spike Protein of SARS-CoV-2. Science 2020, 369, 650–655. [Google Scholar] [CrossRef]
- Liu, L.; Wang, P.; Nair, M.S.; Yu, J.; Rapp, M.; Wang, Q.; Luo, Y.; Chan, J.F.W.; Sahi, V.; Figueroa, A.; et al. Potent Neutralizing Antibodies against Multiple Epitopes on SARS-CoV-2 Spike. Nature 2020, 584, 450–456. [Google Scholar] [CrossRef]
- Papageorgiou, A.C.; Mohsin, I. The SARS-CoV-2 Spike Glycoprotein as a Drug and Vaccine Target: Structural Insights into Its Complexes with ACE2 and Antibodies. Cells 2020, 9, 2343. [Google Scholar] [CrossRef]
- Yi, C.; Sun, X.; Ye, J.; Ding, L.; Liu, M.; Yang, Z.; Lu, X.; Zhang, Y.; Ma, L.; Gu, W.; et al. Key Residues of the Receptor Binding Motif in the Spike Protein of SARS-CoV-2 That Interact with ACE2 and Neutralizing Antibodies. Cell. Mol. Immunol. 2020, 17, 621–630. [Google Scholar] [CrossRef]
- Ju, B.; Zhang, Q.; Ge, J.; Wang, R.; Sun, J.; Ge, X.; Yu, J.; Shan, S.; Zhou, B.; Song, S.; et al. Human Neutralizing Antibodies Elicited by SARS-CoV-2 Infection. Nature 2020, 584, 115–119. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Gil, C.; Ginex, T.; Maestro, I.; Nozal, V.; Barrado-Gil, L.; Cuesta-Geijo, M.Á.; Urquiza, J.; Ramírez, D.; Alonso, C.; Campillo, N.E.; et al. COVID-19: Drug Targets and Potential Treatments. J. Med. Chem. 2020, 63, 12359–12386. [Google Scholar] [CrossRef]
- Hensel, A.; Bauer, R.; Heinrich, M.; Spiegler, V.; Kayser, O.; Hempel, G.; Kraft, K. Challenges at the Time of COVID-19: Opportunities and Innovations in Antivirals from Nature. Planta Med. 2020, 86, 659–664. [Google Scholar] [CrossRef]
- Islam, M.T.; Sarkar, C.; El-Kersh, D.M.; Jamaddar, S.; Uddin, S.J.; Shilpi, J.A.; Mubarak, M.S. Natural Products and Their Derivatives against Coronavirus: A Review of the Non-clinical and Pre-clinical Data. Phyther. Res. 2020, 34, 2471–2492. [Google Scholar] [CrossRef]
- Mani, J.S.; Johnson, J.B.; Steel, J.C.; Broszczak, D.A.; Neilsen, P.M.; Walsh, K.B.; Naiker, M. Natural Product-Derived Phytochemicals as Potential Agents against Coronaviruses: A Review. Virus Res. 2020, 284, 197989. [Google Scholar] [CrossRef]
- Romeo, I.; Mesiti, F.; Lupia, A.; Alcaro, S. Current Updates on Naturally Occurring Compounds Recognizing SARS-CoV-2 Druggable Targets. Molecules 2021, 26, 632. [Google Scholar] [CrossRef]
- Quideau, S.; Deffieux, D.; Douat-Casassus, C.; Pouységu, L. Plant Polyphenols: Chemical Properties, Biological Activities, and Synthesis. Angew. Chemie Int. Ed. 2011, 50, 586–621. [Google Scholar] [CrossRef]
- Gambini, J.; Inglés, M.; Olaso, G.; Lopez-Grueso, R.; Bonet-Costa, V.; Gimeno-Mallench, L.; Mas-Bargues, C.; Abdelaziz, K.M.; Gomez-Cabrera, M.C.; Vina, J.; et al. Properties of Resveratrol: In Vitro and In Vivo Studies about Metabolism, Bioavailability, and Biological Effects in Animal Models and Humans. Oxid. Med. Cell. Longev. 2015, 2015, 837042. [Google Scholar] [CrossRef] [Green Version]
- Du, Q.H.; Peng, C.; Zhang, H. Polydatin: A Review of Pharmacology and Pharmacokinetics. Pharm. Biol. 2013, 51, 1347–1354. [Google Scholar] [CrossRef]
- Zorzete, P.; Reis, T.A.; Felício, J.D.; Baquião, A.C.; Makimoto, P.; Corrêa, B. Fungi, Mycotoxins and Phytoalexin in Peanut Varieties, during Plant Growth in the Field. Food Chem. 2011, 129, 957–964. [Google Scholar] [CrossRef]
- Regev-Shoshani, G.; Shoseyov, O.; Bilkis, I.; Kerem, Z. Glycosylation of Resveratrol Protects It from Enzymic Oxidation. Biochem. J. 2003, 374, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Fabris, S.; Momo, F.; Ravagnan, G.; Stevanato, R. Antioxidant Properties of Resveratrol and Piceid on Lipid Peroxidation in Micelles and Monolamellar Liposomes. Biophys. Chem. 2008, 135, 76–83. [Google Scholar] [CrossRef]
- Platella, C.; Guida, S.; Bonmassar, L.; Aquino, A.; Bonmassar, E.; Ravagnan, G.; Montesarchio, D.; Roviello, G.N.; Musumeci, D.; Fuggetta, M.P. Antitumour Activity of Resveratrol on Human Melanoma Cells: A Possible Mechanism Related to Its Interaction with Malignant Cell Telomerase. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2843–2851. [Google Scholar] [CrossRef] [PubMed]
- Platella, C.; Raucci, U.; Rega, N.; D’Atri, S.; Levati, L.; Roviello, G.N.; Fuggetta, M.P.; Musumeci, D.; Montesarchio, D. Shedding Light on the Interaction of Polydatin and Resveratrol with G-Quadruplex and Duplex DNA: A Biophysical, Computational and Biological Approach. Int. J. Biol. Macromol. 2020, 151, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Francioso, A.; Mastromarino, P.; Masci, A.; D’Erme, M.; Mosca, L. Chemistry, Stability and Bioavailability of Resveratrol. Med. Chem. 2014, 10, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Şöhretoğlu, D.; Baran, M.Y.; Arroo, R.; Kuruüzüm-Uz, A. Recent Advances in Chemistry, Therapeutic Properties and Sources of Polydatin. Phytochem. Rev. 2018, 17, 973–1005. [Google Scholar] [CrossRef]
- Magrone, T.; Magrone, M.; Russo, M.A.; Jirillo, E. Recent Advances on the Anti-Inflammatory and Antioxidant Properties of Red Grape Polyphenols: In Vitro and In Vivo Studies. Antioxidants 2019, 9, 35. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.; Hu, S.; Gao, J. Discovering Drugs to Treat Coronavirus Disease 2019 (COVID-19). Drug Discov. Ther. 2020, 14, 58–60. [Google Scholar] [CrossRef] [Green Version]
- Wahedi, H.M.; Ahmad, S.; Abbasi, S.W. Stilbene-Based Natural Compounds as Promising Drug Candidates against COVID-19. J. Biomol. Struct. Dyn. 2020, 39, 3225–3234. [Google Scholar] [CrossRef]
- Yang, M.; Wei, J.; Huang, T.; Lei, L.; Shen, C.; Lai, J.; Yang, M.; Liu, L.; Yang, Y.; Liu, G.; et al. Resveratrol Inhibits the Replication of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) in Cultured Vero Cells. Phyther. Res. 2020, 35, 1127–1129. [Google Scholar] [CrossRef]
- Feng, H.; Choi, H.Y.; Cui, C.; Xu, H.; Jiang, J.; Yan, G.; Jin, M. Effects of Polydatin on Oleic Acid-Induced Acute Respiratory Distress Syndrome in Rats. Int. J. Clin. Exp. Med. 2018, 11, 12106–12114. [Google Scholar]
- Filardo, S.; Di Pietro, M.; Mastromarino, P.; Sessa, R. Therapeutic Potential of Resveratrol against Emerging Respiratory Viral Infections. Pharmacol. Ther. 2020, 214, 107613. [Google Scholar] [CrossRef]
- Jiang, Q.; Yi, M.; Guo, Q.; Wang, C.; Wang, H.; Meng, S.; Liu, C.; Fu, Y.; Ji, H.; Chen, T. Protective Effects of Polydatin on Lipopolysaccharide-Induced Acute Lung Injury through TLR4-MyD88-NF-ΚB Pathway. Int. Immunopharmacol. 2015, 29, 370–376. [Google Scholar] [CrossRef]
- Khalil, A.; Tazeddinova, D. The Upshot of Polyphenolic Compounds on Immunity amid COVID-19 Pandemic and Other Emerging Communicable Diseases: An Appraisal. Nat. Products Bioprospect. 2020, 10, 411–429. [Google Scholar] [CrossRef]
- Li, X.H.; Gong, X.; Zhang, L.; Jiang, R.; Li, H.Z.; Wu, M.J.; Wan, J.Y. Protective Effects of Polydatin on Septic Lung Injury in Mice via Upregulation of HO-1. Mediators Inflamm. 2013, 2013, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Lanzilli, G.; Cottarelli, A.; Nicotera, G.; Guida, S.; Ravagnan, G.; Fuggetta, M.P. Anti-Inflammatory Effect of Resveratrol and Polydatin by in Vitro IL-17 Modulation. Inflammation 2012, 35, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Lo Muzio, L.; Bizzoca, M.E.; Ravagnan, G. New Intriguing Possibility for Prevention of Coronavirus Pneumonitis: Natural Purified Polyphenols. Oral Dis. 2020, odi.13518. [Google Scholar] [CrossRef]
- Yan, X.-D.; Wang, Q.-M.; Tie, C.; Jin, H.-T.; Han, Y.-X.; Zhang, J.-L.; Yu, X.-M.; Hou, Q.; Zhang, P.-P.; Wang, A.-P.; et al. Polydatin Protects the Respiratory System from PM2.5 Exposure. Sci. Rep. 2017, 7, 40030. [Google Scholar] [CrossRef] [Green Version]
- Ravagnan, G.; De Filippis, A.; Cartenì, M.; De Maria, S.; Cozza, V.; Petrazzuolo, M.; Tufano, M.A.; Donnarumma, G. Polydatin, A Natural Precursor of Resveratrol, Induces β-Defensin Production and Reduces Inflammatory Response. Inflammation 2013, 36, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Pasquereau, S.; Nehme, Z.; Haidar Ahmad, S.; Daouad, F.; Van Assche, J.; Wallet, C.; Schwartz, C.; Rohr, O.; Morot-Bizot, S.; Herbein, G. Resveratrol Inhibits HCoV-229E and SARS-CoV-2 Coronavirus Replication In Vitro. Viruses 2021, 13, 354. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM Structure of the 2019-NCoV Spike in the Prefusion Conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural Basis for the Recognition of SARS-CoV-2 by Full-Length Human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Dennington, R.; Keith, T.; Millam, J. GaussView, Version 6; Semichem Inc.: Shawnee, KS, USA, 2009. [Google Scholar]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Schmidtke, P.; Le Guilloux, V.; Maupetit, J.; Tufféry, P. Fpocket: Online Tools for Protein Ensemble Pocket Detection and Tracking. Nucleic Acids Res. 2010, 38, W582–W589. [Google Scholar] [CrossRef] [Green Version]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An Open Source Platform for Ligand Pocket Detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef] [Green Version]
- Schmidtke, P.; Barril, X. Understanding and Predicting Druggability. A High-Throughput Method for Detection of Drug Binding Sites. J. Med. Chem. 2010, 53, 5858–5867. [Google Scholar] [CrossRef]
- Schmidtke, P.; Souaille, C.; Estienne, F.; Baurin, N.; Kroemer, R.T. Large-Scale Comparison of Four Binding Site Detection Algorithms. J. Chem. Inf. Model. 2010, 50, 2191–2200. [Google Scholar] [CrossRef]
- Ezzat, A.; Kwoh, C.K. Comparison of Structure-Based Tools for the Prediction of Ligand Binding Site Residues in Apo-Structures. Procedia Comput. Sci. 2012, 11, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Windshügel, B. Structural Insights into Ligand-Binding Pocket Formation in Nurr1 by Molecular Dynamics Simulations. J. Biomol. Struct. Dyn. 2019, 37, 4651–4657. [Google Scholar] [CrossRef]
- Pradeepkiran, J.A.; Reddy, P.H. Structure Based Design and Molecular Docking Studies for Phosphorylated Tau Inhibitors in Alzheimer’s Disease. Cells 2019, 8, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, P.T.V.; Yu, H.; Keller, P.A. Molecular Docking Studies to Explore Potential Binding Pockets and Inhibitors for Chikungunya Virus Envelope Glycoproteins. Interdiscip. Sci. Comput. Life Sci. 2018, 10, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.-H.; Huang, C.-X.; Chen, Y.-J.; Chuang, Y.-C.; Huang, B.-Y.; Yang, C.-N. Molecular Modeling for Structural Insights Concerning the Activation Mechanisms of F1174L and R1275Q Mutations on Anaplastic Lymphoma Kinase. Molecules 2018, 23, 1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llorach-Pares, L.; Rodriguez-Urgelles, E.; Nonell-Canals, A.; Alberch, J.; Avila, C.; Sanchez-Martinez, M.; Giralt, A. Meridianins and Lignarenone B as Potential GSK3β Inhibitors and Inductors of Structural Neuronal Plasticity. Biomolecules 2020, 10, 639. [Google Scholar] [CrossRef] [Green Version]
- Vittorio, S.; Seidel, T.; Garon, A.; Gitto, R.; Langer, T.; De Luca, L. In Silico Identification of Potential Druggable Binding Sites on CIN85 SH3 Domain. Int. J. Mol. Sci. 2021, 22, 534. [Google Scholar] [CrossRef]
- Hetényi, C.; van der Spoel, D. Blind Docking of Drug-Sized Compounds to Proteins with up to a Thousand Residues. FEBS Lett. 2006, 580, 1447–1450. [Google Scholar] [CrossRef] [Green Version]
- Hassan, N.M.; Alhossary, A.A.; Mu, Y.; Kwoh, C.-K. Protein-Ligand Blind Docking Using QuickVina-W with Inter-Process Spatio-Temporal Integration. Sci. Rep. 2017, 7, 15451. [Google Scholar] [CrossRef] [Green Version]
- Ghersi, D.; Sanchez, R. Improving Accuracy and Efficiency of Blind Protein-Ligand Docking by Focusing on Predicted Binding Sites. Proteins Struct. Funct. Bioinform. 2009, 74, 417–424. [Google Scholar] [CrossRef] [Green Version]
- Jofily, P.; Pascutti, P.G.; Torres, P.H.M. Improving Blind Docking in DOCK6 through an Automated Preliminary Fragment Probing Strategy. Molecules 2021, 26, 1224. [Google Scholar] [CrossRef]
- Zhou, M.; Luo, H.; Li, R.; Ding, Z. Exploring the Binding Mode of HIV-1 Vif Inhibitors by Blind Docking, Molecular Dynamics and MM/GBSA. RSC Adv. 2013, 3, 22532–22543. [Google Scholar] [CrossRef]
- Iorga, B.; Herlem, D.; Barré, E.; Guillou, C. Acetylcholine Nicotinic Receptors: Finding the Putative Binding Site of Allosteric Modulators Using the “Blind Docking” Approach. J. Mol. Model. 2006, 12, 366–372. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand-Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Walls, A.C.; Tortorici, M.A.; Snijder, J.; Xiong, X.; Bosch, B.-J.; Rey, F.A.; Veesler, D. Tectonic Conformational Changes of a Coronavirus Spike Glycoprotein Promote Membrane Fusion. Proc. Natl. Acad. Sci. USA 2017, 114, 11157–11162. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Gui, M.; Wang, X.; Xiang, Y. Cryo-EM Structure of the SARS Coronavirus Spike Glycoprotein in Complex with Its Host Cell Receptor ACE2. PLoS Pathog. 2018, 14, e1007236. [Google Scholar] [CrossRef]
- Walls, A.C.; Xiong, X.; Park, Y.-J.; Tortorici, M.A.; Snijder, J.; Quispe, J.; Cameroni, E.; Gopal, R.; Dai, M.; Lanzavecchia, A.; et al. Unexpected Receptor Functional Mimicry Elucidates Activation of Coronavirus Fusion. Cell 2019, 176, 1026–1039.e15. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Tomlinson, A.C.A.; Wong, A.H.M.; Zhou, D.; Desforges, M.; Talbot, P.J.; Benlekbir, S.; Rubinstein, J.L.; Rini, J.M. The Human Coronavirus HCoV-229E S-Protein Structure and Receptor Binding. eLife 2019, 8, e51230. [Google Scholar] [CrossRef]
- Walls, A.C.; Tortorici, M.A.; Frenz, B.; Snijder, J.; Li, W.; Rey, F.A.; DiMaio, F.; Bosch, B.-J.; Veesler, D. Glycan Shield and Epitope Masking of a Coronavirus Spike Protein Observed by Cryo-Electron Microscopy. Nat. Struct. Mol. Biol. 2016, 23, 899–905. [Google Scholar] [CrossRef]
- López-Nicolás, J.M.; Pérez-Gilabert, M.; García-Carmona, F. Effect of Protonation and Aggregation State of (E)-Resveratrol on Its Hydroperoxidation by Lipoxygenase. J. Agric. Food Chem. 2009, 57, 4630–4635. [Google Scholar] [CrossRef]
- López-Nicolás, J.M.; García-Carmona, F. Aggregation State and PKa Values of (E)-Resveratrol As Determined by Fluorescence Spectroscopy and UV−Visible Absorption. J. Agric. Food Chem. 2008, 56, 7600–7605. [Google Scholar] [CrossRef]
- Polidase®, the Bioavailable Resveratrol, Sherman Tree Nutraceuticals. Available online: https://shermantree.it/en/polidase/ (accessed on 9 July 2021).
- Izzo, G.M.; Suffritti, G. Polydatin and Atopic Dermatitis in Adults: Clinical Study. J. Cosmetol. Trichol. 2017, 3, 122. [Google Scholar] [CrossRef]
- Cremon, C.; Stanghellini, V.; Barbaro, M.R.; Cogliandro, R.F.; Bellacosa, L.; Santos, J.; Vicario, M.; Pigrau, M.; Alonso Cotoner, C.; Lobo, B.; et al. Randomised Clinical Trial: The Analgesic Properties of Dietary Supplementation with Palmitoylethanolamide and Polydatin in Irritable Bowel Syndrome. Aliment. Pharmacol. Ther. 2017, 45, 909–922. [Google Scholar] [CrossRef]
- Bonucci, M.; Raggi, R.; Vacca, R.A. Polydatin and Its Potential Protective Effect on COVID-19. Clin. Nutr. 2020, 39, 3850–3851. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PD | RESV | |
|---|---|---|
| Spike RBD | −6.9 | −6.5 |
| Spike A/B interface | −7.3 | >−6.5 |
| Spike A/C interface | −7.3 | >−6.5 |
| ACE2 | −8.4 | −6.9 |
| S:ACE2 region I | −8.1 | −7.6 |
| S:ACE2 region II | −6.9 | −6.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perrella, F.; Coppola, F.; Petrone, A.; Platella, C.; Montesarchio, D.; Stringaro, A.; Ravagnan, G.; Fuggetta, M.P.; Rega, N.; Musumeci, D. Interference of Polydatin/Resveratrol in the ACE2:Spike Recognition during COVID-19 Infection. A Focus on Their Potential Mechanism of Action through Computational and Biochemical Assays. Biomolecules 2021, 11, 1048. https://doi.org/10.3390/biom11071048
Perrella F, Coppola F, Petrone A, Platella C, Montesarchio D, Stringaro A, Ravagnan G, Fuggetta MP, Rega N, Musumeci D. Interference of Polydatin/Resveratrol in the ACE2:Spike Recognition during COVID-19 Infection. A Focus on Their Potential Mechanism of Action through Computational and Biochemical Assays. Biomolecules. 2021; 11(7):1048. https://doi.org/10.3390/biom11071048
Chicago/Turabian StylePerrella, Fulvio, Federico Coppola, Alessio Petrone, Chiara Platella, Daniela Montesarchio, Annarita Stringaro, Giampietro Ravagnan, Maria Pia Fuggetta, Nadia Rega, and Domenica Musumeci. 2021. "Interference of Polydatin/Resveratrol in the ACE2:Spike Recognition during COVID-19 Infection. A Focus on Their Potential Mechanism of Action through Computational and Biochemical Assays" Biomolecules 11, no. 7: 1048. https://doi.org/10.3390/biom11071048
APA StylePerrella, F., Coppola, F., Petrone, A., Platella, C., Montesarchio, D., Stringaro, A., Ravagnan, G., Fuggetta, M. P., Rega, N., & Musumeci, D. (2021). Interference of Polydatin/Resveratrol in the ACE2:Spike Recognition during COVID-19 Infection. A Focus on Their Potential Mechanism of Action through Computational and Biochemical Assays. Biomolecules, 11(7), 1048. https://doi.org/10.3390/biom11071048