
Protein Phosphorylation in Cancer: Role of Nitric Oxide Signaling Pathway



Abstract
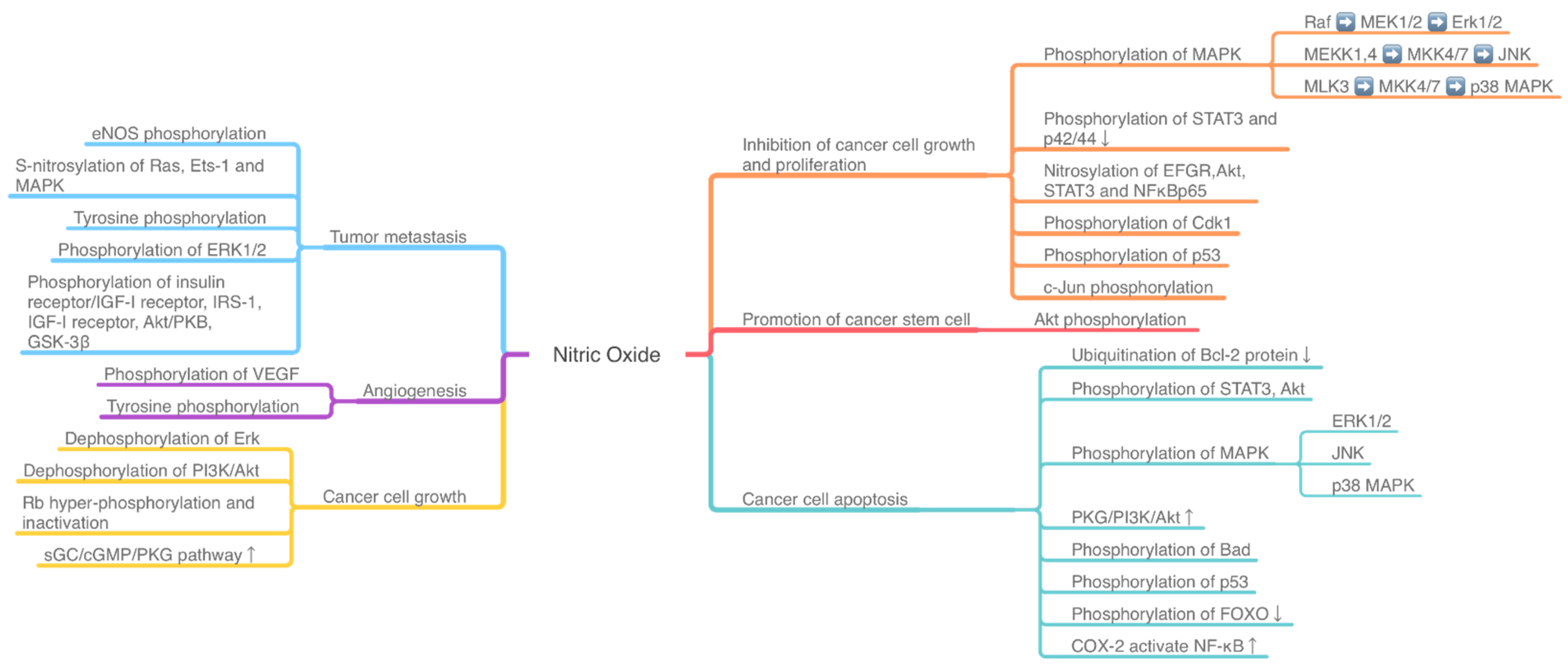
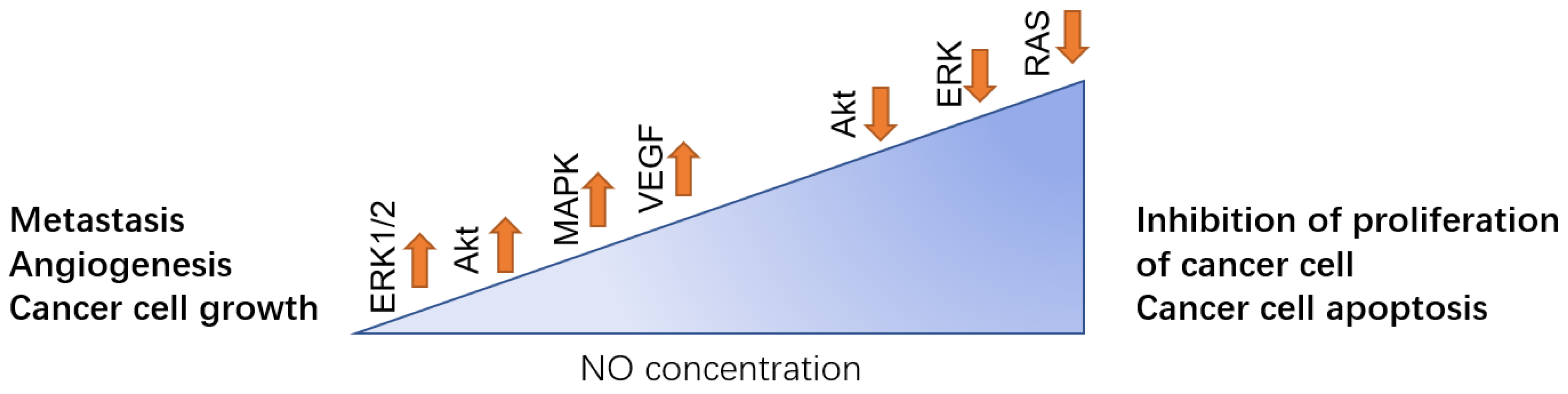
1. Introduction
2. Protein Phosphorylation in Different Cancer Induced by NO
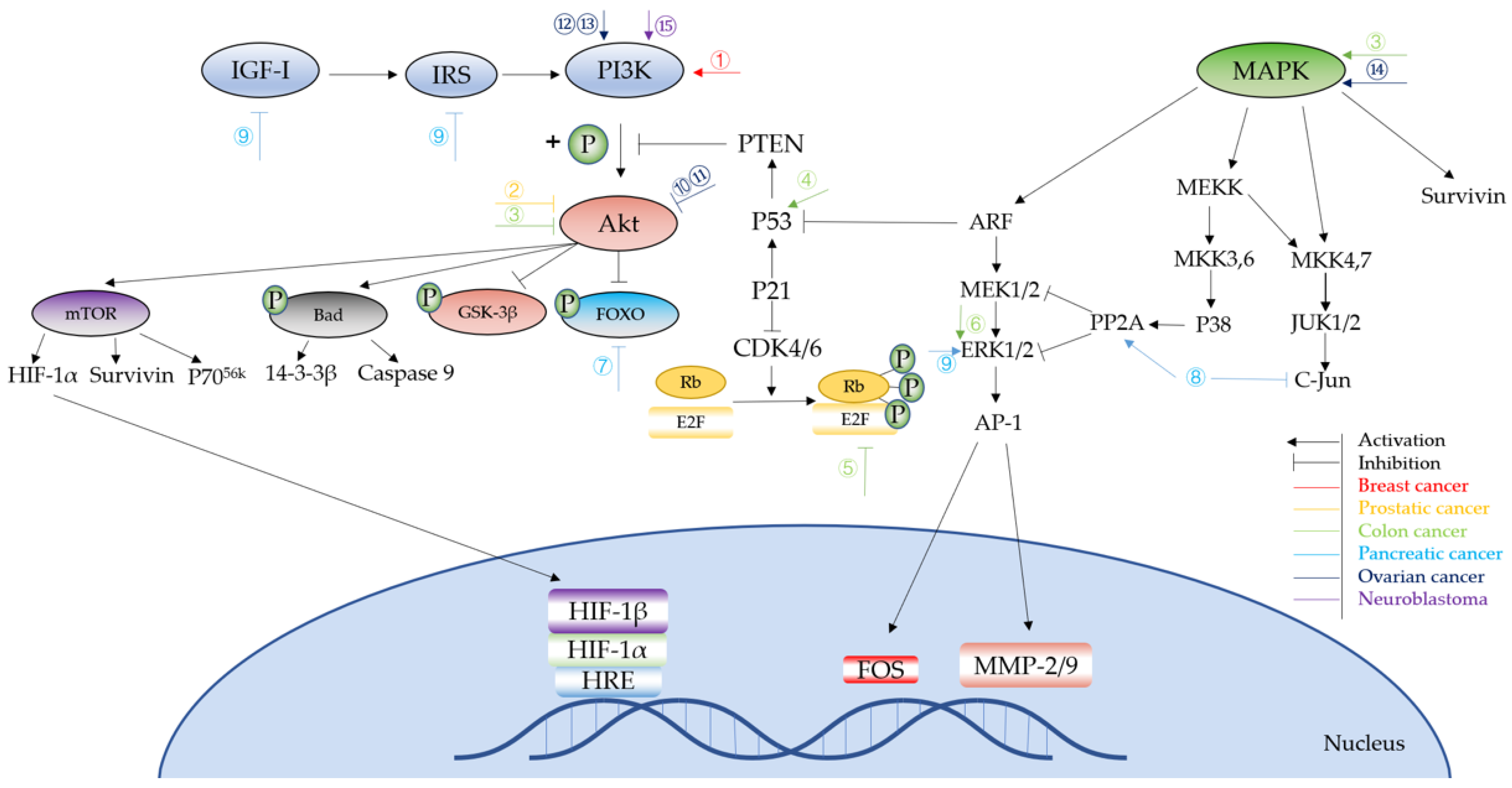
2.1. Breast Cancer
2.2. Lung Cancer
2.3. Prostate Cancer
2.4. Colon Cancer
2.5. Gastric Cancer
2.6. Pancreatic Cancer
2.7. Ovarian Cancer
2.8. Neuroblastoma
3. The Therapeutic Effect of Nitric Oxide
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
| Cancer Types | Experimental Cells | Signaling Pathway/Protein | Pre-Treatment | NO/iNOS/eNOS Level | Result | Ref. |
|---|---|---|---|---|---|---|
| Colon cancer | HT-29 Colon Cancer Cell | ↑Phosphorylation of JNK, p38 MAP kinases, cJun and ATF-2; Activates AP-1 Complex | NO-aspirin | NO↑ | Inhibit cancer cell growth | [14] |
| Breast cancer | ZR-75-30, BT-474, MCF-12F | Akt and ERK1/2 phosphorylation | Inhibitor of NO synthesis | NOS↑ | Tumor cell growth and proliferation | [18] |
| Breast cancer | 4T1 murine breast cancer cells | Akt phosphorylation | 4T1 cancer cell inoculation | NOS2↑ | Tumor cell growth and proliferation | [19] |
| eNOS phosphorylation | NO↓ | Tumor metastasis | ||||
| Lung cancer | Decreasing activity and phosphorylation of eNOS | 4T1 cancer cell inoculation | NO↓ | Metastasis | ||
| Breast cancer | MDA-MB-231 breast cancer cells | Akt phosphorylation | TIMP-1 silencing | NO↑ | Tumor cell growth and proliferation | [20] |
| Breast cancer | MDA-MB-468 breast cancer cells | S-nitrosylation of Ras, Ets-1, MAPK | - | NO↑ | Tumor metastasis | [21] |
| Breast cancer | The murine cell line 66CL4, human breast cancer cell line MCF-7, human triple negative breast cancer (TNBC) cell lines | VEGF, phosphorylation of γ-H2AX foci and RAD51 foci | Glucocorticoids | NO↑ | Angiogenesis | [23] |
| Breast cancer | Human breast cancer cell lines MDA-MB-468, ZR 75-30 and MDA-MB-231 | Dephosphorylation of ERK and Akt | Sodium orthovanadate and antisense oligonucleotides | NO↑ | Cancer cell growth | [24] |
| Breast cancer | Human breast cancer cell lines MDA-MB-231, MCF-7, and MDA-MB-468 | Raf/MEK/ERK and PI-3 kinase/Akt pathways | diethylenetriamine-NONOate (DETA-NONOate) | NO↑ | Proliferation of cancer cell | [25] |
| Lung cancer | C10 and E10 cells, LM1 and LM2 cells | ERK1/2 kinase phosphorylation | ERK 1/2 kinase inhibitor U0126 | NO↑ | Tumor proliferation and growth | [27] |
| Lung cancer | Human colon carcinoma cell line and human lung adenocarcinoma cell line | Tyrosine phosphorylation and p53 gene accumulation | Retroviral vector DFG-iNOS | NO↑NOS↑ | Metastasis | [28] |
| Lung cancer | Bronchiolar Clara cells | Vascular endothelial growth factor | iNOS - | NO↑iNOS↑ | Angiogenesis | [29] |
| Lung cancer | NCI-H157 and NCI-H522 | Tyrosine phosphorylation and MMP expression | - | NO↑NOS↑ | Angiogenesis | [30] |
| Lung cancer | Human non-small lung cancer (NSCLC)-derived cell line H460, H23, and H292 | Akt phosphorylation | - | NO↑ | Promote cancer stem cell | [31] |
| Lung cancer | NCI-H460 cells | Inhibition the ubiquitination of Bcl-2 protein | Sodium nitroprusside, dipropylenetriamine NONOate and cisplatin | NO↑ | increase cell death resistance | [33] |
| Ovarian cancer | SKOV-3 and OVCAR-3 cell lines | ↓Phosphorylation of STAT3, Akt | Spermine nitric oxide complex hydrate (SPER/NO), diethylenetriamine nitric oxide adduct (DETA/NO) | NO↑ | Cancer cell apoptosis | [54] |
| Ovarian cancer | SKOV-3 and OVCAR-3 cell lines | ↑Phosphorylation of STAT3 | Arctigenin, S-nitroso-N-acetylpenicillamine (SNAP) | NO↑ | Inhibit arctigenin- induced cell apoptosis | [55] |
| Ovarian caner | SKOV-3 and OVCAR-3 cell lines | ↑Phosphorylation of p38MAPK, ↓expression of survivin | High concentration of NO donors (SNP, SNAP) or overexpression of iNOS | NO↑ | Cancer cell apoptosis | [57] |
| - | ↑Activation of PI3K/Akt/survivin signaling pathway | Low concentration of NO donors (SNP, SNAP) or ectopic expression of low amounts of iNOS | NO↑ | Cytoprotection | ||
| Ovarian cancer | SKOV-3,OVCARs, A278, C200, OVCAR4, PE01 and PE04 cell lines | ↓Phosphorylation of Akt, STAT3 and p42/44; ↑Nitrosylation of EFGR, Akt, STAT3 and NFκBp65 | S-nitrosoglutathione (GSNO) | NO↑ | Inhibition of cancer cell growth | [59] |
| Ovarian cancer | SKOV-3 cell line | ↑Phosphorylation of ERK, PI3K/Akt and mTOR/p70S6K signaling pathway | Sepiapterin | NO↑ | Increased cancer cell growth and migration | [60] |
| Neuroblastoma | SH-Sy5y and SHEP cell lines | ↑Phosphorylation of JNK | SNP | NO↑ | Cancer cell apoptosis | [62] |
| Neuroblastoma | SH-EP1and SH-SY5Y cell lines | ↑Phosphrylation of ERK1/2 and p38 MAPK | SNP | NO↑ | Cancer cell apoptosis | [63,64] |
| Neuroblastoma | SK-N-MC cells | ↑Activation of PKG/PI3K/Akt signaling pathway, ↑Phosphorylation of Bad | SNAP | NO↑ | Inhibit H2O2- induced cell apoptosis | [66] |
| Gastric cancer | BGC-823 cell line | ↓Phosphorylation of Akt | SNP | NO↑ | Inhibition of cancer cell growth | [44] |
| Gastric cancer | AGS cell line | ↓Phosphorylation of EGFR and ERK1/2 | SNP, L-arginine and overexpression of PKG II | NO↑ | Inhibition of cancer cell growth | [45] |
| Colon adenocarcinoma; Pancreatic adenocarcinoma; Skin carcinoma; cervix adenocarcinoma; Breast adenocarcinoma | SW480, HT-29, HCT-15, and LoVo human colon adenocarcinoma cells; BxPC-3 human pancreatic adenocarcinoma cell; A431 human skin carcinoma cell; HeLa human cervix adenocarcinoma cell; MCF-7 human breast adenocarcinoma cell | ↑Cyclin B1, phosphorylation of Cdk1, oxidative stress, ROS; ↓cyclin D1 and Cdc25C | NO-aspirin | NO↑ | Inhibit cancer cell growth | [37] |
| Colon cancer | Human colon cancer cell lines RKO and RKO-E6; RKOβ-cat cell | ↓NO–induced release of cytochrome c, p53; ↑NO–induced suppression of the antiapoptotic protein, Bcl-xL and Akt phosphorylation | Overexpression β-catenin; DETA- NONOate; sodium nitroprusside | - | Blocks NO–induced cancer cell apoptosis | [39] |
| Colorectal adenocarcinoma | SNU-1040 cell line and HCT-116 cell line | ↑Phosphorylation of p53 | AdiNOS; SNAP | iNOS↑, NO↑ | Cancer cell apoptosis; Inhibit cancer cell growth | [40] |
| Colon cancer | HCT116, HT29, DLD-1, and HCT15 colon cancer cell line; iNOS+/+ and iNOS−/− mice | Rb hyper-phosphorylation and inactivation; ↑activation of sGC/cGMP/PKG pathway, PI3K/AKT and MEK/ERK1/2 pathways | Spermine NONOate | NO↑ | Increase cancer cell growth; reduce cancer cell apoptosis | [41] |
| Colon adenocarcinoma | WiDr, SW 480, SW 620 colon adenocarcinoma cells | ↑Phosphorylation of ERK1/2, activation of AP-1, nuclear translocation of Fra-1/Fra-2, MMP-2/9, RhoB, Rac-1, β-catenin and cyclin D1, cGMP; ↓caspase-8, caspase-3/9 activation and PARP cleavage | SNAP | NO↑ | Increase cancer proliferation, metastasis and invasion; reduce cancer cell apoptosis | [43] |
| Pancreatic ductal adenocarcinoma | LSL-KrasG12D; LSL-Trp53R172H/+; Pdx-1-Cre (KPC) pancreatic cancer mouse model and NOS2-deficient KPC (NKPC) mice; primary pancreatic tumor tissue | ↓Phosphorylation of FOXO, activation of ERK and PI3K/AKT signaling pathway; | iNOS deficiency | iNOS↓ NO↓ | Decrease cancer cell growth; cancer cell apoptosis | [46] |
| Pancreatic cancer | Seventy-two pancreatic adenocarcinoma tissue specimens | ↑iNOS-induced NO | - | - | Cancer cell apoptosis | [48] |
| ↑COX-2 activate NF-κB | Reduce cancer cell apoptosis | |||||
| Pancreatic ductal adenocarcinoma | MiaPaCa II, AsPC-1, and 293T cells; four-week-old SCID-beige mice; Balb/c NU\NU mice | ↑NO, PP2A; ↓MEK/ERK pathway, recruitment of phosphorylated c-Jun, integrin β4 expression, ITGB4 | Netrin-1 | NO↑ | Cancer cell apoptosis; inhibit cancer cell growth | [49] |
| Pancreatic Cancer | MIAPaCa-2, Panc-1, MCF-7, MB 468, and HepG2 cells | ↓Phosphorylation of insulin receptor/IGF-I receptor, IRS-1, IGF-I receptor, Akt/PKB, GSK-3β; ↑phosphorylation of ERK-1/2 | SNAP, GSNO, 1400W | NO↑ | Inhibit cancer proliferation and invasion | [51] |
| BPH, low- and high-grade PIN and prostatic carcinoma | Tissue samples of BPH, low-grade PIN, high-grade PIN and primary prostatic adenocarcinomas | ↑iNOS, iNOS-induced NO | - | NO↑ iNOS↑ | Induce cancer cell metastasis and tumorigenesis | [36] |
| Prostate cancer | PC-3 prostate cancer cell line | ↓Activity of HRE promoter, Akt phosphorylation; inhibit HIF-1α translation | NO-sulindac | NO↑ | Inhibit cancer cell growth and invasion; Cancer cell apoptosis | [34] |
References
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef]
- Pieroni, L.; Iavarone, F.; Olianas, A.; Greco, V.; Desiderio, C.; Martelli, C.; Manconi, B.; Sanna, M.T.; Messana, I.; Castagnola, M.; et al. Enrichments of post-translational modifications in proteomic studies. J. Sep. Sci. 2020, 43, 313–336. [Google Scholar] [CrossRef]
- Humphrey, S.; James, D.E.; Mann, M. Protein Phosphorylation: A Major Switch Mechanism for Metabolic Regulation. Trends Endocrinol. Metab. 2015, 26, 676–687. [Google Scholar] [CrossRef]
- Arrington, J.V.; Hsu, C.-C.; Elder, S.G.; Tao, W.A. Recent advances in phosphoproteomics and application to neurological diseases. Analyst 2017, 142, 4373–4387. [Google Scholar] [CrossRef]
- Singh, V.; Ram, M.; Kumar, R.; Prasad, R.; Roy, B.K.; Singh, K.K. Phosphorylation: Implications in Cancer. Protein J. 2017, 36, 1–6. [Google Scholar] [CrossRef]
- Crane, B.R.; Arvai, A.S.; Ghosh, D.K.; Wu, C.; Getzoff, E.D.; Stuehr, D.J.; Tainer, J.A. Structure of nitric oxide synthase oxygenase dimer with pterin and substrate. Science 1998, 279, 2121–2126. [Google Scholar] [CrossRef]
- Kamm, A.; Przychodzen, P.; Kuban-Jankowska, A.; Jacewicz, D.; Dabrowska, A.M.; Nussberger, S.; Wozniak, M.; Gorska-Ponikowska, M. Nitric oxide and its derivatives in the cancer battlefield. Nitric Oxide 2019, 93, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Crowell, A.J.; Steele, E.V.; Sigman, C.C.; Fay, J.R. Is inducible nitric oxide synthase a target for chemoprevention? Mol. Cancer Ther. 2003, 2, 815–823. [Google Scholar] [PubMed]
- Fukumura, D.; Kashiwagi, S.; Jain, R.K. The role of nitric oxide in tumor progression. Nat. Rev. Cancer 2006, 6, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Vannini, F.; Kashfi, K.; Nath, N. The dual role of iNOS in cancer. Redox Biol. 2015, 6, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Kashfi, K. Nitric oxide in cancer and beyond. Biochem. Pharmacol. 2020, 176, 114006. [Google Scholar] [CrossRef]
- Fukumura, D.; Jain, R.K. Role of nitric oxide in angiogenesis and microcirculation in tumors. Cancer Metastasis Rev. 1998, 17, 77–89. [Google Scholar] [CrossRef]
- Alimoradi, H.; Greish, K.; Gamble, A.B.; Giles, G.I. Controlled Delivery of Nitric Oxide for Cancer Therapy. Pharm. Nanotechnol. 2019, 7, 279–303. [Google Scholar] [CrossRef]
- Hundley, T.R.; Rigas, B. Nitric Oxide-Donating Aspirin Inhibits Colon Cancer Cell Growth via Mitogen-Activated Protein Kinase Activation. J. Pharmacol. Exp. Ther. 2005, 316, 25–34. [Google Scholar] [CrossRef]
- Narayanankutty, A. PI3K/ Akt/ mTOR Pathway as a Therapeutic Target for Colorectal Cancer: A Review of Preclinical and Clinical Evidence. Curr. Drug Targets 2019, 20, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Mishra, D.; Patel, V.; Banerjee, D. Nitric Oxide and S-Nitrosylation in Cancers: Emphasis on Breast Cancer. Breast Cancer: Basic Clin. Res. 2020, 14. [Google Scholar] [CrossRef]
- Basudhar, D.; Somasundaram, V.; de Oliveira, G.A.; Kesarwala, A.; Heinecke, J.L.; Cheng, R.Y.; Glynn, S.A.; Ambs, S.; Wink, D.A.; Ridnour, L.A. Nitric Oxide Synthase-2-Derived Nitric Oxide Drives Multiple Pathways of Breast Cancer Progression. Antioxidants Redox Signal. 2017, 26, 1044–1058. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Kawahara, B.; Chaudhuri, G. Mitochondrial-associated nitric oxide synthase activity inhibits cytochrome c oxidase: Im-plications for breast cancer. Free Radic. Biol. Med. 2013, 57, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Smeda, M.; Kieronska, A.; Adamski, M.G.; Proniewski, B.; Sternak, M.; Mohaissen, T.; Przyborowski, K.; Derszniak, K.; Kaczor, D.; Stojak, M.; et al. Nitric oxide deficiency and endothelial–mesenchymal transition of pulmonary endothelium in the progression of 4T1 metastatic breast cancer in mice. Breast Cancer Res. 2018, 20, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ridnour, L.A.; Barasch, K.M.; Windhausen, A.N.; Dorsey, T.H.; Lizardo, M.M.; Yfantis, H.G.; Lee, N.H.; Switzer, C.H.; Cheng, R.Y.S.; Heinecke, J.L.; et al. Nitric Oxide Synthase and Breast Cancer: Role of TIMP-1 in NO-mediated Akt Activation. PLoS ONE 2012, 7, e44081. [Google Scholar] [CrossRef]
- Marshall, E.H.; Foster, M.W. S-nitrosylation of Ras in breast cancer. Breast Cancer Res. 2012, 14, 1–2. [Google Scholar] [CrossRef]
- Monteiro, H.P.; Rodrigues, E.G.; Reis, A.K.A.; Longo, L.S.; Ogata, F.T.; Moretti, A.I.; da Costa, P.E.; Teodoro, A.C.; Toledo, M.S.; Stern, A. Nitric oxide and interactions with reactive oxygen species in the development of melanoma, breast, and colon cancer: A redox signaling perspective. Nitric Oxide 2019, 89, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, R.L.; Intabli, H.; Falcinelli, M.; Bucca, G.; Hesketh, A.; Patel, B.A.; Allen, M.; Smith, C.; Flint, M.S. Stress hormone-mediated acceleration of breast cancer metastasis is halted by inhibition of nitric oxide synthase. Cancer Lett. 2019, 459, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Pervin, S.; Singh, R.; Freije, A.W.; Chaudhuri, G. MKP-1-induced dephosphorylation of extracellular signal-regulated kinase is essential for triggering nitric oxide-induced apoptosis in human breast cancer cell lines: Implications in breast cancer. Cancer Res. 2003, 63, 8853–8860. [Google Scholar] [PubMed]
- Pervin, S.; Singh, R.; Hernandez, E.; Wu, G.; Chaudhuri, G. Nitric Oxide in Physiologic Concentrations Targets the Translational Machinery to Increase the Proliferation of Human Breast Cancer Cells: Involvement of Mammalian Target of Rapamycin/eIF4E Pathway. Cancer Res. 2007, 67, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.; Singh, R.P.; Ramasamy, K.; Raina, K.; Redente, E.F.; Dwyer-Nield, L.D.; Radcliffe, R.A.; Malkinson, A.M.; Agarwal, R. Growth inhibition and regression of lung tumors by silibinin: Modulation of angiogenesis by macrophage-associated cytokines and nuclear factor-kappaB and signal transducers and activators of transcription 3. Cancer Prev. Res. 2009, 2, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.L.; Barrett, B.S.; Fritz, J.M.; Srebernak, M.C.; Kisley, L.R.; Malkinson, A.M.; Dwyer-Nield, L.D. Regulation of cytokine-induced prostanoid and nitric oxide synthesis by extracellular signal–regulated kinase 1/2 in lung epithelial cells. Exp. Lung Res. 2010, 36, 558–571. [Google Scholar] [CrossRef]
- Forrester, K.; Ambs, S.; Lupold, S.E.; Kapust, R.B.; Spillare, E.A.; Weinberg, W.C.; Felley-Bosco, E.; Wang, X.W.; Geller, D.A.; Tzeng, E.; et al. Nitric oxide-induced p53 accumulation and regulation of inducible nitric oxide synthase expression by wild-type p53. Proc. Natl. Acad. Sci. USA 1996, 93, 2442–2447. [Google Scholar] [CrossRef]
- Kisley, L.R.; Barrett, B.S.; Bauer, A.K.; Dwyer-Nield, L.D.; Barthel, B.; Meyer, A.M.; Thompson, D.C.; Malkinson, A.M. Genetic ablation of inducible nitric oxide synthase decreases mouse lung tumorigenesis. Cancer Res. 2002, 62, 6850–6856. [Google Scholar]
- Phillips, P.G.; Birnby, L.M.; Narendran, A.; Milonovich, W.L. Nitric oxide modulates capillary formation at the endothelial cell-tumor cell interface. Am. J. Physiol. Cell. Mol. Physiol. 2001, 281, L278–L290. [Google Scholar] [CrossRef]
- Maiuthed, A.; Bhummaphan, N.; Luanpitpong, S.; Mutirangura, A.; Aporntewan, C.; Meeprasert, A.; Rungrotmongkol, T.; Rojanasakul, Y.; Chanvorachote, P. Nitric oxide promotes cancer cell dedifferentiation by disrupting an Oct4:caveolin-1 complex: A new regulatory mechanism for cancer stem cell formation. J. Biol. Chem. 2018, 293, 13534–13552. [Google Scholar] [CrossRef]
- Tyryshkin, A.; Gorgun, F.; Fattah, E.A.; Mazumdar, T.; Pandit, L.; Zeng, S.; Eissa, N. Src Kinase-mediated Phosphorylation Stabilizes Inducible Nitric-oxide Synthase in Normal Cells and Cancer Cells. J. Biol. Chem. 2010, 285, 784–792. [Google Scholar] [CrossRef]
- Chanvorachote, P.; Nimmannit, U.; Stehlik, C.; Wang, L.; Jiang, B.-H.; Ongpipatanakul, B.; Rojanasakul, Y. Nitric Oxide Regulates Cell Sensitivity to Cisplatin-Induced Apoptosis through S-Nitrosylation and Inhibition of Bcl-2 Ubiquitination. Cancer Res. 2006, 66, 6353–6360. [Google Scholar] [CrossRef]
- Stewart, G.D.; Nanda, J.; Brown, D.J.; Riddick, A.C.; Ross, J.A.; Habib, F.K. NO-sulindac inhibits the hypoxia response of PC-3 prostate cancer cells via the Akt signalling pathway. Int. J. Cancer 2009, 124, 223–232. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Baltaci, S.; Orhan, D.; Gögüs, Ç.; Türkölmez, K.; Tulunay, Ö.; Gögüs, O. Inducible nitric oxide synthase expression in benign prostatic hyperplasia, low- and high-grade prostatic intraepithelial neoplasia and prostatic carcinoma. BJU Int. 2001, 88, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Williams, J.L. Abstract 3817: Nitric oxide-donating aspirin induces G2/M phase cell cycle arrest in human cancer cells by regulating phase transition proteins. Exp. Mol. Ther. 2012, 72, 3817. [Google Scholar] [CrossRef]
- Menon, S.G.; Goswami, P.C. A redox cycle within the cell cycle: Ring in the old with the new. Oncogene 2006, 26, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; MacNaughton, W.K. Overexpressed beta-catenin blocks nitric oxide-induced apoptosis in colonic cancer cells. Cancer Res. 2005, 65, 8604–8607. [Google Scholar] [CrossRef] [PubMed]
- Cook, T.; Wang, Z.; Alber, S.; Liu, K.; Watkins, S.C.; Vodovotz, Y.; Billiar, T.R.; Blumberg, D. Nitric oxide and ionizing radiation syner-gistically promote apoptosis and growth inhibition of cancer by activating p53. Cancer Res. 2004, 64, 8015–8021. [Google Scholar] [CrossRef]
- Stern, A.; Costa, P.; Monteiro, H.; Reis, A. Nitric oxide: Protein tyrosine phosphorylation and protein S-nitrosylation in cancer. Biomed. J. 2015, 38, 380. [Google Scholar] [CrossRef]
- Ying, L.; Hofseth, A.B.; Browning, D.D.; Nagarkatti, M.; Nagarkatti, P.S.; Hofseth, L.J. Nitric Oxide Inactivates the Retinoblastoma Pathway in Chronic Inflammation. Cancer Res. 2007, 67, 9286–9293. [Google Scholar] [CrossRef]
- Babykutty, S.; Suboj, P.; Srinivas, P.; Nair, A.S.; Chandramohan, K.; Gopala, S. Insidious role of nitric oxide in migration/invasion of colon cancer cells by upregulating MMP-2/9 via activation of cGMP-PKG-ERK signaling pathways. Clin. Exp. Metastasis 2012, 29, 471–472. [Google Scholar] [CrossRef] [PubMed]
- Sang, J.; Chen, Y.; Tao, Y. Nitric oxide inhibits gastric cancer cell growth through the modulation of the Akt pathway. Mol. Med. Rep. 2011, 4, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Wu, Y.; Zhu, M.; Qian, H.; Chen, Y. Nitric oxide/cyclic guanosine monophosphate inducers sodium nitroprusside and L-arginine inhibit the proliferation of gastric cancer cells via the activation of type II cyclic guanosine monophos-phate-dependent protein kinase. Oncol. Lett. 2015, 10, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; He, P.; Gaida, M.; Yang, S.; Schetter, A.J.; Gaedcke, J.; Ghadimi, B.M.; Ried, T.; Yfantis, H.; Lee, D.; et al. Inducible nitric oxide synthase enhances disease aggressiveness in pancreatic cancer. Oncotarget 2016, 7, 52993–53004. [Google Scholar] [CrossRef] [PubMed]
- Paik, J.H.; Kollipara, R.; Chu, G.; Ji, H.; Xiao, Y.; Ding, Z.; Miao, L.; Tothova, Z.; Horner, J.W.; Carrasco, D.R.; et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell 2007, 128, 309–323. [Google Scholar] [CrossRef]
- Kong, G.; Kim, E.K.; Kim, W.S.; Lee, K.T.; Lee, Y.W.; Lee, J.K.; Paik, S.W.; Rhee, J.C. Role of cyclooxygenase-2 and inducible nitric oxide synthase in pancreatic cancer. J. Gastroenterol. Hepatol. 2002, 17, 914–921. [Google Scholar] [CrossRef] [PubMed]
- An, X.-Z.; Zhao, Z.-G.; Luo, Y.; Zhang, R.; Tang, X.-Q.; Hao, D.-L.; Zhao, X.; Lv, X.; Liu, D.-P. Netrin-1 suppresses the MEK/ERK pathway and ITGB4 in pancreatic cancer. Oncotarget 2016, 7, 24719–24733. [Google Scholar] [CrossRef][Green Version]
- Bergmann, U.; Funatomi, H.; Kornmann, M.; Beger, H.G.; Korc, M. Increased Expression of Insulin Receptor Substrate-1 in Human Pancreatic Cancer. Biochem. Biophys. Res. Commun. 1996, 220, 886–890. [Google Scholar] [CrossRef]
- Sugita, H.; Kaneki, M.; Furuhashi, S.; Hirota, M.; Takamori, H.; Baba, H. Nitric oxide inhibits the proliferation and invasion of pan-creatic cancer cells through degradation of insulin receptor substrate-1 protein. Mol. Cancer Res. 2010, 8, 1152–1163. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.; Ralyea, C.; Lockwood, S. Ovarian Cancer: An Integrated Review. Semin. Oncol. Nurs. 2019, 35, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shen, Y.; Wang, S.; Shen, Q.; Zhou, X. The role of STAT3 in leading the crosstalk between human cancers and the immune system. Cancer Lett. 2018, 415, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Kielbik, M.; Klink, M.; Brzezinska, M.; Szulc, I.; Sulowska, Z. Nitric oxide donors: Spermine/NO and diethylenetriamine/NO induce ovarian cancer cell death and affect STAT3 and AKT signaling proteins. Nitric Oxide 2013, 35, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Li, L.A.; Meng, Y.G.; You, Y.Q.; Fu, X.Y.; Song, L. Arctigenin promotes apoptosis in ovarian cancer cells via the iN-OS/NO/STAT3/survivin signalling. Basic Clin. Pharmacol. Toxicol. 2014, 115, 507–511. [Google Scholar] [CrossRef]
- Ferrandina, G.; Legge, F.; Martinelli, E.; Ranelletti, O.F.; Zannoni, G.F.; Lauriola, L.; Gessi, M.; Gallotta, V.; Scambia, G. Survivin expression in ovarian cancer and its correlation with clinico-pathological, surgical and apoptosis-related parameters. Br. J. Cancer 2005, 92, 271–277. [Google Scholar] [CrossRef]
- Engels, K.; Knauer, S.K.; Loibl, S.; Fetz, V.; Harter, P.; Schweitzer, A.; Fisseler-Eckhoff, A.; Kommoss, F.; Hanker, L.; Nekljudova, V.; et al. NO Signaling Confers Cytoprotectivity through the Survivin Network in Ovarian Carcinomas. Cancer Res. 2008, 68, 5159–5166. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.; Chan, S.L.; Chan, S.S.L.; Fiscus, R.R.; Tsang, B.K. Regulation of p53 and suppression of apoptosis by the soluble guanylyl cyclase/cGMP pathway in human ovarian cancer cells. Oncogene 2005, 25, 2203–2212. [Google Scholar] [CrossRef] [PubMed]
- Gir, S.; Rattan, R.; Deshpande, M.; Maguire, J.L.; Johnson, Z.; Graham, R.P.; Shridhar, V. Preclinical therapeutic potential of a nitro-sylating agent in the treatment of ovarian cancer. PLoS ONE 2014, 9, e97897. [Google Scholar]
- Choi, S.W.; Cho, Y.-R.; Kim, S.H.; Ko, H.Y.; Kim, M.-D.; Seo, D.-W. Sepiapterin inhibits cell proliferation and migration of ovarian cancer cells via down-regulation of p70S6K-dependent VEGFR-2 expression. Oncol. Rep. 2011, 26, 861–867. [Google Scholar] [CrossRef]
- Behrens, A.; Sibilia, M.; Wagner, E.F. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat. Genet. 1999, 21, 326–329. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Feng, Z.; Porter, A.G. JNK-dependent Phosphorylation of c-Jun on Serine 63 Mediates Nitric Oxide-induced Apoptosis of Neuroblastoma Cells. J. Biol. Chem. 2004, 279, 4058–4065. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Guo, D.; Zhang, W.; Xie, Y.; Yang, H.; Cheng, B.; Wang, L.; Yang, R.; Bi, J.; Feng, Z. Biglycan, a Nitric Oxide-Downregulated Proteoglycan, Prevents Nitric Oxide-Induced Neuronal Cell Apoptosis via Targeting Erk1/2 and p38 Signaling Pathways. J. Mol. Neurosci. 2018, 66, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Nashida, T.; Takuma, K.; Fukuda, S.; Kawasaki, T.; Takahashi, T.; Baba, A.; Ago, Y.; Matsuda, T. The specific Na+/Ca2+ exchange inhibitor SEA0400 prevents nitric oxide-induced cytotoxicity in SH-SY5Y cells. Neurochem. Int. 2011, 59, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Ha, K.S.; Kim, K.M.; Kwon, Y.G.; Bai, S.K.; Nam, W.D.; Yoo, Y.M.; Kim, P.K.; Chung, H.T.; Billiar, T.R.; Kim, Y.M. Nitric oxide prevents 6-hydroxydopamine-induced apoptosis in PC12 cells through cGMP-dependent PI3 kinase/Akt activation. FASEB J. 2003, 17, 1036–1047. [Google Scholar] [CrossRef]
- Yoo, Y.M.; Jung, E.M.; Ahn, C.; Jeung, E.B. Nitric oxide prevents H2O2-induced apoptosis in SK-N-MC human neuroblastoma cells. Int. J. Biol. Sci. 2018, 14, 1974–1984. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-F.; Diers, A.R.; Hogg, N. Cancer cell metabolism and the modulating effects of nitric oxide. Free. Radic. Biol. Med. 2015, 79, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Choudhari, S.K.; Chaudhary, M.; Bagde, S.; Gadbail, A.R.; Joshi, V. Nitric oxide and cancer: A review. World J. Surg. Oncol. 2013, 11, 118. [Google Scholar] [CrossRef]
- Yasuda, H. Solid tumor physiology and hypoxia-induced chemo/radio-resistance: Novel strategy for cancer therapy: Nitric oxide donor as a therapeutic enhancer. Nitric Oxide 2008, 19, 205–216. [Google Scholar] [CrossRef]
- Kang, Y.; Kim, J.; Park, J.; Lee, Y.M.; Saravanakumar, G.; Park, K.M.; Choi, W.; Kim, K.; Lee, E.; Kim, C.; et al. Tumor vasodilation by N-Heterocyclic carbene-based nitric oxide delivery triggered by high-intensity focused ultrasound and enhanced drug homing to tumor sites for anti-cancer therapy. Biomaterials 2019, 217, 119297. [Google Scholar] [CrossRef]
- Huerta, S. Nitric oxide for cancer therapy. Futur. Sci. OA 2015, 1, 44. [Google Scholar] [CrossRef]
- Frederiksen, L.J.; Sullivan, R.; Maxwell, L.R.; MacDonald-Goodfellow, S.K.; Adams, M.A.; Bennett, B.M.; Siemens, D.R.; Graham, C.H. Chemosensitization of Cancer In vitro and In vivo by Nitric Oxide Signaling. Clin. Cancer Res. 2007, 13, 2199–2206. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Cai, A.; Peng, Z.; Liang, W.; Xi, H.; Li, P.; Chen, G.; Yu, J.; Chen, L. JS-K induces reactive oxygen species-dependent anti-cancer effects by targeting mitochondria respiratory chain complexes in gastric cancer. J. Cell. Mol. Med. 2019, 23, 2489–2504. [Google Scholar] [CrossRef] [PubMed]
- Ishima, Y.; Inoue, A.; Fang, J.; Kinoshita, R.; Ikeda, M.; Watanabe, H.; Maeda, H.; Otagiri, M.; Maruyama, T. Poly-S-nitrosated human albumin enhances the antitumor and antimetastasis effect of bevacizumab, partly by inhibiting autophagy through the gen-eration of nitric oxide. Cancer Sci. 2015, 106, 194–200. [Google Scholar] [CrossRef]
- Wang, L.; Chang, Y.; Feng, Y.; Li, X.; Cheng, Y.; Jian, H.; Ma, X.; Zheng, R.; Wu, X.; Xu, K.; et al. Nitric Oxide Stimulated Programmable Drug Release of Nanosystem for Multidrug Resistance Cancer Therapy. Nano Lett. 2019, 19, 6800–6811. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Song, X.; Zhu, W.; Chen, Y.; Zhu, R.; Wang, L.; Chen, X.; Song, J.; Yang, H. Light-Switchable Yolk-Mesoporous Shell UCNPs@MgSiO3 for Nitric Oxide-Evoked Multidrug Resistance Reversal in Cancer Therapy. ACS Appl. Mater. Interfaces 2020, 12, 30066–30076. [Google Scholar] [CrossRef]
- Chegaev, K.; Fraix, A.; Gazzano, E.; Abd-Ellatef, G.E.F.; Blangetti, M.; Rolando, B.; Conoci, S.; Riganti, C.; Fruttero, R.; Gasco, A.; et al. Light-Regulated NO Release as a Novel Strategy to Overcome Doxorubicin Multidrug Resistance. ACS Med. Chem. Lett. 2017, 8, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yung, B.; Kim, W.J.; Chen, X. Combination of nitric oxide and drug delivery systems: Tools for overcoming drug resistance in chemotherapy. J. Control. Release 2017, 263, 223–230. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Zhang, Y.; Wang, Y.; Yang, M.; Hong, F.; Yang, S. Protein Phosphorylation in Cancer: Role of Nitric Oxide Signaling Pathway. Biomolecules 2021, 11, 1009. https://doi.org/10.3390/biom11071009
Liu X, Zhang Y, Wang Y, Yang M, Hong F, Yang S. Protein Phosphorylation in Cancer: Role of Nitric Oxide Signaling Pathway. Biomolecules. 2021; 11(7):1009. https://doi.org/10.3390/biom11071009
Chicago/Turabian StyleLiu, Xinran, Yiping Zhang, Yijie Wang, Meiwen Yang, Fenfang Hong, and Shulong Yang. 2021. "Protein Phosphorylation in Cancer: Role of Nitric Oxide Signaling Pathway" Biomolecules 11, no. 7: 1009. https://doi.org/10.3390/biom11071009
APA StyleLiu, X., Zhang, Y., Wang, Y., Yang, M., Hong, F., & Yang, S. (2021). Protein Phosphorylation in Cancer: Role of Nitric Oxide Signaling Pathway. Biomolecules, 11(7), 1009. https://doi.org/10.3390/biom11071009