
Phosphorylation of RCC1 on Serine 11 Facilitates G1/S Transition in HPV E7-Expressing Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Western Blot
2.3. Nuclear and Cytoplasmic Protein Extraction
2.4. siRNAs and Plasmid Transfection
- siRCC1, 5′- TGGAGATGATGGGCAAACA-3′;
- siCDK1, 5′-GATCAACTCTTCAGGATTT-3′;
- siAKT, 5′-GAAGGAAGUCAUCGUGGCC-3′;
- E6, 5′-UACAACAAACCGUUGUGUG-3′;
- E7, 5′-GCACACACGUAGACAUUCG-3′.
2.5. Flow Cytometric (FACS) Analysis
2.6. BrdU Labeling
2.7. Statistical Analysis
3. Results
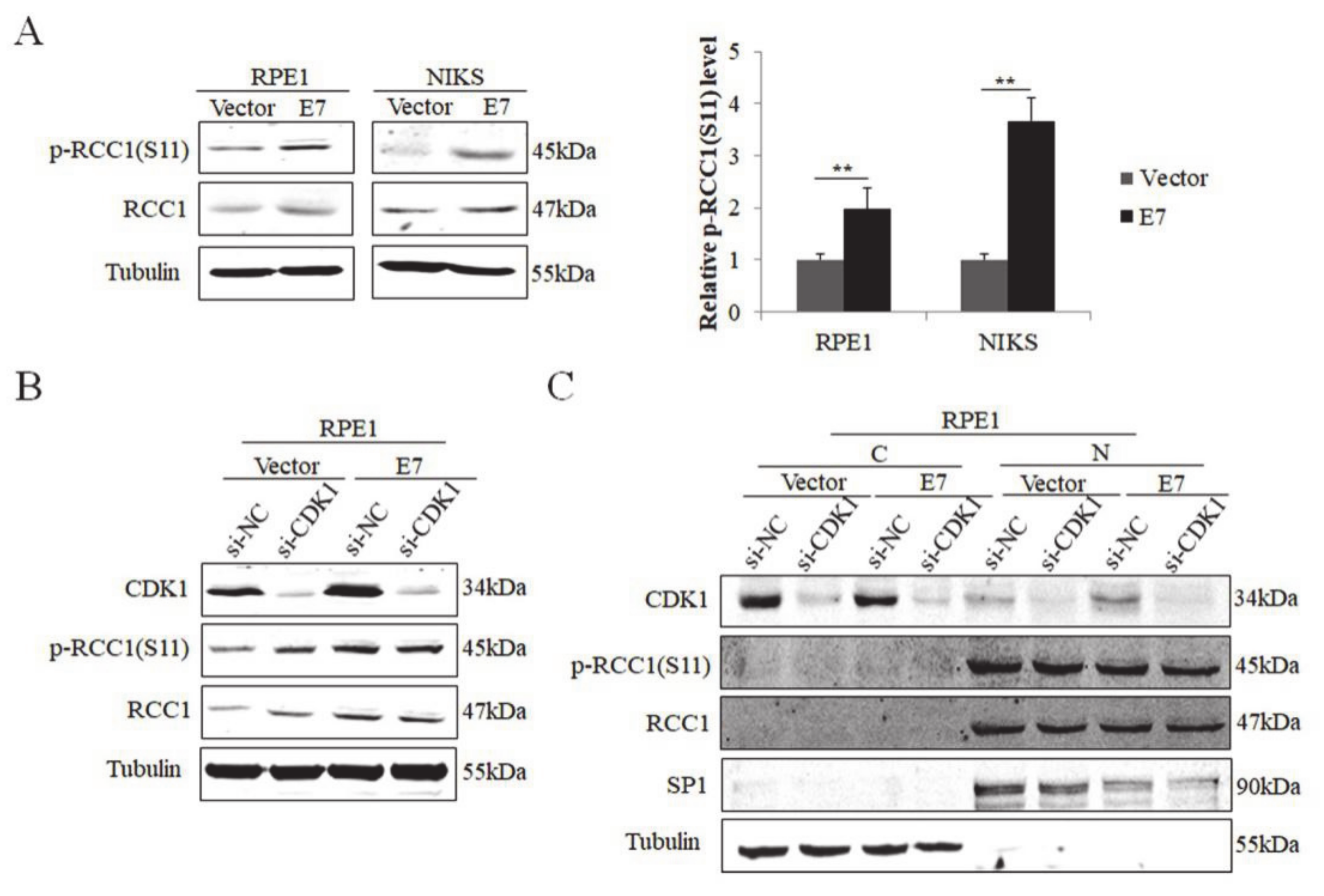
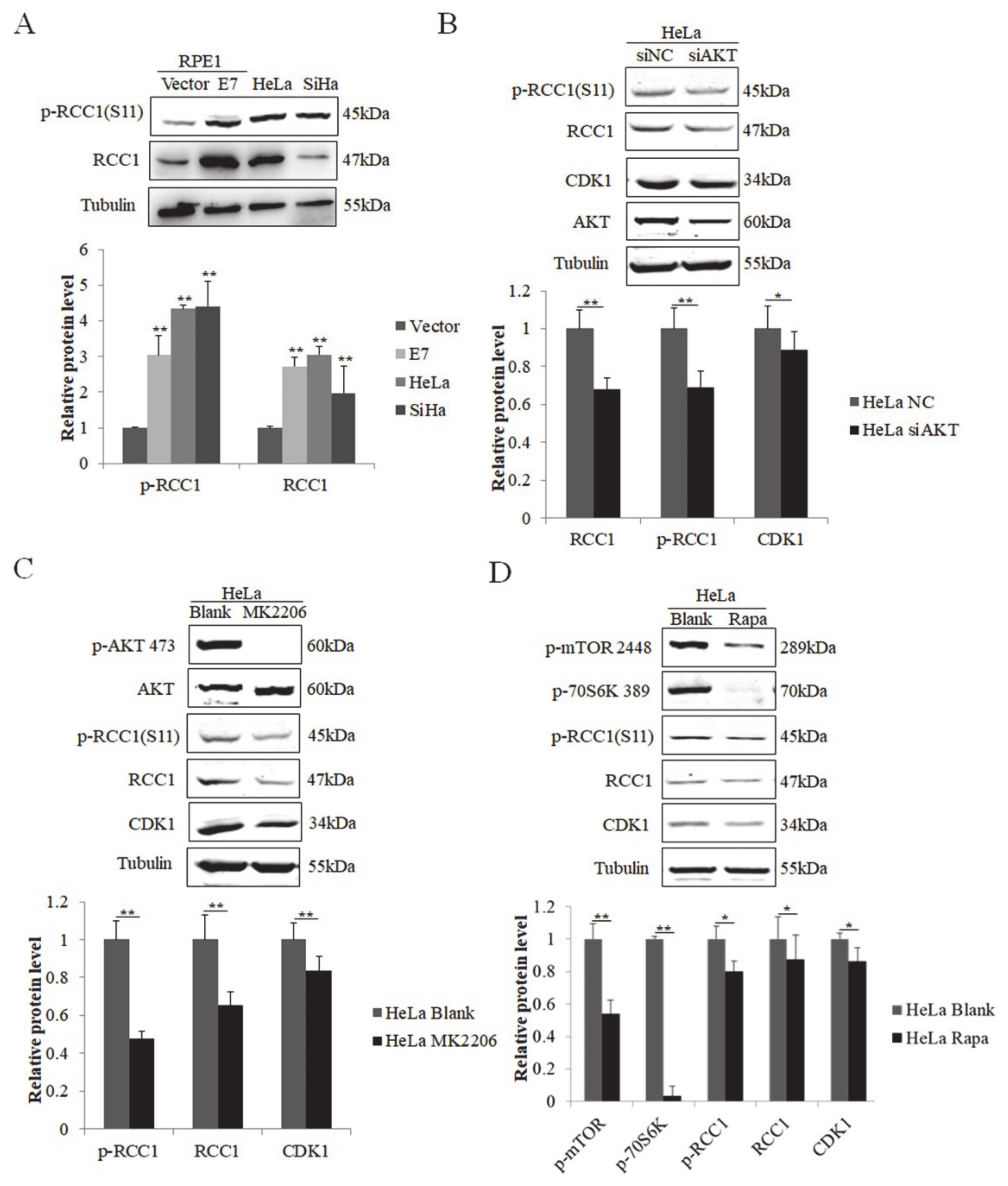
3.1. S11 of RCC1 Is Phosphorylated in HPV16 E7-Expressing Cells
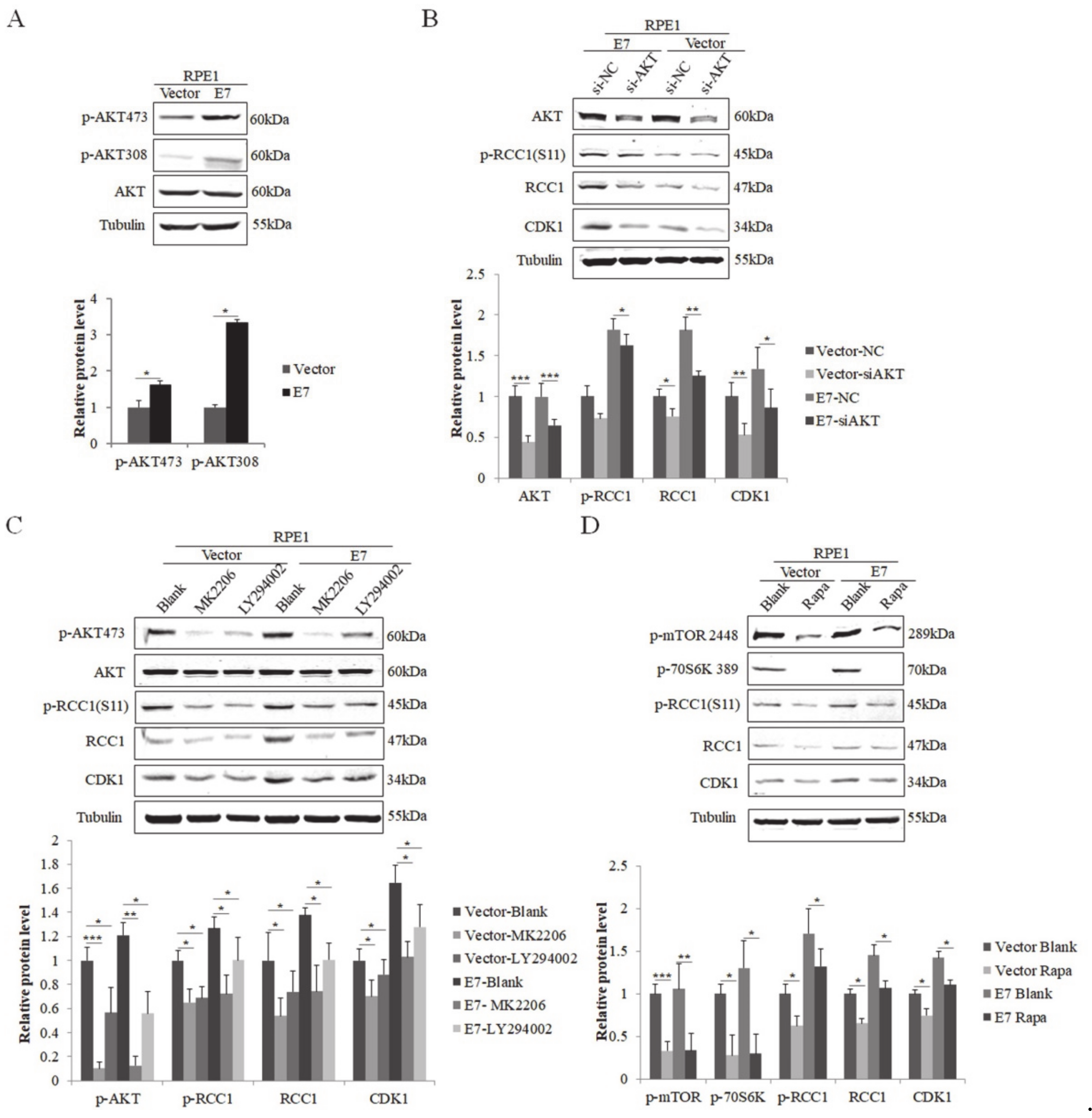
3.2. HPV E7 Phosphorylates RCC1 by the PI3K/AKT/mTOR Signaling Pathway
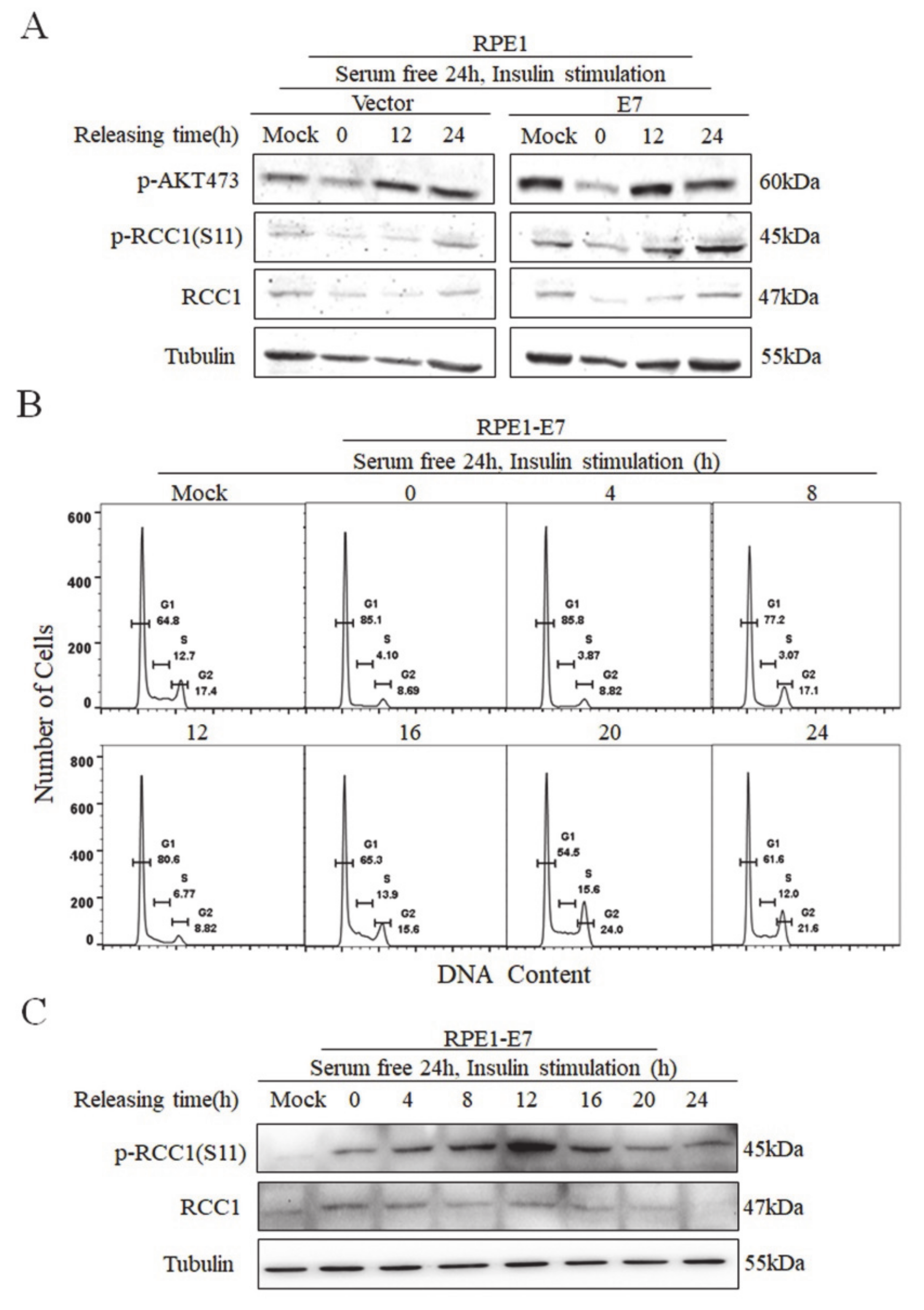
3.3. Phosphorylation of RCC1 S11 Occurs throughout the Cell Cycle
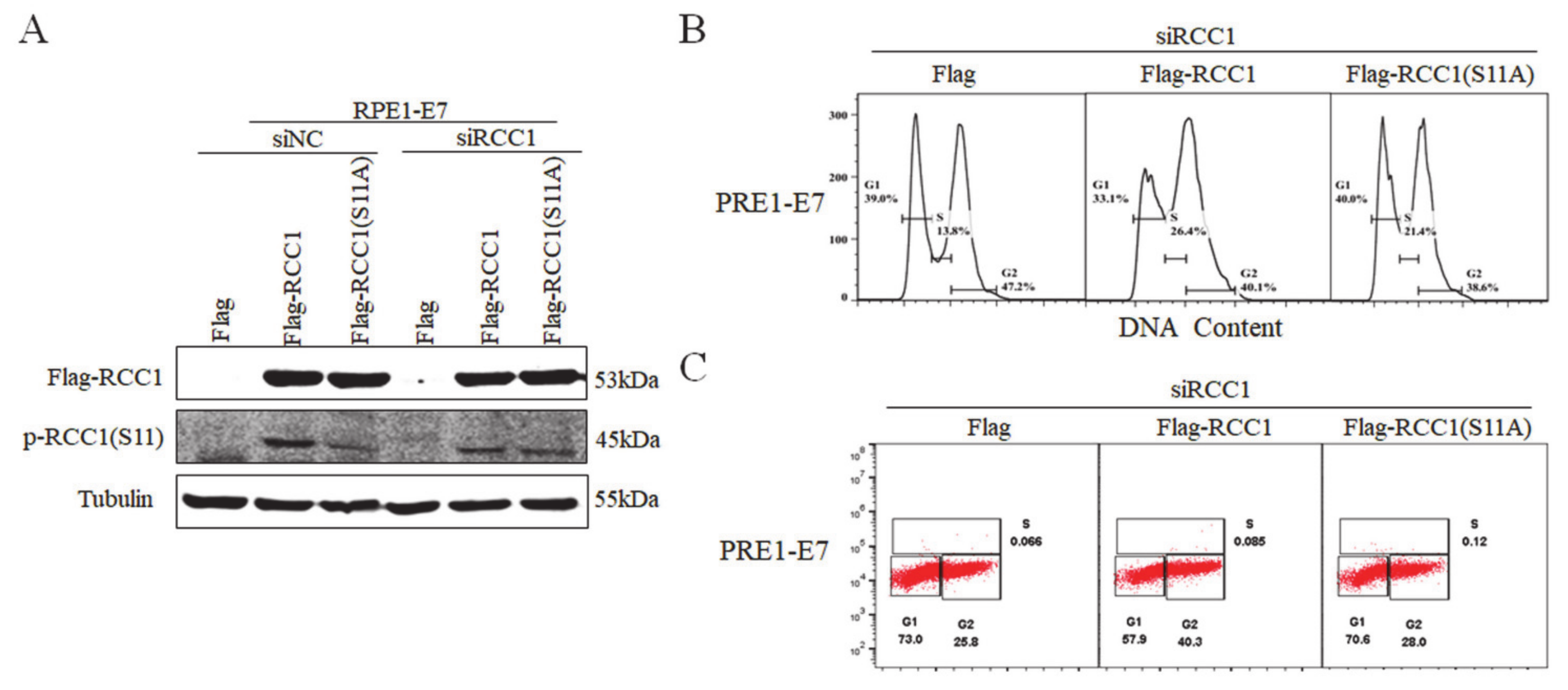
3.4. Non-Phosphorylation of RCC1 on S11 Inhibits the G1/S Transition
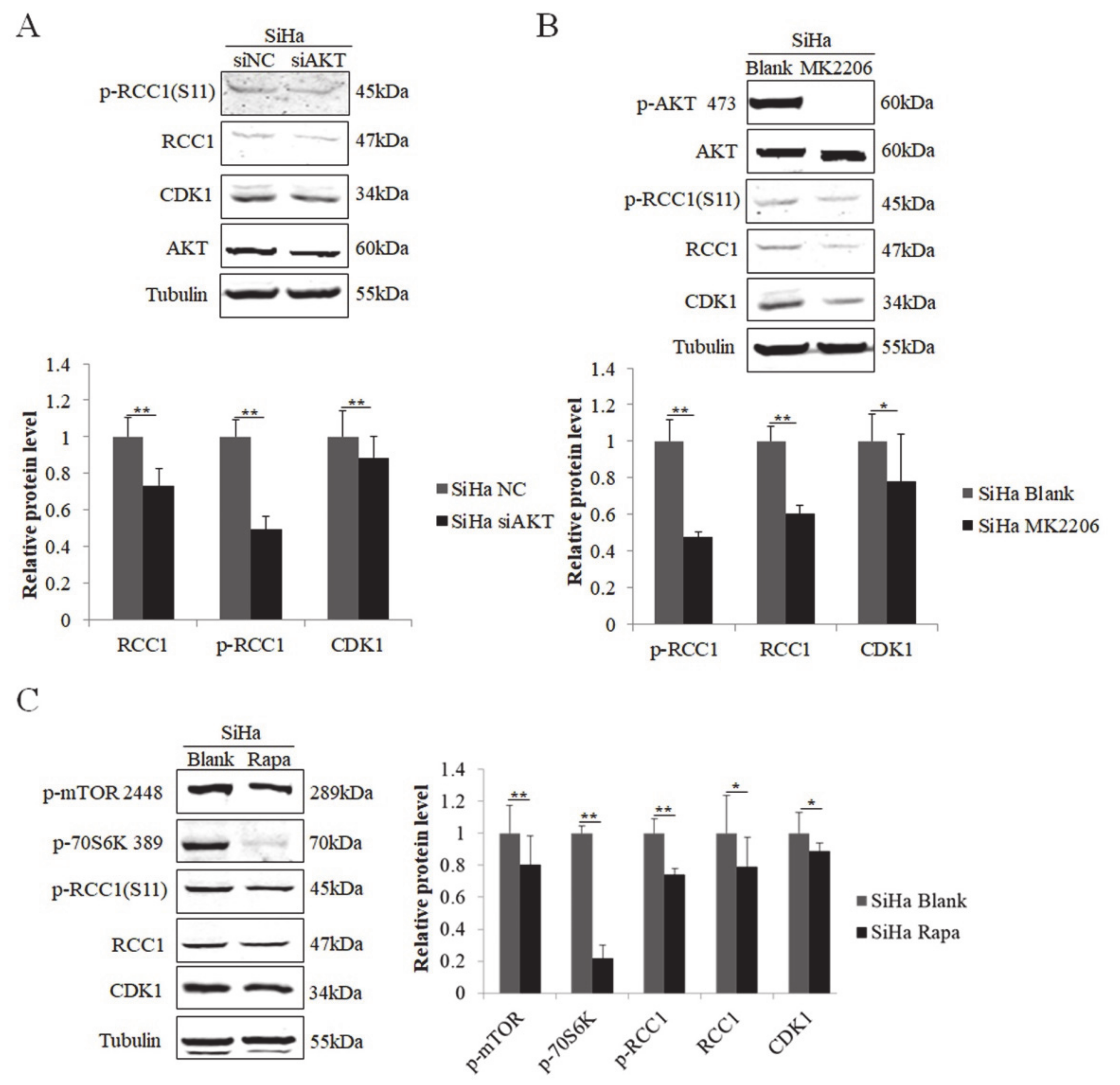
3.5. S11 of RCC1 Is Phosphorylated by the PI3K/AKT/mTOR Pathway in HPV-Positive Cervical Cancer Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ibeanu, O.A. Molecular pathogenesis of cervical cancer. Cancer Biol. Ther. 2011, 11, 295–306. [Google Scholar] [CrossRef]
- Buck, C.B.; Thompson, C.D. Production of papillomavirus-based gene transfer vectors. Curr. Protoc. Cell Biol. 2007, 26, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Zur, H.H.; de Villiers, E.M. Human papillomaviruses. Annu. Rev. Microbiol. 1994, 48, 427–447. [Google Scholar]
- Chen, J.J.; Reid, C.E.; Band, V.; Androphy, E.J. Interaction of papillomavirus E6 oncoproteins with a putative calcium-binding protein. Science 1995, 269, 529–531. [Google Scholar] [CrossRef] [PubMed]
- Dyson, N.; Howley, P.M.; Munger, K.; Harlow, E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 1989, 243, 934–937. [Google Scholar] [CrossRef]
- McLaughlin-Drubin, M.E.; Munger, K. The human papillomavirus E7 oncoprotein. Virology 2009, 384, 335–344. [Google Scholar] [CrossRef]
- Dasso, M. RCC1 in the cell cycle: The regulator of chromosome condensation takes on new roles. Trends Biochem. Sci. 1993, 18, 96–101. [Google Scholar] [CrossRef]
- Dasso, M.; Nishitani, H.; Kornbluth, S.; Nishimoto, T.; Newport, J.W. RCC1, a regulator of mitosis, is essential for DNA replication. Mol. Cell. Biol. 1992, 12, 3337–3345. [Google Scholar] [CrossRef]
- Ren, M.; Drivas, G.; D’Eustachio, P.; Rush, M.G. Ran/TC4: A small nuclear GTP-binding protein that regulates DNA synthesis. J. Cell Biol. 1993, 120, 313–323. [Google Scholar] [CrossRef]
- Ohtsubo, M.; Kai, R.; Furuno, N.; Sekiguchi, T.; Sekiguchi, M.; Hayashida, H.; Kuma, K.; Miyata, T.; Fukushige, S.; Murotsu, T.; et al. Isolation and characterization of the active cDNA of the human cell cycle gene (RCC1) involved in the regulation of onset of chromosome condensation. Genes Dev. 1987, 1, 585–593. [Google Scholar] [CrossRef]
- Nishijima, H.; Seki, T.; Nishitani, H.; Nishimoto, T. Premature chromatin condensation caused by loss of RCC1. Prog. Cell Cycle Res. 2000, 4, 145–156. [Google Scholar]
- Beaudouin, J.; Mora-Bermudez, F.; Klee, T.; Daigle, N.; Ellenberg, J. Dissecting the contribution of diffusion and interactions to the mobility of nuclear proteins. Biophys. J. 2006, 90, 1878–1894. [Google Scholar] [CrossRef]
- Bierbaum, M.; Bastiaens, P.I. Cell cycle-dependent binding modes of the ran exchange factor RCC1 to chromatin. Biophys. J. 2013, 104, 1642–1651. [Google Scholar] [CrossRef]
- Cekan, P.; Hasegawa, K.; Pan, Y.; Tubman, E.; Odde, D.; Chen, J.Q.; Herrmann, M.A.; Kumar, S.; Kalab, P. RCC1-dependent activation of Ran accelerates cell cycle and DNA repair, inhibiting DNA damage-induced cell senescence. Mol. Biol. Cell 2016, 27, 1346–1357. [Google Scholar] [CrossRef]
- Moore, J.D. The Ran-GTPase and cell-cycle control. Bioessays 2001, 23, 77–85. [Google Scholar] [CrossRef]
- Ghobrial, I.M.; McCormick, D.J.; Kaufmann, S.H.; Leontovich, A.A.; Loegering, D.A.; Dai, N.T.; Krajnik, K.L.; Stenson, M.J.; Melhem, M.F.; Novak, A.J.; et al. Proteomic analysis of mantle-cell lymphoma by protein microarray. Blood 2005, 105, 3722–3730. [Google Scholar] [CrossRef]
- Hsu, C.H.; Hsu, C.W.; Hsueh, C.; Wang, C.L.; Wu, Y.C.; Wu, C.C.; Liu, C.C.; Yu, J.S.; Chang, Y.S.; Yu, C.J. Identification and characterization of potential biomarkers by quantitative tissue proteomics of primary lung adenocarcinoma. Mol. Cell. Proteom. 2016, 15, 2396–2410. [Google Scholar] [CrossRef]
- Qiao, L.; Zheng, J.; Tian, Y.; Zhang, Q.; Wang, X.; Chen, J.J.; Zhang, W. Regulator of chromatin condensation 1 abrogates the G1 cell cycle checkpoint via Cdk1 in human papillomavirus E7-expressing epithelium and cervical cancer cells. Cell Death Dis. 2018, 9, 583. [Google Scholar] [CrossRef]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR signaling in cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef]
- Sarris, E.G.; Saif, M.W.; Syrigos, K.N. The biological role of PI3K pathway in lung cancer. Pharmaceuticals 2012, 5, 1236–1264. [Google Scholar] [CrossRef]
- Zvelebil, M.J.; MacDougall, L.; Leevers, S.; Volinia, S.; Vanhaesebroeck, B.; Gout, I.; Panayotou, G.; Domin, J.; Stein, R.; Pages, F.; et al. Structural and functional diversity of phosphoinositide 3-kinases. Philos. Trans. R. Soc. B Biol. Sci. 1996, 351, 217–223. [Google Scholar]
- Vanhaesebroeck, B.; Whitehead, M.A.; Pineiro, R. Molecules in medicine mini-review: Isoforms of PI3K in biology and disease. J. Mol. Med. 2016, 94, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Revathidevi, S.; Munirajan, A.K. Akt in cancer: Mediator and more. Semin. Cancer Biol. 2019, 59, 80–91. [Google Scholar] [CrossRef]
- Burke, J.E. Structural basis for regulation of phosphoinositide kinases and their involvement in human disease. Mol. Cell 2018, 71, 653–673. [Google Scholar] [CrossRef] [PubMed]
- Abeyrathna, P.; Su, Y. The critical role of Akt in cardiovascular function. Vascul. Pharmacol. 2015, 74, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, M.G.; Fairn, G.D.; Antonescu, C.N. Akt-ing up just about everywhere: Compartment-specific Akt activation and function in receptor tyrosine kinase signaling. Front. Cell Dev. Biol. 2019, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Mackay, H.J.; Eisenhauer, E.A.; Kamel-Reid, S.; Tsao, M.; Clarke, B.; Karakasis, K.; Werner, H.M.; Trovik, J.; Akslen, L.A.; Salvesen, H.B.; et al. Molecular determinants of outcome with mammalian target of rapamycin inhibition in endometrial cancer. Cancer Am. Cancer Soc. 2014, 120, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Altomare, D.A.; Lyons, G.E.; Mitsuuchi, Y.; Cheng, J.Q.; Testa, J.R. Akt2 mRNA is highly expressed in embryonic brown fat and the AKT2 kinase is activated by insulin. Oncogene 1998, 16, 2407–2411. [Google Scholar] [CrossRef]
- Brodbeck, D.; Cron, P.; Hemmings, B.A. A human protein kinase Bgamma with regulatory phosphorylation sites in the activation loop and in the C-terminal hydrophobic domain. J. Biol. Chem. 1999, 274, 9133–9136. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef]
- Rosner, M.; Hengstschlager, M. Cytoplasmic and nuclear distribution of the protein complexes mTORC1 and mTORC2: Rapamycin triggers dephosphorylation and delocalization of the mTORC2 components rictor and sin1. Hum. Mol. Genet. 2008, 17, 2934–2948. [Google Scholar] [CrossRef] [PubMed]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zeng, J.; Shen, K. PI3K/AKT/mTOR signaling pathway as a therapeutic target for ovarian cancer. Arch. Gynecol. Obstet. 2014, 290, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Tapia, O.; Riquelme, I.; Leal, P.; Sandoval, A.; Aedo, S.; Weber, H.; Letelier, P.; Bellolio, E.; Villaseca, M.; Garcia, P.; et al. The PI3K/AKT/mTOR pathway is activated in gastric cancer with potential prognostic and predictive significance. Virchows Arch. 2014, 465, 25–33. [Google Scholar] [CrossRef]
- Staal, S.P. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: Amplification of AKT1 in a primary human gastric adenocarcinoma. Proc. Natl. Acad. Sci. USA 1987, 84, 5034–5037. [Google Scholar] [CrossRef]
- Cheng, J.Q.; Ruggeri, B.; Klein, W.M.; Sonoda, G.; Altomare, D.A.; Watson, D.K.; Testa, J.R. Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc. Natl. Acad. Sci. USA 1996, 93, 3636–3641. [Google Scholar] [CrossRef]
- Ruggeri, B.A.; Huang, L.; Wood, M.; Cheng, J.Q.; Testa, J.R. Amplification and overexpression of the AKT2 oncogene in a subset of human pancreatic ductal adenocarcinomas. Mol. Carcinog. 1998, 21, 81–86. [Google Scholar] [CrossRef]
- Fan, X.; Liu, Y.; Heilman, S.A.; Chen, J.J. Human papillomavirus E7 induces rereplication in response to DNA damage. J. Virol. 2013, 87, 1200–1210. [Google Scholar] [CrossRef]
- Hutchins, J.R.; Moore, W.J.; Hood, F.E.; Wilson, J.S.; Andrews, P.D.; Swedlow, J.R.; Clarke, P.R. Phosphorylation regulates the dynamic interaction of RCC1 with chromosomes during mitosis. Curr. Biol. 2004, 14, 1099–1104. [Google Scholar] [CrossRef]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef]
- David, O.; Jett, J.; LeBeau, H.; Dy, G.; Hughes, J.; Friedman, M.; Brody, A.R. Phospho-Akt overexpression in non-small cell lung cancer confers significant stage-independent survival disadvantage. Clin. Cancer Res. 2004, 10, 6865–6871. [Google Scholar] [CrossRef]
- Tsurutani, J.; Fukuoka, J.; Tsurutani, H.; Shih, J.H.; Hewitt, S.M.; Travis, W.D.; Jen, J.; Dennis, P.A. Evaluation of two phosphorylation sites improves the prognostic significance of Akt activation in non-small-cell lung cancer tumors. J. Clin. Oncol. 2006, 24, 306–314. [Google Scholar] [CrossRef]
- Hung, C.M.; Garcia-Haro, L.; Sparks, C.A.; Guertin, D.A. mTOR-dependent cell survival mechanisms. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Zhou, Y.; Pan, Y.; Zhang, S.; Shi, X.; Ning, T.; Ke, Y. Increased phosphorylation of p70 S6 kinase is associated with HPV16 infection in cervical cancer and esophageal cancer. Br. J. Cancer 2007, 97, 218–222. [Google Scholar] [CrossRef]
- Hood, F.E.; Clarke, P.R. RCC1 isoforms differ in their affinity for chromatin, molecular interactions and regulation by phosphorylation. J. Cell Sci. 2007, 120, 3436–3445. [Google Scholar] [CrossRef]
- Li, H.Y.; Zheng, Y. Phosphorylation of RCC1 in mitosis is essential for producing a high RanGTP concentration on chromosomes and for spindle assembly in mammalian cells. Genes Dev. 2004, 18, 512–527. [Google Scholar] [CrossRef]
- Stern, P.L.; van der Burg, S.H.; Hampson, I.N.; Broker, T.R.; Fiander, A.; Lacey, C.J.; Kitchener, H.C.; Einstein, M.H. Therapy of human papillomavirus-related disease. Vaccine 2012, 30 (Suppl. S5), F71–F82. [Google Scholar] [CrossRef]
- Lim, H.J.; Crowe, P.; Yang, J.L. Current clinical regulation of PI3K/PTEN/Akt/mTOR signalling in treatment of human cancer. J. Cancer Res. Clin. Oncol. 2015, 141, 671–689. [Google Scholar] [CrossRef]
- Hoppe-Seyler, K.; Bossler, F.; Braun, J.A.; Herrmann, A.L.; Hoppe-Seyler, F. The HPV E6/E7 oncogenes: Key factors for viral carcinogenesis and therapeutic targets. Trends Microbiol. 2018, 26, 158–168. [Google Scholar] [CrossRef]
- Pim, D.; Massimi, P.; Dilworth, S.M.; Banks, L. Activation of the protein kinase B pathway by the HPV-16 E7 oncoprotein occurs through a mechanism involving interaction with PP2A. Oncogene 2005, 24, 7830–7838. [Google Scholar] [CrossRef]
- Menges, C.W.; Baglia, L.A.; Lapoint, R.; McCance, D.J. Human papillomavirus type 16 E7 up-regulates AKT activity through the retinoblastoma protein. Cancer Res. 2006, 66, 5555–5559. [Google Scholar] [CrossRef]
- Contreras-Paredes, A.; De la Cruz-Hernandez, E.; Martinez-Ramirez, I.; Duenas-Gonzalez, A.; Lizano, M. E6 variants of human papillomavirus 18 differentially modulate the protein kinase B/phosphatidylinositol 3-kinase (akt/PI3K) signaling pathway. Virology 2009, 383, 78–85. [Google Scholar] [CrossRef]
- Henken, F.E.; Banerjee, N.S.; Snijders, P.J.; Meijer, C.J.; De-Castro, A.J.; Rosl, F.; Broker, T.R.; Chow, L.T.; Steenbergen, R.D. PIK3CA-mediated PI3-kinase signalling is essential for HPV-induced transformation in vitro. Mol. Cancer 2011, 10, 71. [Google Scholar] [CrossRef]
- Schwarz, J.K.; Payton, J.E.; Rashmi, R.; Xiang, T.; Jia, Y.; Huettner, P.; Rogers, B.E.; Yang, Q.; Watson, M.; Rader, J.S.; et al. Pathway-specific analysis of gene expression data identifies the PI3K/Akt pathway as a novel therapeutic target in cervical cancer. Clin. Cancer Res. 2012, 18, 1464–1471. [Google Scholar] [CrossRef] [PubMed]
- Faried, L.S.; Faried, A.; Kanuma, T.; Sano, T.; Nakazato, T.; Tamura, T.; Kuwano, H.; Minegishi, T. Predictive and prognostic role of activated mammalian target of rapamycin in cervical cancer treated with cisplatin-based neoadjuvant chemotherapy. Oncol. Rep. 2006, 16, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Beitner-Johnson, D.; Rust, R.T.; Hsieh, T.C.; Millhorn, D.E. Hypoxia activates Akt and induces phosphorylation of GSK-3 in PC12 cells. Cell Signal. 2001, 13, 23–27. [Google Scholar] [CrossRef]
- Hartwell, L.H.; Kastan, M.B. Cell cycle control and cancer. Science 1994, 266, 1821–1828. [Google Scholar] [CrossRef] [PubMed]
- Lanni, J.S.; Jacks, T. Characterization of the p53-dependent postmitotic checkpoint following spindle disruption. Mol. Cell. Biol. 1998, 18, 1055–1064. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef]
- Aleem, E.; Kiyokawa, H.; Kaldis, P. Cdc2-cyclin E complexes regulate the G1/S phase transition. Nat. Cell Biol. 2005, 7, 831–836. [Google Scholar] [CrossRef]
- Tian, Y.; Chen, H.; Qiao, L.; Zhang, W.; Zheng, J.; Zhao, W.; Chen, J.J.; Zhang, W. CIP2A facilitates the G1/S cell cycle transition via B-Myb in human papillomavirus 16 oncoprotein E6-expressing cells. J. Cell. Mol. Med. 2018, 22, 4150–4160. [Google Scholar] [CrossRef]
- Santamaria, D.; Barriere, C.; Cerqueira, A.; Hunt, S.; Tardy, C.; Newton, K.; Caceres, J.F.; Dubus, P.; Malumbres, M.; Barbacid, M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2007, 448, 811–815. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, X.; Qiao, L.; Liu, R.; Han, X.; Zhang, W. Phosphorylation of RCC1 on Serine 11 Facilitates G1/S Transition in HPV E7-Expressing Cells. Biomolecules 2021, 11, 995. https://doi.org/10.3390/biom11070995
Hou X, Qiao L, Liu R, Han X, Zhang W. Phosphorylation of RCC1 on Serine 11 Facilitates G1/S Transition in HPV E7-Expressing Cells. Biomolecules. 2021; 11(7):995. https://doi.org/10.3390/biom11070995
Chicago/Turabian StyleHou, Xiaoyan, Lijun Qiao, Ruijuan Liu, Xuechao Han, and Weifang Zhang. 2021. "Phosphorylation of RCC1 on Serine 11 Facilitates G1/S Transition in HPV E7-Expressing Cells" Biomolecules 11, no. 7: 995. https://doi.org/10.3390/biom11070995
APA StyleHou, X., Qiao, L., Liu, R., Han, X., & Zhang, W. (2021). Phosphorylation of RCC1 on Serine 11 Facilitates G1/S Transition in HPV E7-Expressing Cells. Biomolecules, 11(7), 995. https://doi.org/10.3390/biom11070995