
Micro-Aqueous Organic System: A Neglected Model in Computational Lipase Design?


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
2.1. System Setup and MD Simulations
2.2. Analytical Methods
2.3. Protein, Expression and Assay of Lipase Synthetic Activity
3. Results and Discussion
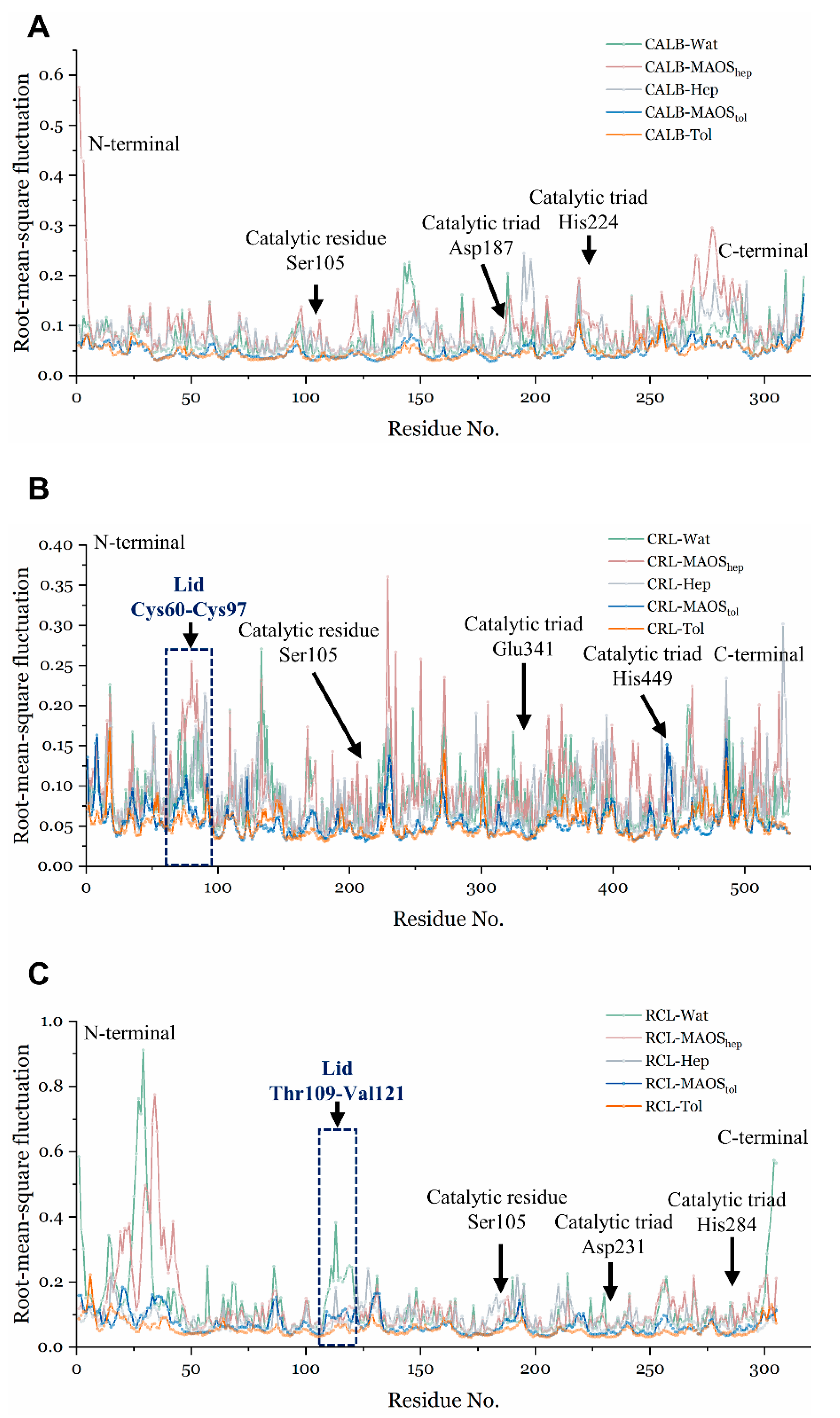
3.1. Overall Conformations of Lipases in Different Solvent Environments
3.2. The Property of the Lipase Surface
3.3. The Dynamic Behavior of Water in MAOS Systems
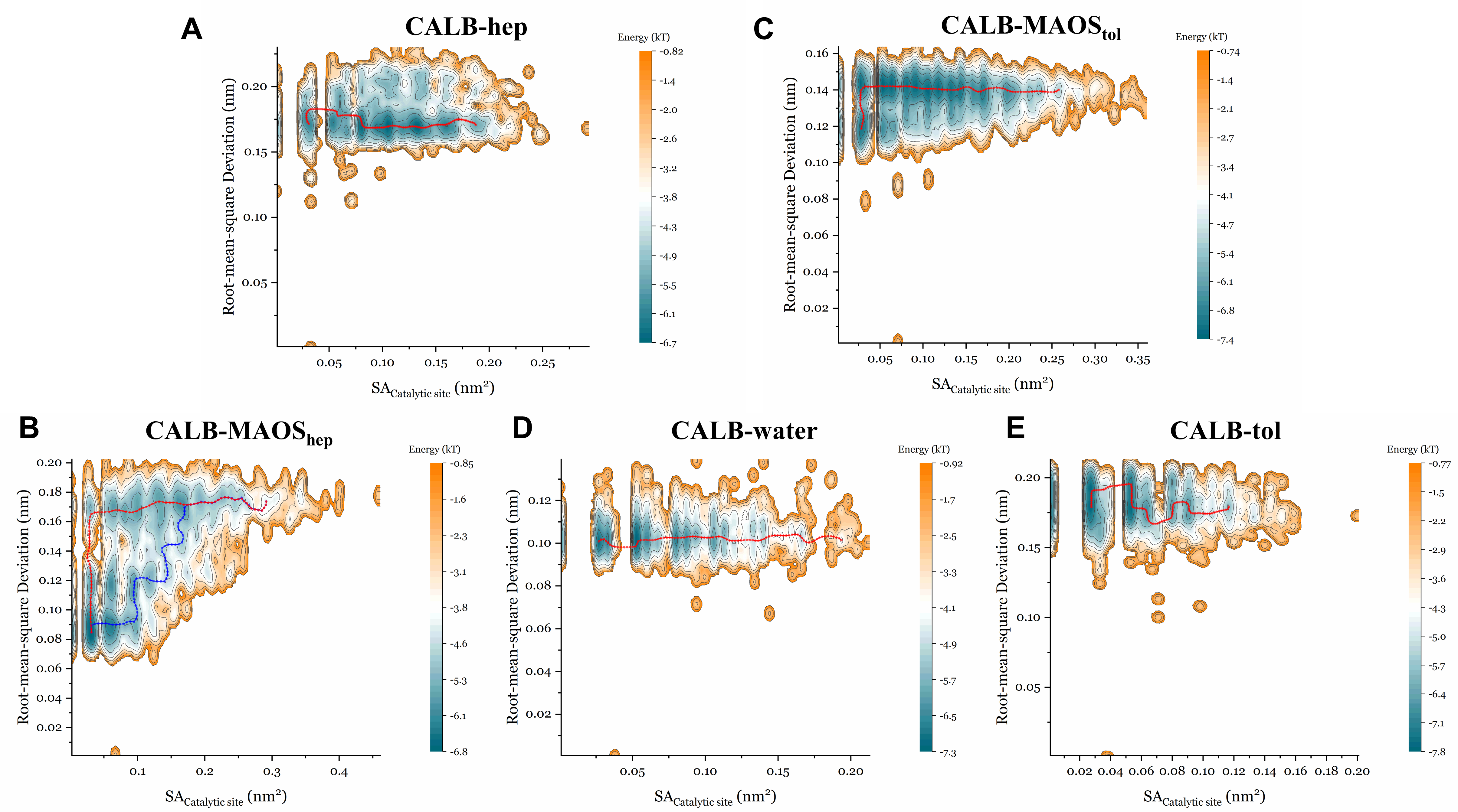
3.4. The Energy Landscape and the Minimum Energy Path in the Presence of Different Solvents
3.5. The Effect of Water Content on Lipase Synthetic Activities
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MD | Molecular dynamic |
| CALB | Candida antarctica lipase B |
| CRL | Candida rugosa lipase |
| RCL | Rhizopus chinensis lipase |
| MAOS | Micro-aqueous organic solvents |
| RDF | Radial distribution function |
| RMSF | Root-mean-square fluctuation |
| RMSD | Root-mean-square deviation |
| SASA | Solvent accessible surface area |
| RCF | Rotation correlation function |
| MSD | Mean-square displacement |
| HBs | Hydrogen bonds |
| EL | Energy landscapes |
| FID | Flame ionization detector |
| MEP | Minimum energy path |
| PCM | Polarizable continuum model |
| SMD | Solvation Model Based on Density |
| GAFF | General Amber force field |
References
- Villeneuve, P. Lipases in lipophilization reactions. Biotechnol. Adv. 2007, 25, 515–536. [Google Scholar] [CrossRef]
- Schmid, R.D.; Verger, R. Lipases: Interfacial enzymes with attractive applications. Angew. Chem. Int. Ed. Engl. 1998, 37, 1608–1633. [Google Scholar] [CrossRef]
- Jaeger, K.-E.; Reetz, M.T. Microbial lipases form versatile tools for biotechnology. Trends Biotechnol. 1998, 16, 396–403. [Google Scholar] [CrossRef]
- Rodrigues, R.C.; Virgen-Ortíz, J.J.; Santos, J.C.S.D.; Berenguer-Murcia, Á.; Alcantara, A.R.; Barbosa, O.; Ortiz, C.; Fernandez-Lafuente, R. Immobilization of lipases on hydrophobic supports: Immobilization mechanism, advantages, problems, and solutions. Biotechnol. Adv. 2019, 37, 746–770. [Google Scholar] [CrossRef] [PubMed]
- Sankar, S.; Ponnuraj, K. Less explored plant lipases: Modeling and molecular dynamics simulations of plant lipases in different solvents and temperatures to understand structure-function relationship. Int. J. Biol. Macromol. 2020, 164, 3546–3558. [Google Scholar] [CrossRef]
- Chen, Z.S.; Wu, Y.D.; Hao, J.H.; Liu, Y.J.; He, K.P.; Jiang, W.H.; Xiong, M.J.; Lv, Y.S.; Cao, S.L.; Zhu, J. Molecular dynamic simulation of the porcine pancreatic lipase in non-aqueous organic solvents. Front. Bioeng. Biotechnol. 2020, 8. [Google Scholar] [CrossRef]
- Mohtashami, M.; Fooladi, J.; Haddad-Mashadrizeh, A.; Housaindokht, M.R.; Monhemi, H. Molecular mechanism of enzyme tolerance against organic solvents: Insights from molecular dynamics simulation. Int. J. Biol. Macromol. 2019, 122, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, L.; Zhang, H.; Feng, W.; Tan, T. Lid closure mechanism of Yarrowia lipolytica lipase in methanol investigated by molecular dynamics simulation. J. Chem. Inf. Model. 2014, 54, 2033–2041. [Google Scholar] [CrossRef]
- Ravelo, M.; Esteban, J.; Ladero, M.; García-Ochoa, F. Enzymatic synthesis of ibuprofen monoglycerides catalyzed by free Candida antarctica lipase B in a toluene–glycerol biphasic medium. RSC Adv. 2016, 6, 69658–69669. [Google Scholar] [CrossRef]
- Herbst, D.; Peper, S.; Niemeyer, B. Enzyme catalysis in organic solvents: Influence of water content, solvent composition and temperature on Candida rugosa lipase catalyzed transesterification. J. Biotechnol. 2012, 162, 398–403. [Google Scholar] [CrossRef]
- Zaks, A.; Klibanov, A.M. The effect of water on enzyme action in organic media. J. Biol. Chem. 1988, 263, 8017–8021. [Google Scholar] [CrossRef]
- Kahlow, U.H.; Schmid, R.D.; Pleiss, J. A model of the pressure dependence of the enantioselectivity of Candida rugosa lipase towards (+/−)-menthol. Protein Sci. 2001, 10, 1942–1952. [Google Scholar] [CrossRef]
- Khan, F.I.; Lan, D.; Durrani, R.; Huan, W.; Zhao, Z.; Wang, Y. The lid domain in lipases: Structural and functionaldeterminant of enzymatic properties. Front. Bioeng. Biotechnol. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Rostkowski, M.; Olsson, M.H.; Sondergaard, C.R.; Jensen, J.H. Graphical analysis of pH-dependent properties of proteins predicted using PROPKA. BMC Struct. Biol. 2011, 11. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham, T.E.; Debolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. AMBER, a package of computer-programs for applying molecular mechanics, normal-mode analysis, molecular-dynamics and free-energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Petersson, G.A.; Al-Laham, M.A. A complete basis set model chemistry. II. Open-shell systems and the total energies of the first-row atoms. J. Chem. Phys. 1991, 94, 6081–6090. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functional. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. PCCP 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.; Tomasi, J. Geometry optimization of molecular structures in solution by the polarizable continuum model. J. Comput. Chem. 1998, 19, 404–417. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. Gromacs: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theor. Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 27–28, 33–38. [Google Scholar] [CrossRef]
- Joosten, R.P.; Beek, T.A.t.; Krieger, E.; Hekkelman, M.L.; Hooft, R.W.; Schneider, R.; Sander, C.; Vriend, G. A series of PDB related databases for everyday needs. Nucleic Acids Res. 2011, 39, D411–D419. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- EWeinan, E.; Ren, W.; Vanden-Eijnden, E. String method for the study of rare events. Phys. Rev. B 2002, 66. [Google Scholar] [CrossRef]
- Zhang, M.; Yu, X.W.; Xu, Y.; Guo, R.T.; Swapna, G.V.T.; Szyperski, T.; Hunt, J.F.; Montelione, G.T. Structural basis by which the N-terminal polypeptide segment of Rhizopus chinensis lipase regulates its substrate binding affinity. Biochemistry 2019, 58, 3943–3954. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.B.G.; Lawton, M. Drying of Organic Solvents: Quantitative Evaluation of the Efficiency of Several Desiccants. J. Org. Chem. 2010, 75, 8351–8354. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Qin, D.; Wu, Z.; Sun, B.; Sun, X.; Huang, M.; Sun, J.; Zheng, F. Characterization of key aroma compounds in Chinese Guojing sesame-flavor Baijiu by means of molecular sensory science. Food Chem. 2019, 284, 100–107. [Google Scholar] [CrossRef]
- Wang, D.; Yan, X.; Teng, Y. Synthetic activity enhancement of membrane-bound lipase from Rhizopus chinensis by pretreatment with isooctane. Bioprocess Biosyst. Eng. 2007, 30, 147–155. [Google Scholar] [CrossRef]
- Wang, S.; Xu, Y.; Yu, X.W. Propeptide in Rhizopus chinensis lipase: New insights into its mechanism of activity and substrate selectivity by computational design. J. Agric. Food. Chem. 2021, 69, 4263–4275. [Google Scholar] [CrossRef] [PubMed]
- Kulschewski, T.; Sasso, F.; Secundo, F.; Lotti, M.; Pleiss, J. Molecular mechanism of deactivation of C. antarctica lipase B by methanol. J. Biotechnol. 2013, 168, 462–469. [Google Scholar] [CrossRef]
- Wang, S.; Xu, Y.; Yu, X.W. A phenylalanine dynamic switch controls the interfacial activation of Rhizopus chinensis lipase. Int. J. Biol. Macromol. 2021, 173, 1–12. [Google Scholar] [CrossRef]
- Nienhuys, H.-K.; Woutersen, S.; van Santen, R.A.; Bakker, H.J. Mechanism for vibrational relaxation in water investigated by femtosecond infrared spectroscopy. J. Chem. Phys. 1999, 111, 1494–1500. [Google Scholar] [CrossRef]
- Montrose, C.J.; Bucaro, J.A.; Marshall-Coakley, J.; Litovitz, T.A. Depolarized Rayleigh scattering and hydrogen bonding in liquid water. J. Chem. Phys. 1974, 60, 5025–5029. [Google Scholar] [CrossRef]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids, 2nd ed.; Oxford University Press: Oxford, UK, 2017. [Google Scholar]
- Chen, X.; Weber, I.; Harrison, R.W. Hydration water and bulk water in proteins have distinct properties in radial distributions calculated from 105 atomic resolution crystal structures. J. Phys. Chem. B 2008, 112, 12073–12080. [Google Scholar] [CrossRef]
- Ballesteros, A.; Plou, F.; Iborra, J.; Halling, P. Engineering stability of enzymes in systems with organic solvents. In Stability and Stabilization of Biocatalysts, Proceedings of the an International Symposium Organized under Auspices of the Working Party on Applied Biocatalysis of the European Federation of Biotechnology, the University of Cordoba, Spain, and the Spanish Society of Biotechnology, Cordoba, Spain, 19–22 April 1998; Elsevier Science Limited: New York, NY, USA, 1998; p. 355. [Google Scholar]
- Chu, X.; Liu, F.; Maxwell, B.A.; Wang, Y.; Suo, Z.; Wang, H.; Han, W.; Wang, J. Dynamic conformational change regulates the protein-DNA recognition: An investigation on binding of a Y-family polymerase to its target DNA. PLoS Comput. Biol. 2014, 10, e1003804. [Google Scholar] [CrossRef]
- Wang, Y.; Gan, L.; Wang, E.; Wang, J. Exploring the dynamic functional landscape of adenylate kinase modulated by substrates. J. Chem. Theory Comput. 2013, 9, 84–95. [Google Scholar] [CrossRef]
- Jaeger, K.E.; Dijkstra, B.W.; Reetz, M.T. Bacterial Biocatalysts: Molecular biology, three-dimensional structures, and biotechnological applications of lipases. Annu. Rev. Microbiol. 1999, 53, 315–351. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Perez, A.; Dill, K.A.; Maccallum, J.L. Computing the relative stabilities and the per-residue components in protein conformational changes. Structure 2014, 22, 168–175. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shah, S.; Gupta, M.N. Lipase catalyzed preparation of biodiesel from Jatropha oil in a solvent free system. Process Biochem. 2007, 42, 409–414. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Xu, Y.; Yu, X.-W. Micro-Aqueous Organic System: A Neglected Model in Computational Lipase Design? Biomolecules 2021, 11, 848. https://doi.org/10.3390/biom11060848
Wang S, Xu Y, Yu X-W. Micro-Aqueous Organic System: A Neglected Model in Computational Lipase Design? Biomolecules. 2021; 11(6):848. https://doi.org/10.3390/biom11060848
Chicago/Turabian StyleWang, Shang, Yan Xu, and Xiao-Wei Yu. 2021. "Micro-Aqueous Organic System: A Neglected Model in Computational Lipase Design?" Biomolecules 11, no. 6: 848. https://doi.org/10.3390/biom11060848
APA StyleWang, S., Xu, Y., & Yu, X.-W. (2021). Micro-Aqueous Organic System: A Neglected Model in Computational Lipase Design? Biomolecules, 11(6), 848. https://doi.org/10.3390/biom11060848