
Nucleoside Analogues Are Potent Inducers of Pol V-mediated Mutagenesis

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Species, Strains and Growth Measurements
2.2. Peptide
2.3. Nucleoside Analogues (NAs)
2.4. Bacterial Cultivation
2.5. MIC Assay
2.6. Rifampicin Mutagenicity Assay (RifR) Assay
2.7. Protein Extraction
2.8. Multiplexed Inhibitor Beads (MIB) Assay and Mass Spectrometry (MS) Analysis of Proteome
2.9. Metabolite Profiling
2.10. Data Analysis
2.11. Mass Spectrometry Data Analysis
3. Results and Discussion
3.1. NAs Induce Mutagenesis in Bacteria at Concentrations That Do Not Affect Bacterial Growth
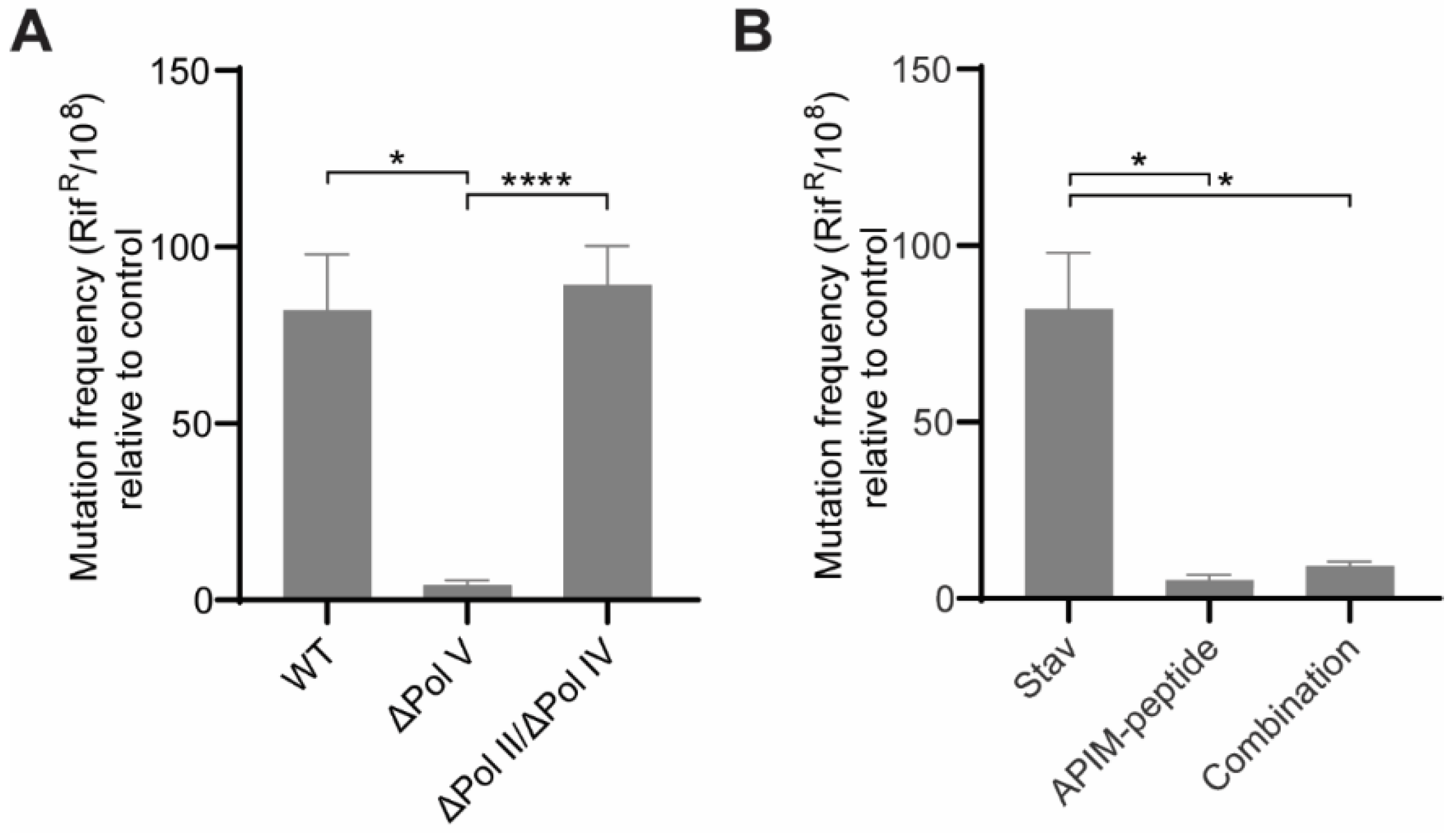
3.2. Pol V Is the TLS Polymerase Responsible for the Mutagenesis Induced by NAs
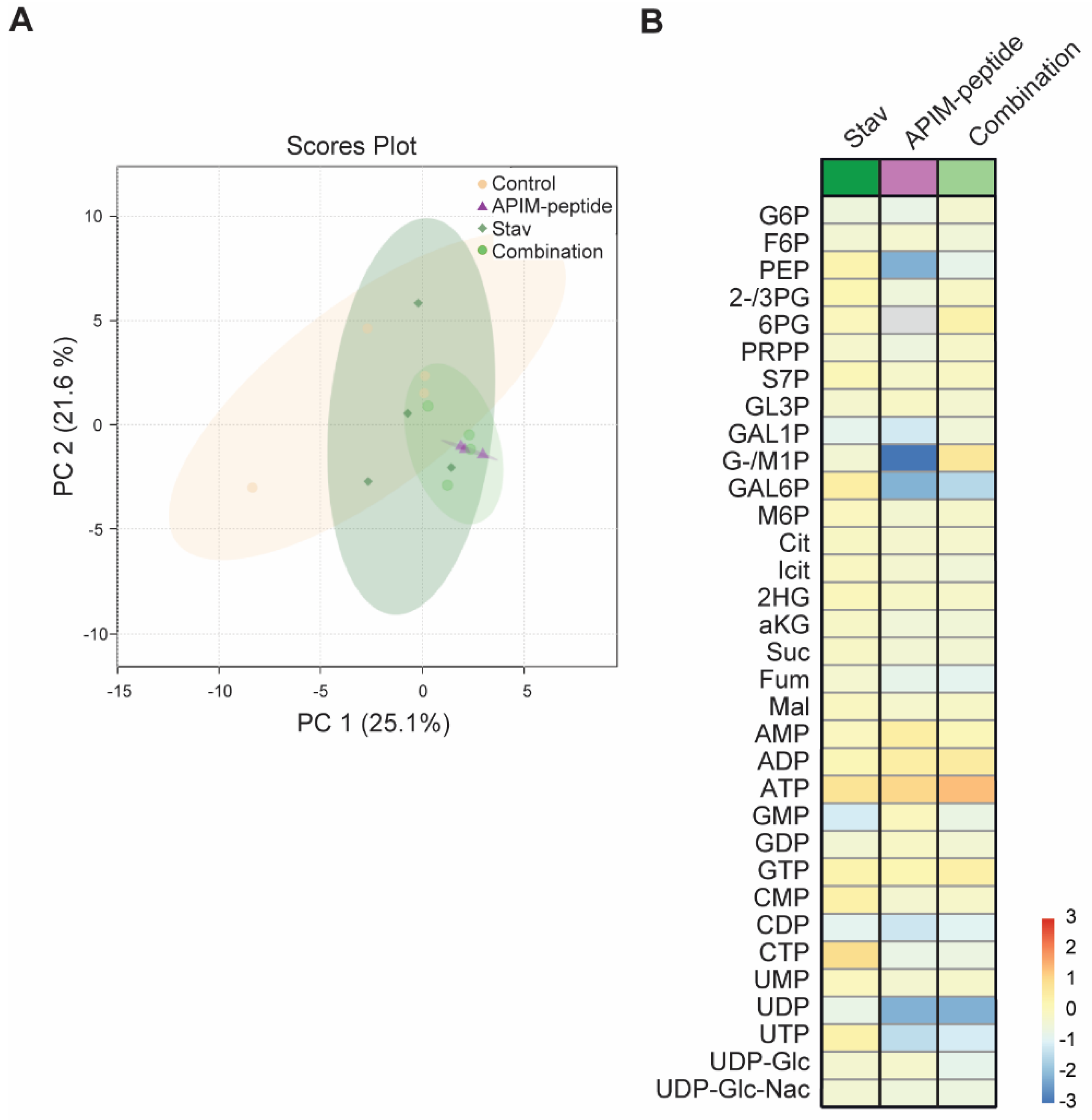
3.3. Stavudine Treatment Induces only Minor Changes in Central Carbon Metabolite Pools
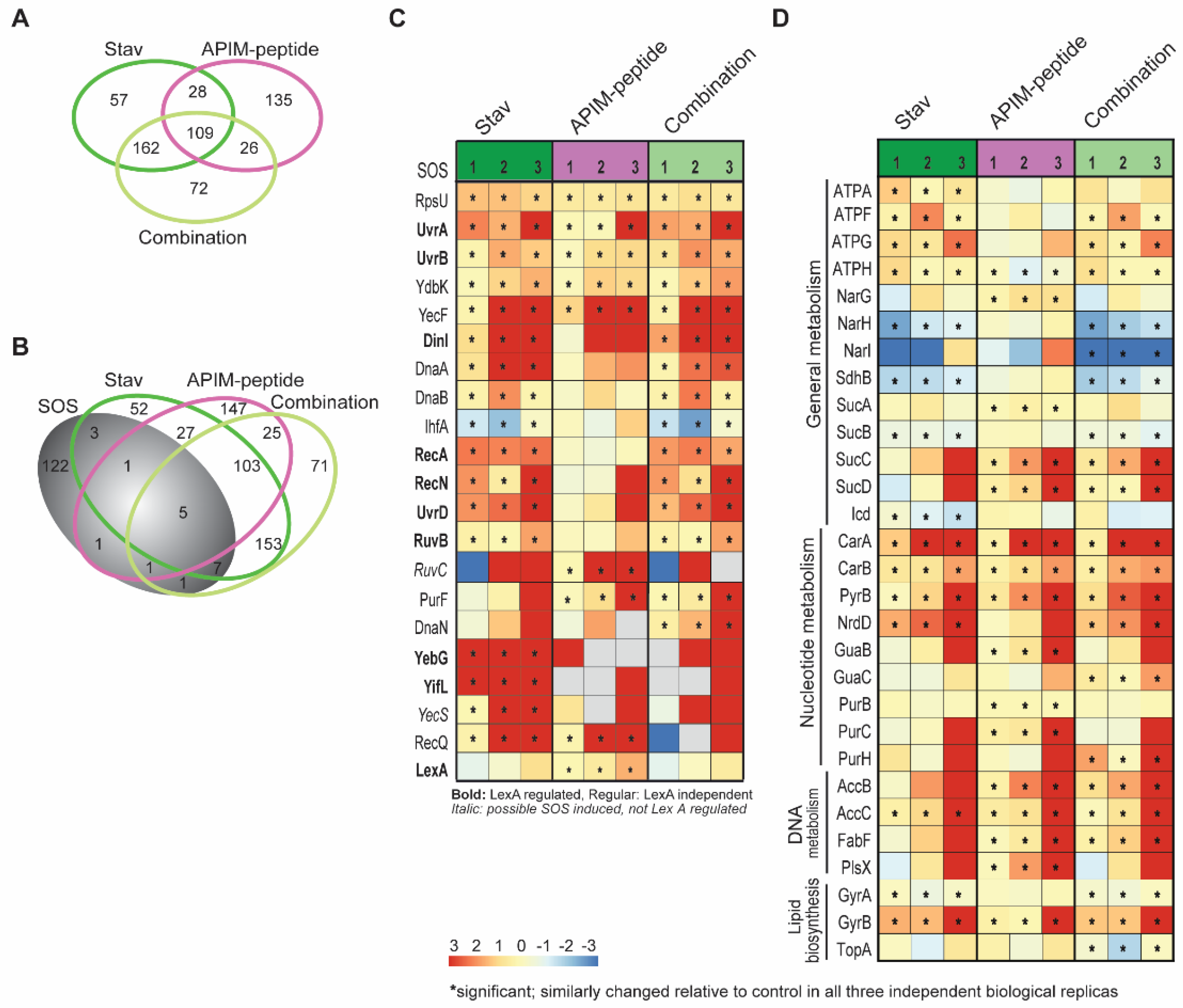
3.4. Stavudine Treatment Activates the SOS System
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- CDCP. Antibiotic Resistance Threats in the United States; US Department of Health and Human Service: Atlanta, GA, USA, 2019.
- European Centre for Disease Prevention and Control. Surveillance of Antimicrobial Resistance in Europe 2018; European Centre for Disease Prevention and Control: Stockholm, Sweden, 2019.
- Tadesse, B.T.; Ashley, E.A.; Ongarello, S.; Havumaki, J.; Wijegoonewardena, M.; González, I.J.; Dittrich, S. Antimicrobial resistance in Africa: A systematic review. BMC Infect. Dis. 2017, 17, 1–17. [Google Scholar] [CrossRef]
- O’Neill, J. Review on Antimicrobial Resistance. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. 2014. Available online: https://amr-review.org (accessed on 4 January 2020).
- Getahun, H.; Smith, I.; Trivedi, K.; Paulin, S.; Balkhy, H.H. Tackling antimicrobial resistance in the COVID-19 pandemic. Bull. World Health Organ. 2020, 98, 442–442A. [Google Scholar] [CrossRef]
- World Health Organization. Clinical Management of COVID-19: Interim Guidance, 27 May 2020; World Health Organization: Geneva, Switzerland , 2020. [Google Scholar]
- Rawson, T.M.; Moore, L.S.; Zhu, N.; Ranganathan, N.; Skolimowska, K.; Gilchrist, M.; Satta, G.; Cooke, G.; Holmes, A. Bacterial and fungal co-infection in individuals with coronavirus: A rapid review to support COVID-19 antimicrobial prescribing. Clin. Infect. Dis. 2020, 71, 2459–2468. [Google Scholar]
- Cirz, R.T.; Chin, J.K.; Andes, D.R.; De Crécy-Lagard, V.; Craig, W.A.; Romesberg, F.E. Inhibition of Mutation and Combating the Evolution of Antibiotic Resistance. PLoS Biol. 2005, 3, e176. [Google Scholar] [CrossRef] [PubMed]
- Merrikh, H.; Kohli, R.M. Targeting evolution to inhibit antibiotic resistance. FEBS J. 2020, 287, 4341–4353. [Google Scholar] [CrossRef] [PubMed]
- Petrosino, J.F.; Galhardo, R.S.; Morales, L.D.; Rosenberg, S.M. Stress-induced beta-lactam antibiotic resistance mutation and sequences of stationary-phase mutations in the Escherichia coli chromosome. J. Bacteriol. 2009, 191, 5881–5889. [Google Scholar] [CrossRef] [PubMed]
- Gawel, D.; Seed, P.C. Urinary tract infection drives genome instability in uropathogenic Escherichia coli and necessitates translesion synthesis DNA polymerase IV for virulence. Virulence 2011, 2, 222–232. [Google Scholar] [CrossRef]
- Altieri, A.S.; Kelman, Z. DNA Sliding Clamps as Therapeutic Targets. Front. Mol. Biosci. 2018, 5, 87. [Google Scholar] [CrossRef]
- Campos, P.E.; Suarez, P.G.; Sánchez, J.; Zavala, D.; Arevalo, J.; Ticona, E.; Nolan, C.M.; Hooton, T.M.; Holmes, K.K. Multidrug-resistant Mycobacterium tuberculosis in HIV-Infected Persons, Peru. Emerg. Infect. Dis. 2003, 9, 1571–1578. [Google Scholar] [CrossRef]
- Marami, D.; Balakrishnan, S.; Seyoum, B. Prevalence, Antimicrobial Susceptibility Pattern of Bacterial Isolates, and Associated Factors of Urinary Tract Infections among HIV-Positive Patients at Hiwot Fana Specialized University Hospital, Eastern Ethiopia. Can. J. Infect. Dis. Med Microbiol. 2019, 2019, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Romana, K.; Rieger, A.; Presterl, E.; Wrba, T.; Diab-Elschahawi, M. Epidemiology of Urinary Tract Infections in HIV Positive Patients at a Tertiary Care University Hospital in Central Europe (2011–2016). Infect. Disord. Drug Targets 2018, 18, 199–206. [Google Scholar]
- Ssengooba, W.; Lukoye, D.; Meehan, C.J.; Kateete, D.P.; Joloba, M.L.; De Jong, B.C.; Cobelens, F.G.; Van Leth, F. Tuberculosis resistance-conferring mutations with fitness cost among HIV-positive individuals in Uganda. Int. J. Tuberc. Lung Dis. 2017, 21, 531–536. [Google Scholar] [CrossRef]
- Adeyemi, F.M.; Ako-nai, K.A.; Adejuyigbe, E.; Ebhodaghe, B.I.; Osho, P.O.; Oyeniyi, T.T.; Kassim, O.O. Molecular characterization and antibiotic resistance profiles of bacterial isolates cultured from HIV seropositive patients. Arch. Clin. Microbiol. 2015, 6, e2. [Google Scholar]
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef]
- Thomson, J.M.; Lamont, I.L. Nucleoside Analogues as Antibacterial Agents. Front. Microbiol. 2019, 10, 952. [Google Scholar] [CrossRef]
- Furman, P.A.; Fyfe, J.A.; Clair, M.H.S.; Weinhold, K.; Rideout, J.L.; Freeman, G.A.; Lehrman, S.N.; Bolognesi, D.P.; Broder, S.; Mitsuya, H. Phosphorylation of 3′-azido-3′-deoxythymidine and selective interaction of the 5′-triphosphate with human immunodeficiency virus reverse transcriptase. Proc. Natl. Acad. Sci. USA 1986, 83, 8333–8337. [Google Scholar] [CrossRef]
- Nedal, A.; Ræder, S.B.; Dalhus, B.; Helgesen, E.; Forstrøm, R.J.; Lindland, K.; Sumabe, B.K.; Martinsen, J.H.; Kragelund, B.B.; Skarstad, K.; et al. Peptides containing the PCNA interacting motif APIM bind to the β-clamp and inhibit bacterial growth and mutagenesis. Nucleic Acids Res. 2020, 48, 5540–5554. [Google Scholar] [CrossRef]
- CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard—Ninth Edition; CLSI Document M07-A9; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2012. [Google Scholar]
- Petrovic, V.; Olaisen, C.; Sharma, A.; Nepal, A.; Bugge, S.; Sundby, E.; Hoff, B.H.; Slupphaug, G.; Otterlei, M. On-column trypsinization allows for re-use of matrix in modified multiplexed inhibitor beads assay. Anal. Biochem. 2017, 523, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Røst, L.M.; Thorfinnsdottir, L.B.; Kumar, K.; Fuchino, K.; Langørgen, I.E.; Bartosova, Z.; Kristiansen, K.A.; Bruheim, P. Absolute Quantification of the Central Carbon Metabolome in Eight Commonly Applied Prokaryotic and Eukaryotic Model Systems. Metabolites 2020, 10, 74. [Google Scholar] [CrossRef] [PubMed]
- Stafsnes, M.H.; Røst, L.M.; Bruheim, P. Improved phosphometabolome profiling applying isotope dilution strategy and capillary ion chromatography-tandem mass spectrometry. J. Chromatogr. B 2018, 1083, 278–283. [Google Scholar] [CrossRef]
- Røst, L.M.; Shafaei, A.; Fuchino, K.; Bruheim, P. Zwitterionic HILIC tandem mass spectrometry with isotope dilution for rapid, sensitive and robust quantification of pyridine nucleotides in biological extracts. J. Chromatogr. B 2020, 1144, 122078. [Google Scholar] [CrossRef]
- Lien, S.K.; Kvitvang, H.F.N.; Bruheim, P. Utilization of a deuterated derivatization agent to synthesize internal standards for gas chromatography–tandem mass spectrometry quantification of silylated metabolites. J. Chromatogr. A 2012, 1247, 118–124. [Google Scholar] [CrossRef]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Geiszler, D.J.; Kong, A.T.; Avtonomov, D.M.; Yu, F.; Leprevost, F.D.V.; Nesvizhskii, A.I. PTM-Shepherd: Analysis and Summarization of Post-Translational and Chemical Modifications From Open Search Results. Mol. Cell. Proteom. 2021, 20, 100018. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate Proteome-wide Label-free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, J.D.; Chakraborti, S. Nonparametric Statistical Inference, 6th ed.; CRC Press: Boca Raton, FL, USA, 2021. [Google Scholar]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
- Sandrini, M.P.B.; Clausen, A.R.; On, S.L.W.; Aarestrup, F.M.; Munch-Petersen, B.; Piškur, J. Nucleoside analogues are activated by bacterial deoxyribonucleoside kinases in a species-specific manner. J. Antimicrob. Chemother. 2007, 60, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Shilaih, M.; Angst, D.C.; Marzel, A.; Bonhoeffer, S.; Günthard, H.F.; Kouyos, R.D. Short communication Antibacterial effects of antiretrovirals, potential implications for microbiome studies in HIV. Antivir. Ther. 2018, 23, 91–94. [Google Scholar] [CrossRef]
- Hurst, M.; Noble, S. Stavudine. Drugs 1999, 58, 919–949. [Google Scholar] [CrossRef]
- Cruciani, M.; Liuzzi, G.; Chirianni, A.; Audagnotto, S.; Bonora, S.; Di Biagio, A.; Sinicco, A.; Bassetti, M.; Gatti, G. Penetration of didanosine in semen of HIV-1-infected men. J. Antimicrob. Chemother. 2006, 57, 1244–1247. [Google Scholar] [CrossRef][Green Version]
- Bang, L.M.; Scott, L.J. Emtricitabine. Drugs 2003, 63, 2413–2424. [Google Scholar] [CrossRef]
- Calcagno, A.; De Requena, D.G.; Simiele, M.; D’Avolio, A.; Tettoni, M.C.; Salassa, B.; Orofino, G.; Bramato, C.; Libanore, V.; Motta, I.; et al. Tenofovir Plasma Concentrations According to Companion Drugs: A Cross-Sectional Study of HIV-Positive Patients with Normal Renal Function. Antimicrob. Agents Chemother. 2013, 57, 1840–1843. [Google Scholar] [CrossRef] [PubMed]
- Capparelli, E.V.; Letendre, S.L.; Ellis, R.J.; Patel, P.; Holland, D.; McCutchan, J.A. Population Pharmacokinetics of Abacavir in Plasma and Cerebrospinal Fluid. Antimicrob. Agents Chemother. 2005, 49, 2504–2506. [Google Scholar] [CrossRef]
- Yuen, G.J.; Lou, Y.; Bumgarner, N.F.; Bishop, J.P.; Smith, G.A.; Otto, V.R.; Hoelscher, D.D. Equivalent Steady-State Pharmacokinetics of Lamivudine in Plasma and Lamivudine Triphosphate within Cells following Administration of Lamivudine at 300 Milligrams Once Daily and 150 Milligrams Twice Daily. Antimicrob. Agents Chemother. 2004, 48, 176–182. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Garré, B.; Shebany, K.; Gryspeerdt, A.; Baert, K.; Van Der Meulen, K.; Nauwynck, H.; Deprez, P.; De Backer, P.; Croubels, S. Pharmacokinetics of Acyclovir after Intravenous Infusion of Acyclovir and after Oral Administration of Acyclovir and Its Prodrug Valacyclovir in Healthy Adult Horses. Antimicrob. Agents Chemother. 2007, 51, 4308–4314. [Google Scholar] [CrossRef]
- Lerner, A.M.; Bailey, E.J. Concentrations of Idoxuridine in Serum, Urine, and Cerebrospinal Fluid of Patients with Suspected Diagnoses of Herpesvirus hominis Encephalitis. J. Clin. Investig. 1972, 51, 45–49. [Google Scholar] [CrossRef][Green Version]
- McEvoy, G.K. American Hospital Formulary Service-Drug Information 92; American Society of Hospital Pharmacists Inc.: Bethesda, MD, USA, 1993. [Google Scholar]
- National Center for Biotechnology Information. In PubChem Compound Summary for CID 21704, Vidarabine; National Center for Biotechnology Information: Bethesda, MD, USA, 2020.
- Robinson, D.M.; Scott, L.J.; Plosker, G.L. Entecavir. Drugs 2006, 66, 1605–1622. [Google Scholar] [CrossRef] [PubMed]
- Kosovec, J.E.; Egorin, M.J.; Gjurich, S.; Beumer, J.H. Quantitation of 5-fluorouracil (5-FU) in human plasma by liquid chromatography/electrospray ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2007, 22, 224–230. [Google Scholar] [CrossRef]
- Gandhi, V.; Plunkett, W. Cellular and Clinical Pharmacology of Fludarabine. Clin. Pharmacokinet. 2002, 41, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Hande, K.R.; Stein, R.S.; McDonough, D.A.; Greco, F.A.; Wolff, S.N. Effects of high-dose cytarabine. Clin. Pharmacol. Ther. 1982, 31, 669–674. [Google Scholar] [CrossRef]
- Sutton, M.D.; Smith, B.T.; Godoy, V.G.; Walker, G.C. The SOS response: Recent insights into umuDC-dependent mutagenesis and DNA damage tolerance. Annu. Rev. Genet. 2000, 34, 479–497. [Google Scholar] [CrossRef] [PubMed]
- Røst, L.M.; Olaisen, C.; Sharma, A.; Nedal, A.; Petrovic, V.; Kvitvang, H.F.N.; Bruheim, P.; Otterlei, M. PCNA has specific functions in regulation of metabolism in haematological cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Simmons, L.A.; Foti, J.J.; Cohen, S.E.; Walker, G.C. The SOS Regulatory Network. EcoSal Plus 2008, 3. [Google Scholar] [CrossRef]
- Cirz, R.T.; Jones, M.B.; Gingles, N.A.; Minogue, T.D.; Jarrahi, B.; Peterson, S.N.; Romesberg, F.E. Complete and SOS-Mediated Response of Staphylococcus aureus to the Antibiotic Ciprofloxacin. J. Bacteriol. 2007, 189, 531–539. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleoside Analogue (NA) | Reported Plasma Concentration (Pl.CR.) (µg/mL) | MIC (µg/mL) | Structure | Use |
|---|---|---|---|---|
| Stavudine (Stav) | ≤0.9 [37] | 64 |  | HIV, NRTI. dTTP analogue |
| Didanosine (Dida) | 0.05–0.86 [38] | 8 |  | HIV NRTI. dATP analogue |
| Emtricitabine (Emtri) | 1.01–11.5 [39] | >256 |  | HIV NRTI. dCTP analogue |
| Tenofovir (Teno) | 0.050–0.077 [40] | >256 |  | HIV NRTI. dTTP analogue |
| Abacavir (Abaca) | 0.04–1.13 [41] | >256 |  | HIV NRTI. dGTP analogue |
| Lamivudine (Lamu) | 0.02–0.11 [42] | >256 |  | HIV-1 NRTI. dTTP analogue |
| Acyclovir (Acyc) | 10.0 [43] | >256 |  | Herpesviruses. dGTP analogue |
| Idoxuridine (Idox) | 10.0–36.0 [44] | >256 |  | Herpesviruses. dUTP analogue. Thymidylate phosphatase inhibitor. |
| Vidarabine (Vida) | 0.2–0.4 [45,46] | >256 |  | Herpesviruses. dATP analogue |
| Entecavir (Ente) | 0.015–0.036 [47] | >256 |  | Hepatitis virus. dGTP analogue |
| 5-Fluoro Uracil (5-FU) | 0.025–8.0 [48] | 4 |  | Multiple cancers. Thymidylate synthase inhibitor |
| Fludarabine (Fluda) | 0.4–2.0 [49] | >256 |  | Leukaemia and lymphoma. dATP analogue |
| Cytarabine (Cyta) | ~486.0 [50] | >256 |  | Leukaemia. dCTP analogue |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sumabe, B.K.; Ræder, S.B.; Røst, L.M.; Sharma, A.; Donkor, E.S.; Mosi, L.; Duodu, S.; Bruheim, P.; Otterlei, M. Nucleoside Analogues Are Potent Inducers of Pol V-mediated Mutagenesis. Biomolecules 2021, 11, 843. https://doi.org/10.3390/biom11060843
Sumabe BK, Ræder SB, Røst LM, Sharma A, Donkor ES, Mosi L, Duodu S, Bruheim P, Otterlei M. Nucleoside Analogues Are Potent Inducers of Pol V-mediated Mutagenesis. Biomolecules. 2021; 11(6):843. https://doi.org/10.3390/biom11060843
Chicago/Turabian StyleSumabe, Balagra Kasim, Synnøve Brandt Ræder, Lisa Marie Røst, Animesh Sharma, Eric S. Donkor, Lydia Mosi, Samuel Duodu, Per Bruheim, and Marit Otterlei. 2021. "Nucleoside Analogues Are Potent Inducers of Pol V-mediated Mutagenesis" Biomolecules 11, no. 6: 843. https://doi.org/10.3390/biom11060843
APA StyleSumabe, B. K., Ræder, S. B., Røst, L. M., Sharma, A., Donkor, E. S., Mosi, L., Duodu, S., Bruheim, P., & Otterlei, M. (2021). Nucleoside Analogues Are Potent Inducers of Pol V-mediated Mutagenesis. Biomolecules, 11(6), 843. https://doi.org/10.3390/biom11060843