
DNMT3A Mutation-Induced CDK1 Overexpression Promotes Leukemogenesis by Modulating the Interaction between EZH2 and DNMT3A


Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmids Construction
2.2. Cell Culture, Transfection, RNA Interference and Immunoprecipitations
2.3. Immunofluorescence and FRET Analysis
2.4. Immunoblotting
2.5. Flow Cytometric Analysis
2.6. Chemical Reagents and Drug Combination Analysis
2.7. Statistical Analysis
3. Results
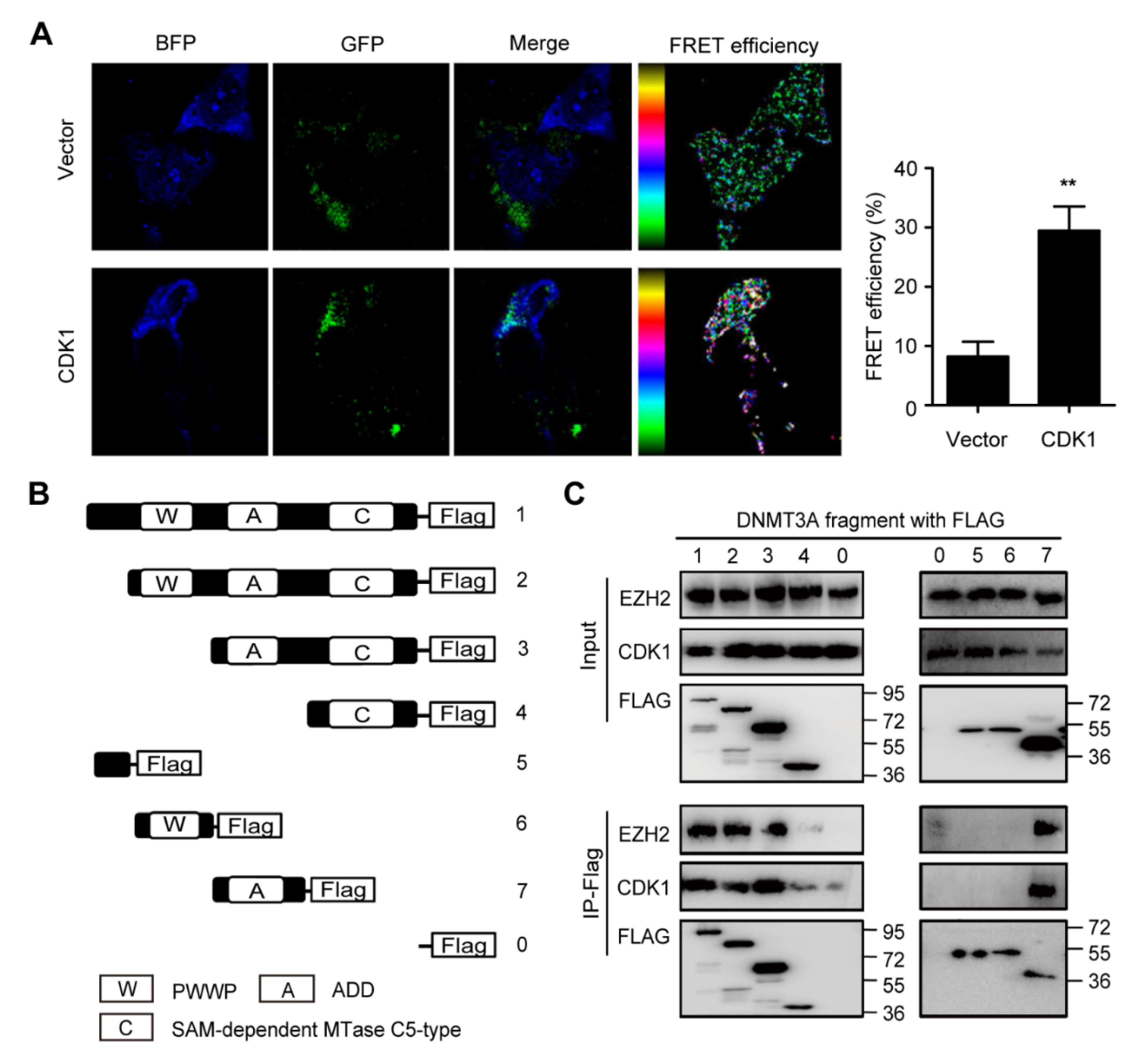
3.1. DNMT3A Protein Bound to CDK1 Protein through the ADD Domain

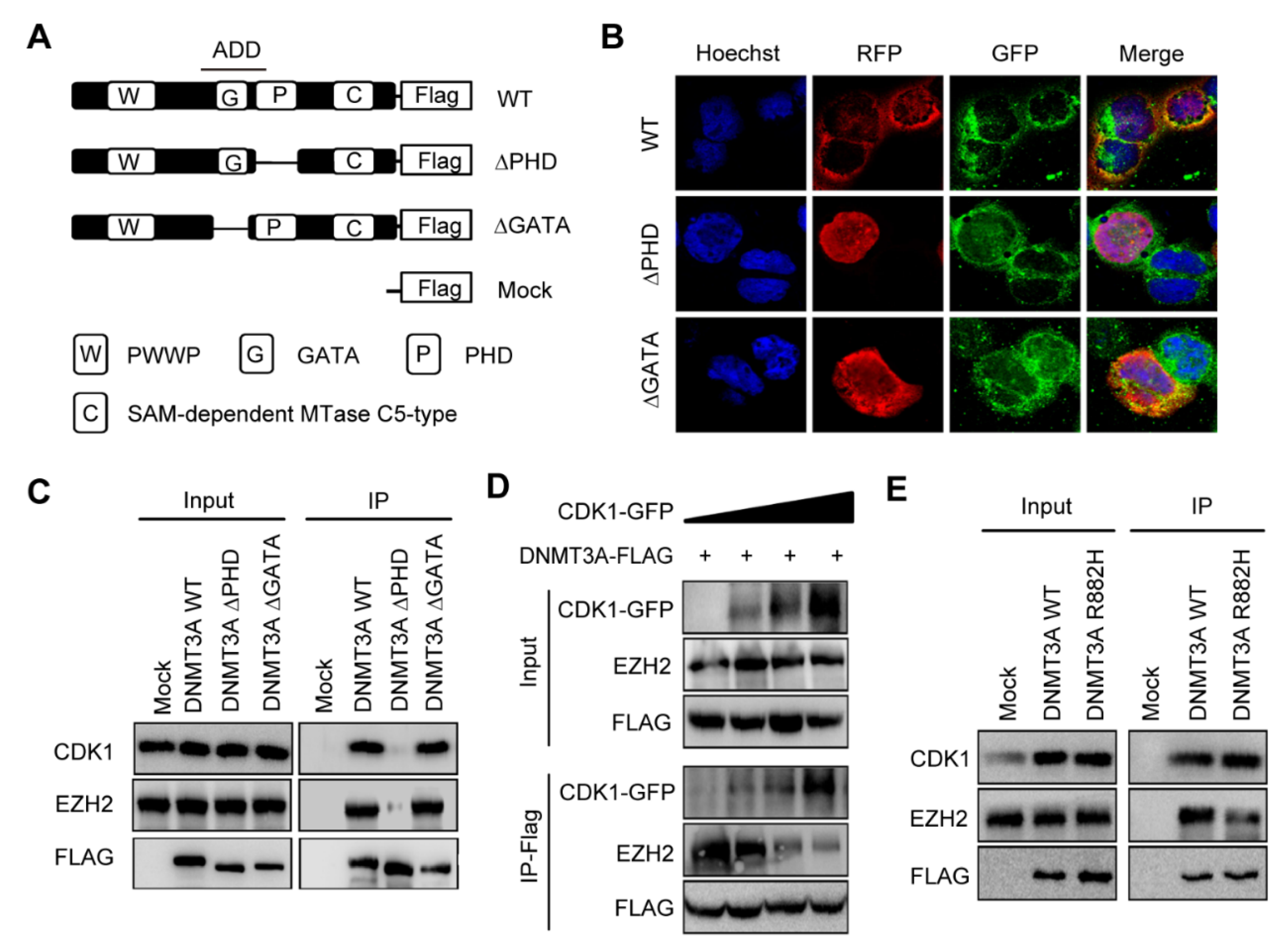
3.2. DNMT3A R882H Mutation-Induced CDK1 Overexpression Interrupted the Binding of EZH2 to PHD Domain of DNMT3A

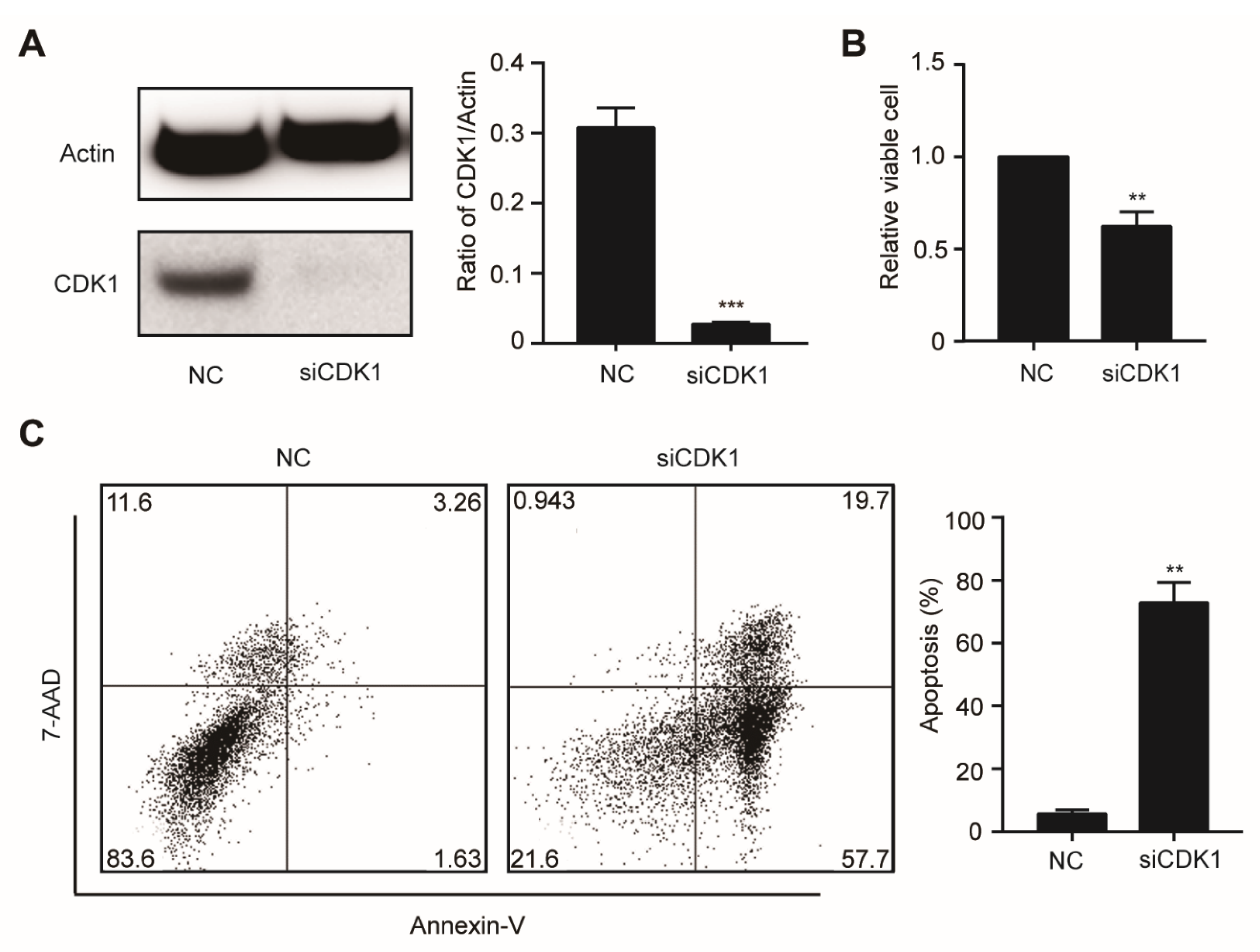
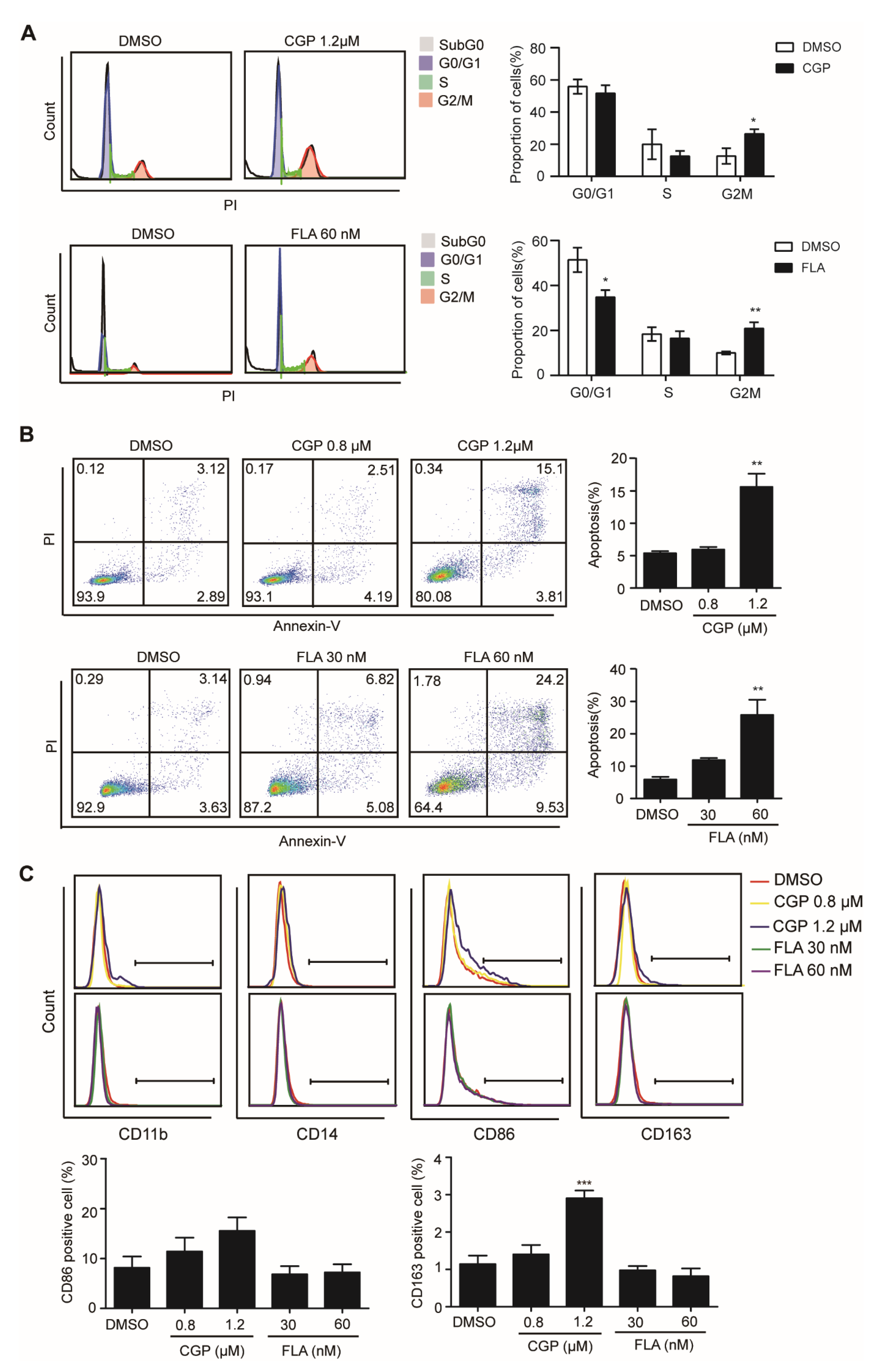
3.3. CDK1 Inhibitor Could Arrest Cells in G2M Phase and Induce Apoptosis in DNMT3A Mutation AML Cells
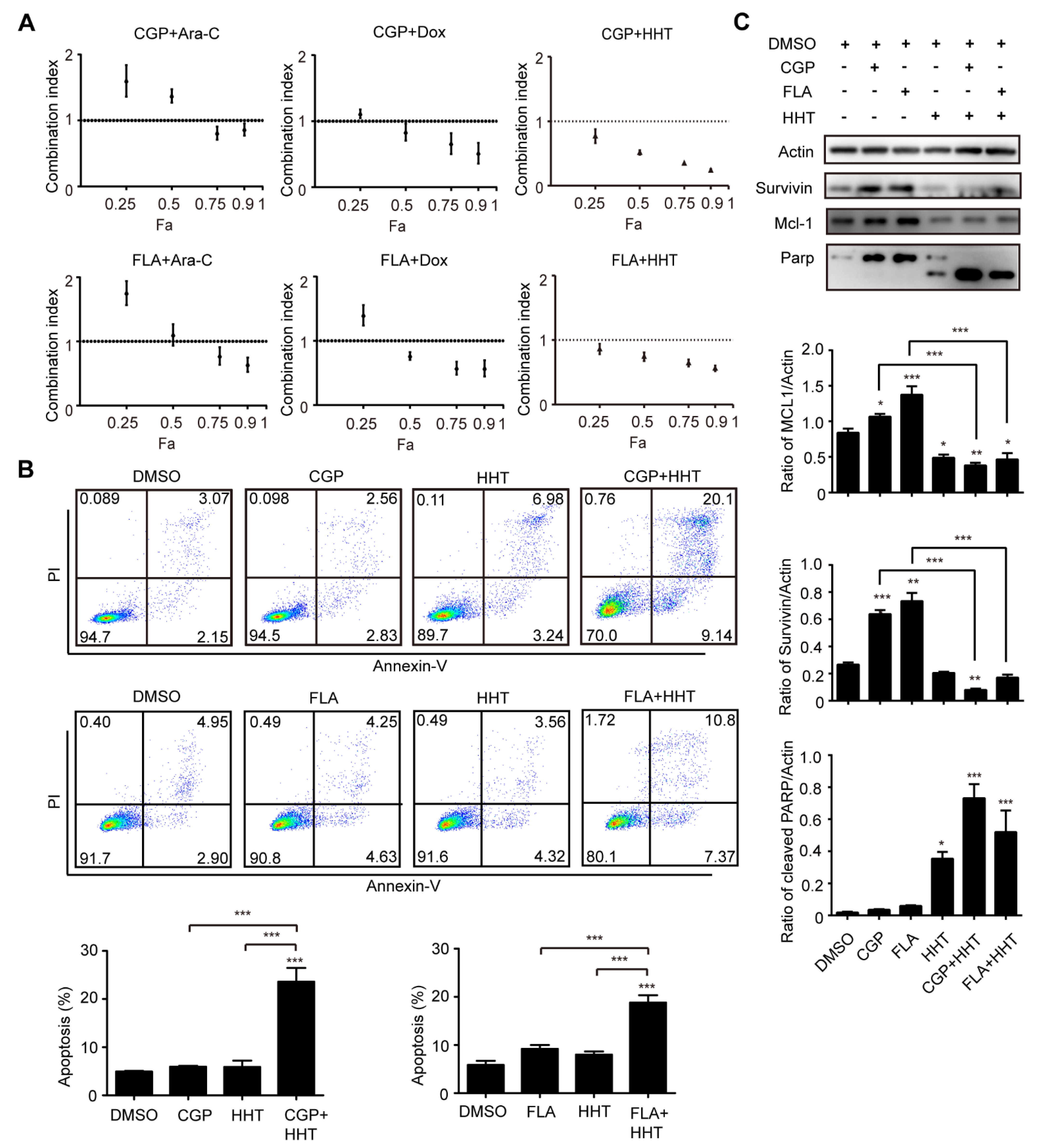
3.4. CDK1 Inhibitor and HHT Synergically Inhibited Proliferation of OCI-AML3 Cells
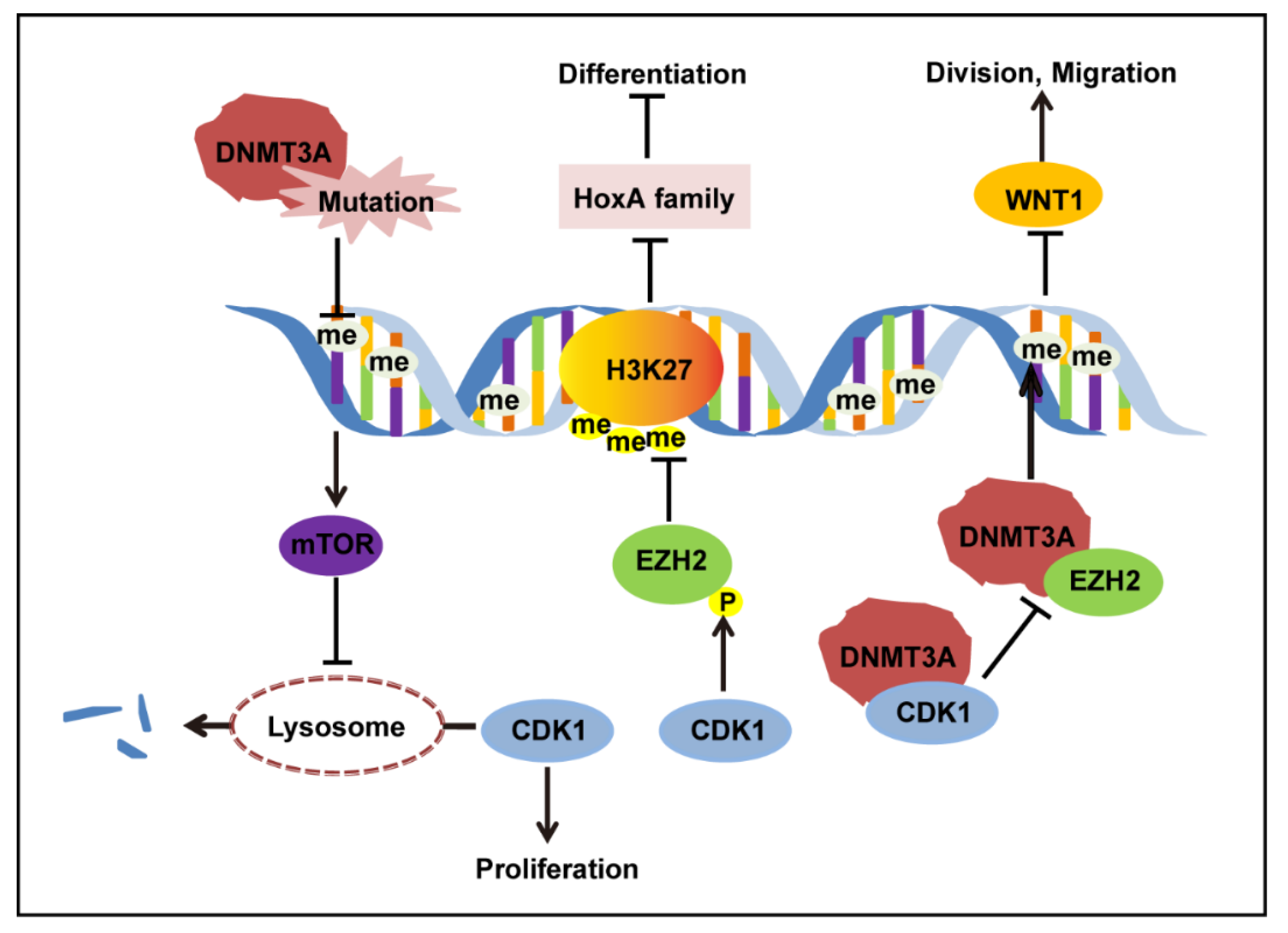
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA Methylation in the Mammalian Genome. Cell 2016, 167, 233–247.e17. [Google Scholar] [CrossRef] [PubMed]
- Norvil, A.B.; AlAbdi, L.; Liu, B.; Tu, Y.H.; Forstoffer, N.E.; Michie, A.R.; Chen, T.; Gowher, H. The acute myeloid leukemia variant DNMT3A Arg882His is a DNMT3B-like enzyme. Nucleic Acids Res. 2020, 48, 3761–3775. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.J.; Xu, J.; Gu, Z.H.; Pan, C.M.; Lu, G.; Shen, Y.; Shi, J.Y.; Zhu, Y.M.; Tang, L.; Zhang, X.W.; et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat. Genet. 2011, 43, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, L.; Gundry, M.C.; Goodell, M.A. DNMT3A in Leukemia. Cold Spring Harb. Perspect. Med. 2017, 7, a030320. [Google Scholar] [CrossRef] [PubMed]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef]
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2011, 44, 23–31. [Google Scholar] [CrossRef]
- Guryanova, O.A.; Lieu, Y.K.; Garrett-Bakelman, F.E.; Spitzer, B.; Glass, J.L.; Shank, K.; Martinez, A.B.; Rivera, S.A.; Durham, B.H.; Rapaport, F.; et al. Dnmt3a regulates myeloproliferation and liver-specific expansion of hematopoietic stem and progenitor cells. Leukemia 2016, 30, 1133–1142. [Google Scholar] [CrossRef]
- Russler-Germain, D.A.; Spencer, D.H.; Young, M.A.; Lamprecht, T.L.; Miller, C.A.; Fulton, R.; Meyer, M.R.; Erdmann-Gilmore, P.; Townsend, R.R.; Wilson, R.K.; et al. The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell 2014, 25, 442–454. [Google Scholar] [CrossRef]
- Lu, R.; Wang, P.; Parton, T.; Zhou, Y.; Chrysovergis, K.; Rockowitz, S.; Chen, W.Y.; Abdel-Wahab, O.; Wade, P.A.; Zheng, D.; et al. Epigenetic Perturbations by Arg882-Mutated DNMT3A Potentiate Aberrant Stem Cell Gene-Expression Program and Acute Leukemia Development. Cancer Cell 2016, 30, 92–107. [Google Scholar] [CrossRef]
- Xu, J.; Wang, Y.Y.; Dai, Y.J.; Zhang, W.; Zhang, W.N.; Xiong, S.M.; Gu, Z.H.; Wang, K.K.; Zeng, R.; Chen, Z.; et al. DNMT3A Arg882 mutation drives chronic myelomonocytic leukemia through disturbing gene expression/DNA methylation in hematopoietic cells. Proc. Natl. Acad. Sci. USA 2014, 111, 2620–2625. [Google Scholar] [CrossRef]
- Dai, Y.J.; Wang, Y.Y.; Huang, J.Y.; Xia, L.; Shi, X.D.; Xu, J.; Lu, J.; Su, X.B.; Yang, Y.; Zhang, W.N.; et al. Conditional knockin of Dnmt3a R878H initiates acute myeloid leukemia with mTOR pathway involvement. Proc. Natl. Acad. Sci. USA 2017, 114, 5237–5242. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Chen, Y.H.; Li, L.Y.; Lang, J.; Yeh, S.P.; Shi, B.; Yang, C.C.; Yang, J.Y.; Lin, C.Y.; Lai, C.C.; et al. CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nat. Cell Biol. 2011, 13, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Rondelet, G.; Dal Maso, T.; Willems, L.; Wouters, J. Structural basis for recognition of histone H3K36me3 nucleosome by human de novo DNA methyltransferases 3A and 3B. J. Struct. Biol. 2016, 194, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.M.; Lu, R.; Wang, P.; Yu, Y.; Chen, D.; Gao, L.; Liu, S.; Ji, D.; Rothbart, S.B.; Wang, Y.; et al. Structural basis for DNMT3A-mediated de novo DNA methylation. Nature 2018, 554, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Wang, L.; Li, J.; Ding, Z.; Xiao, J.; Yin, X.; He, S.; Shi, P.; Dong, L.; Li, G.; et al. Structural insight into autoinhibition and histone H3-induced activation of DNMT3A. Nature 2015, 517, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Viré, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Short, N.; Rytting, M.; Cortes, J. Acute myeloid leukaemia. Lancet (Lond. Engl.) 2018, 392, 593–606. [Google Scholar] [CrossRef]
- Kantarjian, H.; Kadia, T.; DiNardo, C.; Daver, N.; Borthakur, G.; Jabbour, E.; Garcia-Manero, G.; Konopleva, M.; Ravandi, F. Acute myeloid leukemia: Current progress and future directions. Blood Cancer J. 2021, 11, 41. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Bixby, D.; Perl, A.; Bhatt, V.R.; Altman, J.K.; Appelbaum, F.R.; de Lima, M.; Fathi, A.T.; Foran, J.M.; Gojo, I.; et al. NCCN Guidelines Insights: Acute Myeloid Leukemia, Version 2.2021. J. Natl. Compr. Cancer Netw 2021, 19, 16–27. [Google Scholar] [CrossRef]
- Santamaria, D.; Barriere, C.; Cerqueira, A.; Hunt, S.; Tardy, C.; Newton, K.; Caceres, J.F.; Dubus, P.; Malumbres, M.; Barbacid, M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2007, 448, 811–815. [Google Scholar] [CrossRef]
- Hedblom, A.; Laursen, K.B.; Miftakhova, R.; Sarwar, M.; Anagnostaki, L.; Bredberg, A.; Mongan, N.P.; Gudas, L.J.; Persson, J.L. CDK1 interacts with RARgamma and plays an important role in treatment response of acute myeloid leukemia. Cell Cycle 2013, 12, 1251–1266. [Google Scholar] [CrossRef]
- Dai, Y.; Dent, P.; Grant, S. Induction of apoptosis in human leukemia cells by the CDK1 inhibitor CGP74514A. Cell Cycle 2002, 1, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Cyclin-dependent protein serine/threonine kinase inhibitors as anticancer drugs. Pharmacol. Res. 2019, 139, 471–488. [Google Scholar] [CrossRef]
- George, S.; Kasimis, B.S.; Cogswell, J.; Schwarzenberger, P.; Shapiro, G.I.; Fidias, P.; Bukowski, R.M. Phase I study of flavopiridol in combination with Paclitaxel and Carboplatin in patients with non-small-cell lung cancer. Clin. Lung Cancer 2008, 9, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.S.; Ruppert, A.S.; Johnson, A.J.; Fischer, B.; Heerema, N.A.; Andritsos, L.A.; Blum, K.A.; Flynn, J.M.; Jones, J.A.; Hu, W.; et al. Phase II study of flavopiridol in relapsed chronic lymphocytic leukemia demonstrating high response rates in genetically high-risk disease. J. Clin. Oncol. 2009, 27, 6012–6018. [Google Scholar] [CrossRef]
- Lanasa, M.C.; Andritsos, L.; Brown, J.R.; Gabrilove, J.; Caligaris-Cappio, F.; Ghia, P.; Larson, R.A.; Kipps, T.J.; Leblond, V.; Milligan, D.W.; et al. Final results of EFC6663: A multicenter, international, phase 2 study of alvocidib for patients with fludarabine-refractory chronic lymphocytic leukemia. Leuk. Res. 2015, 39, 495–500. [Google Scholar] [CrossRef]
- Karp, J.E.; Smith, B.D.; Resar, L.S.; Greer, J.M.; Blackford, A.; Zhao, M.; Moton-Nelson, D.; Alino, K.; Levis, M.J.; Gore, S.D.; et al. Phase 1 and pharmacokinetic study of bolus-infusion flavopiridol followed by cytosine arabinoside and mitoxantrone for acute leukemias. Blood 2011, 117, 3302–3310. [Google Scholar] [CrossRef]
- Zeidner, J.F.; Foster, M.C.; Blackford, A.L.; Litzow, M.R.; Morris, L.E.; Strickland, S.A.; Lancet, J.E.; Bose, P.; Levy, M.Y.; Tibes, R.; et al. Randomized multicenter phase II study of flavopiridol (alvocidib), cytarabine, and mitoxantrone (FLAM) versus cytarabine/daunorubicin (7+3) in newly diagnosed acute myeloid leukemia. Haematologica 2015, 100, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Litzow, M.R.; Wang, X.V.; Carroll, M.P.; Karp, J.E.; Ketterling, R.P.; Zhang, Y.; Kaufmann, S.H.; Lazarus, H.M.; Luger, S.M.; Paietta, E.M.; et al. A randomized trial of three novel regimens for recurrent acute myeloid leukemia demonstrates the continuing challenge of treating this difficult disease. Am. J. Hematol. 2019, 94, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Youvan, D.C.; Silva, C.M.; Bylina, E.J.; Coleman, W.J.; Dilworth, M.R.; Yang, M.M. Calibration of Fluorescence Resonance Energy Transfer in Microscopy. U.S. Patent 6,661,909, 9 December 2003. [Google Scholar]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharm. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Krishnamurthy, N.; Kurzrock, R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat. Rev. 2018, 62, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, H.; Vertino, P.M.; Cheng, X. Molecular coupling of DNA methylation and histone methylation. Epigenomics 2010, 2, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Grandage, V.L.; Linch, D.C.; Khwaja, A. Constitutive activation of the Wnt/beta-catenin signalling pathway in acute myeloid leukaemia. Oncogene 2005, 24, 2410–2420. [Google Scholar] [CrossRef]
- Ibrahem, L.; Mahfouz, R.; Elhelw, L.; Abdsalam, E.M.; Soliman, R. Prognostic significance of DNMT3A mutations in patients with acute myeloid leukemia. Blood Cells Mol. Dis. 2015, 54, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Gaidzik, V.I.; Weber, D.; Paschka, P.; Kaumanns, A.; Krieger, S.; Corbacioglu, A.; Kronke, J.; Kapp-Schwoerer, S.; Kramer, D.; Horst, H.A.; et al. DNMT3A mutant transcript levels persist in remission and do not predict outcome in patients with acute myeloid leukemia. Leukemia 2018, 32, 30–37. [Google Scholar] [CrossRef]
- Yang, X.; Wang, J. Precision therapy for acute myeloid leukemia. J. Hematol. Oncol. 2018, 11, 3. [Google Scholar] [CrossRef]
- Rau, R.E.; Rodriguez, B.A.; Luo, M.; Jeong, M.; Rosen, A.; Rogers, J.H.; Campbell, C.T.; Daigle, S.R.; Deng, L.; Song, Y.; et al. DOT1L as a therapeutic target for the treatment of DNMT3A-mutant acute myeloid leukemia. Blood 2016, 128, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Radomska, H.S.; Alberich-Jorda, M.; Will, B.; Gonzalez, D.; Delwel, R.; Tenen, D.G. Targeting CDK1 promotes FLT3-activated acute myeloid leukemia differentiation through C/EBPalpha. J. Clin. Investig. 2012, 122, 2955–2966. [Google Scholar] [CrossRef]
- Kantarjian, H.; Ravandi, F.; O’Brien, S.; Cortes, J.; Faderl, S.; Garcia-Manero, G.; Jabbour, E.; Wierda, W.; Kadia, T.; Pierce, S.; et al. Intensive chemotherapy does not benefit most older patients (age 70 years or older) with acute myeloid leukemia. Blood 2010, 116, 4422–4429. [Google Scholar] [CrossRef]
- Gollner, S.; Oellerich, T.; Agrawal-Singh, S.; Schenk, T.; Klein, H.U.; Rohde, C.; Pabst, C.; Sauer, T.; Lerdrup, M.; Tavor, S.; et al. Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia. Nat. Med. 2017, 23, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wang, J. Homoharringtonine and omacetaxine for myeloid hematological malignancies. J. Hematol. Oncol. 2014, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Jiang, D.Z.; Mai, W.Y.; Meng, H.T.; Qian, W.B.; Tong, H.Y.; Huang, J.; Mao, L.P.; Tong, Y.; Wang, L.; et al. Homoharringtonine in combination with cytarabine and aclarubicin resulted in high complete remission rate after the first induction therapy in patients with de novo acute myeloid leukemia. Leukemia 2006, 20, 1361–1367. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, S.; Yang, J.; Song, X.; Chen, L.; Huang, C.; Hou, J.; Zhang, W. A homoharringtonine-based induction regimen for the treatment of elderly patients with acute myeloid leukemia: A single center experience from China. J. Hematol. Oncol. 2009, 2, 32. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yuan, K.; Wang, X.; Dong, H.; Min, W.; Hao, H.; Yang, P. Selective inhibition of CDK4/6: A safe and effective strategy for developing anticancer drugs. Acta Pharm. Sin. B 2021, 11, 30–54. [Google Scholar] [CrossRef]
- Tibes, R.; McDonagh, K.T.; Lekakis, L.; Bogenberger, J.M.; Kim, S.; Frazer, N.; Mohrland, S.; Bassett, D.; Garcia, R.; Schroeder, K.; et al. Phase I study of the novel Cdc2/CDK1 and AKT inhibitor terameprocol in patients with advanced leukemias. Investig. New Drugs 2015, 33, 389–396. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Forward | Reverse |
|---|---|---|
| EZH2-PEGFP-C1 | CCCTCGAGACATGGGCCAGACTGGGAA | CGGGATCCTCAAGGGATTTCCATTTCTCT |
| CDK1-PEGFP-C1 | CGGAATTCGATGGAAGATTATACCAAAATAGAGA | CGGGATCCCTACATCTTCTTAATCTGATTGTCC |
| WT-DNMT3A-Flag(1) | CGGAATTCCATGCCCGCCATGCCCTCCAGCGGCCCC | GAAGATCTTTACACACACGCAAAATACTCCTTCAGC |
| 195–912-DNMT3A-Flag(2) | CGGAATTCCATGCCCTACTACATCAGCAAGCGCAA | GAAGATCTTTACACACACGCAAAATACTCCTTCAGC |
| 431–912-DNMT3A-Flag(3) | CGGAATTCCATGAATCCCTACAAAGAAGTGTACACGG | GAAGATCTTTACACACACGCAAAATACTCCTTCAGC |
| 611–912-DNMT3A-Flag(4) | CGGAATTCCATGGCTAATAACCACGACCAGGAATTTG | GAAGATCTTTACACACACGCAAAATACTCCTTCAGC |
| 1–194-DNMT3A-Flag(5) | CGGAATTCCATGCCCGCCATGCCCTCCAGCGGCCCC | GAAGATCTTTAGTCCCCCGCCTGGAAGGTGAGCCTCG |
| 195–430-DNMT3A-Flag(6) | CGGAATTCCATGCCCTACTACATCAGCAAGCGCAA | GAAGATCTTTACTTCTCTTCTTCTGGTGGCT |
| 431–610-DNMT3A-Flag(7) | CGGAATTCCATGAATCCCTACAAAGAAGTGTACACGG | GAAGATCTTTAGAAGAACATCTGGAGCCGGGAGGG |
| ∆PHD-DNMT3A-Flag | CGACGACGGCTACACCTACGGGCTGC | GCAGCCCGTAGGTGTAGCCGTCGTCG |
| ∆GATA-DNMT3A-Flag | CCGGAACATTGAGGACTGTGCGTACCAGTACG | CGTACTGGTACGCACAGTCCTCAATGTTCCGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Dai, Y.; Yang, X.; Wu, S.; Wang, Y. DNMT3A Mutation-Induced CDK1 Overexpression Promotes Leukemogenesis by Modulating the Interaction between EZH2 and DNMT3A. Biomolecules 2021, 11, 781. https://doi.org/10.3390/biom11060781
Yang Y, Dai Y, Yang X, Wu S, Wang Y. DNMT3A Mutation-Induced CDK1 Overexpression Promotes Leukemogenesis by Modulating the Interaction between EZH2 and DNMT3A. Biomolecules. 2021; 11(6):781. https://doi.org/10.3390/biom11060781
Chicago/Turabian StyleYang, Ying, Yujun Dai, Xuejiao Yang, Songfang Wu, and Yueying Wang. 2021. "DNMT3A Mutation-Induced CDK1 Overexpression Promotes Leukemogenesis by Modulating the Interaction between EZH2 and DNMT3A" Biomolecules 11, no. 6: 781. https://doi.org/10.3390/biom11060781
APA StyleYang, Y., Dai, Y., Yang, X., Wu, S., & Wang, Y. (2021). DNMT3A Mutation-Induced CDK1 Overexpression Promotes Leukemogenesis by Modulating the Interaction between EZH2 and DNMT3A. Biomolecules, 11(6), 781. https://doi.org/10.3390/biom11060781