
Novel Chemically Modified Curcumin (CMC) Derivatives Inhibit Tyrosinase Activity and Melanin Synthesis in B16F10 Mouse Melanoma Cells


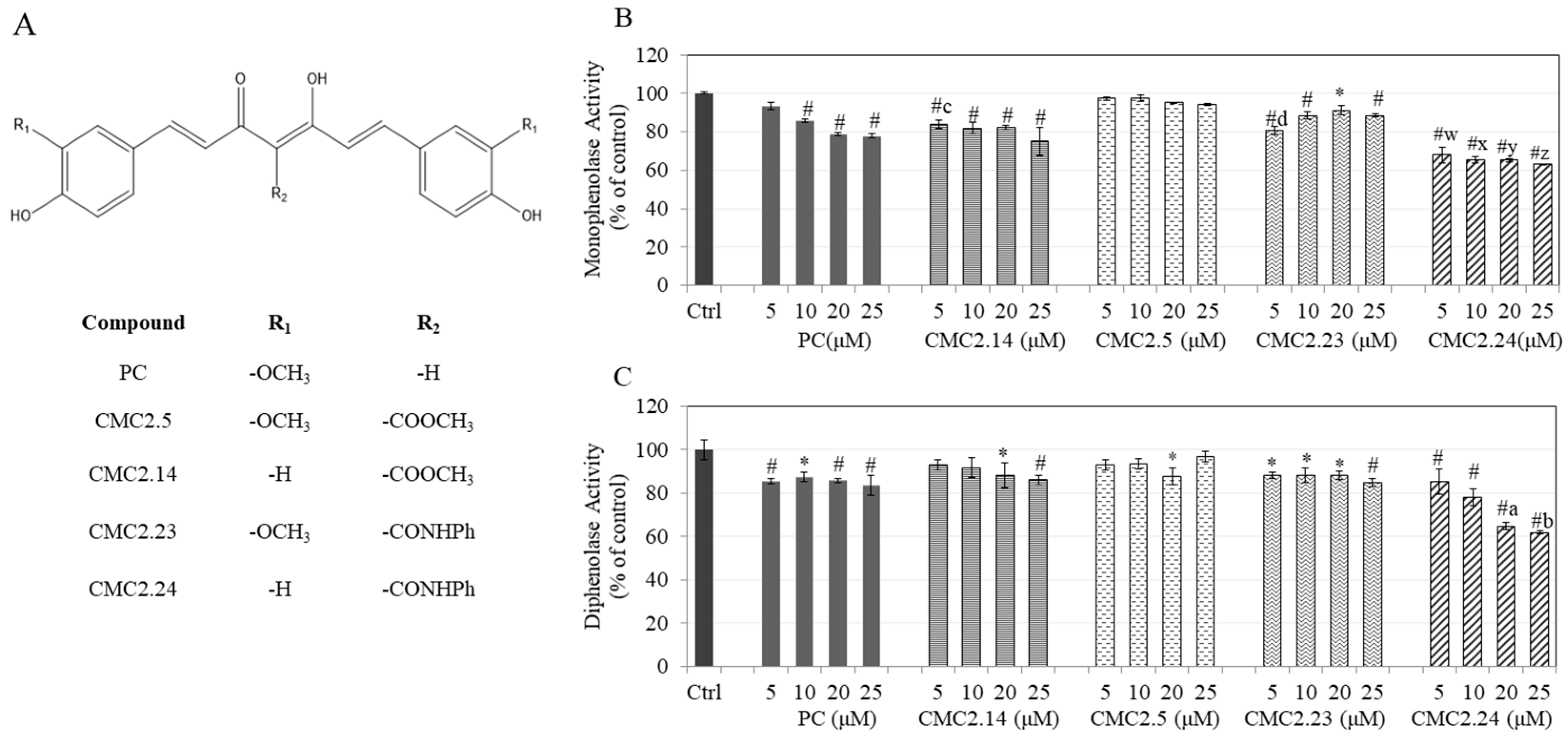{kind=link}
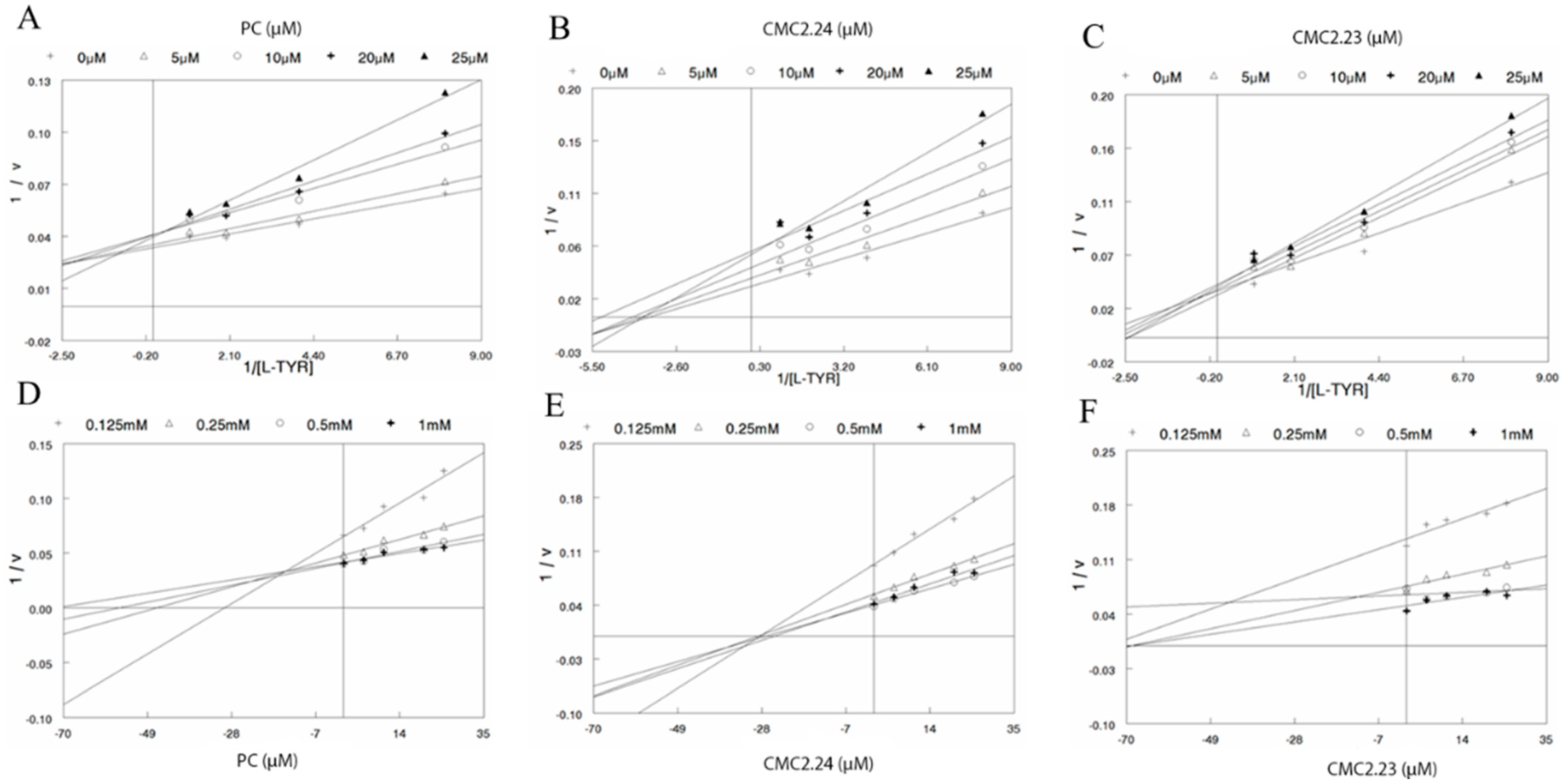{kind=link}
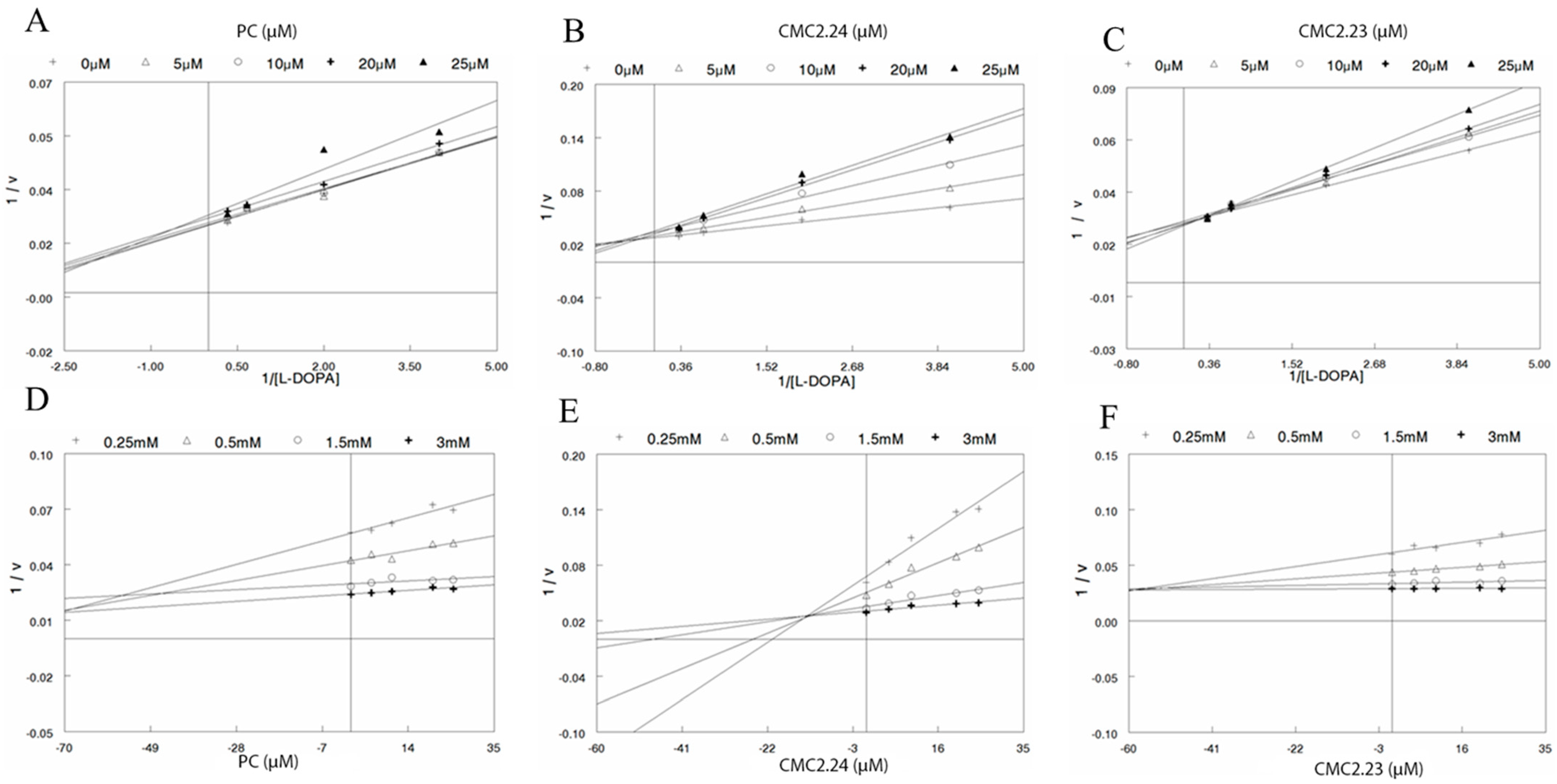{kind=link}
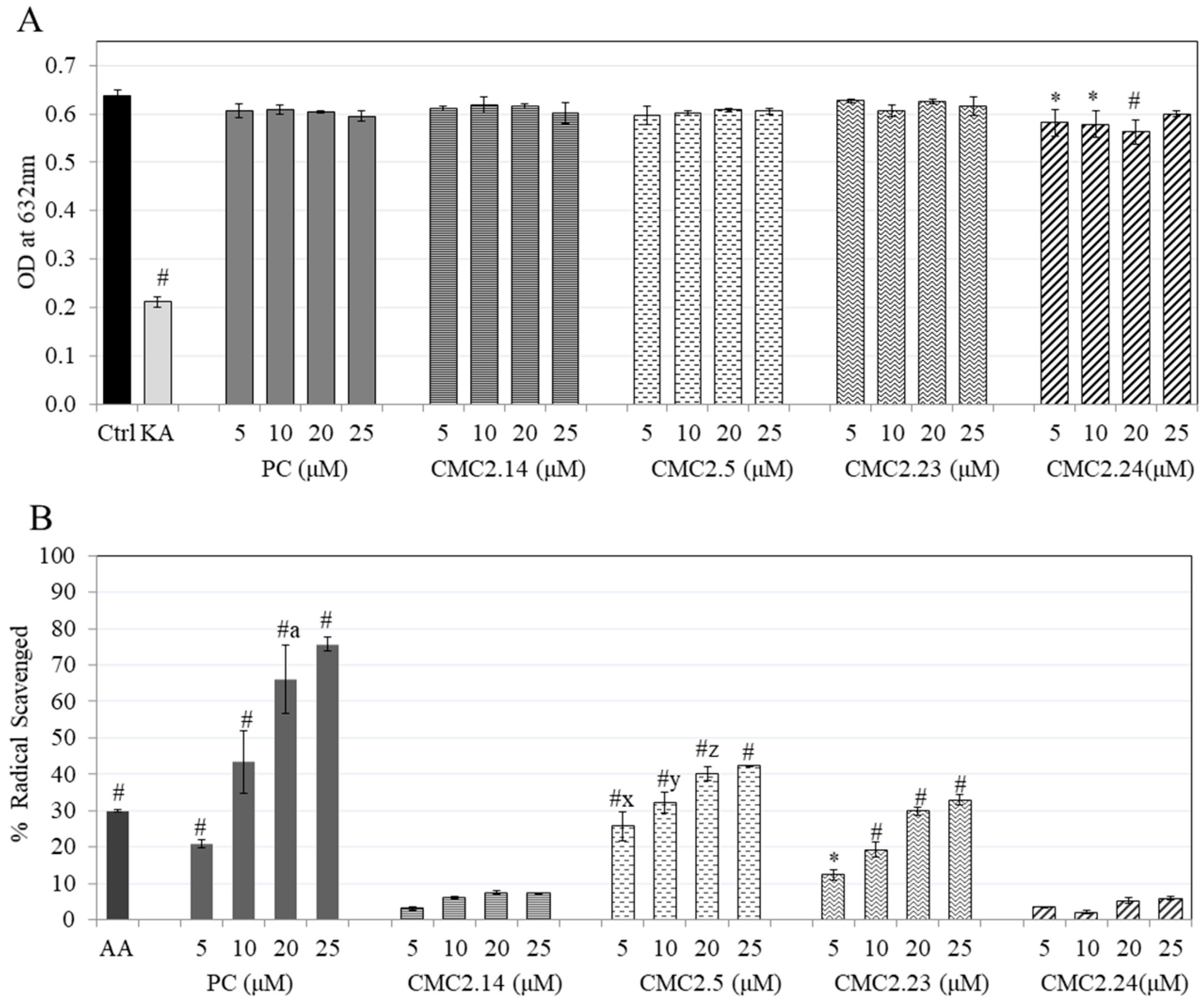{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
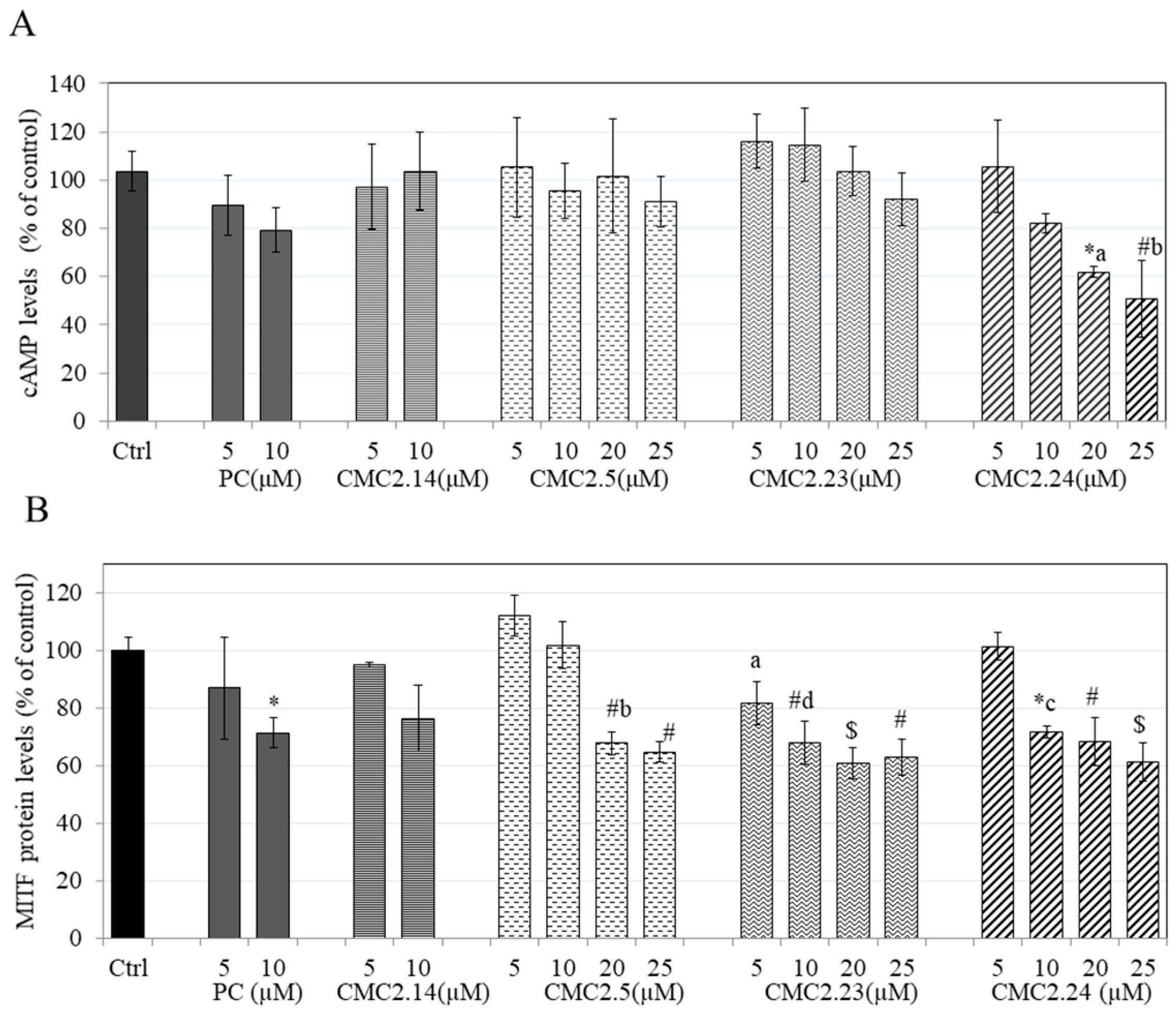
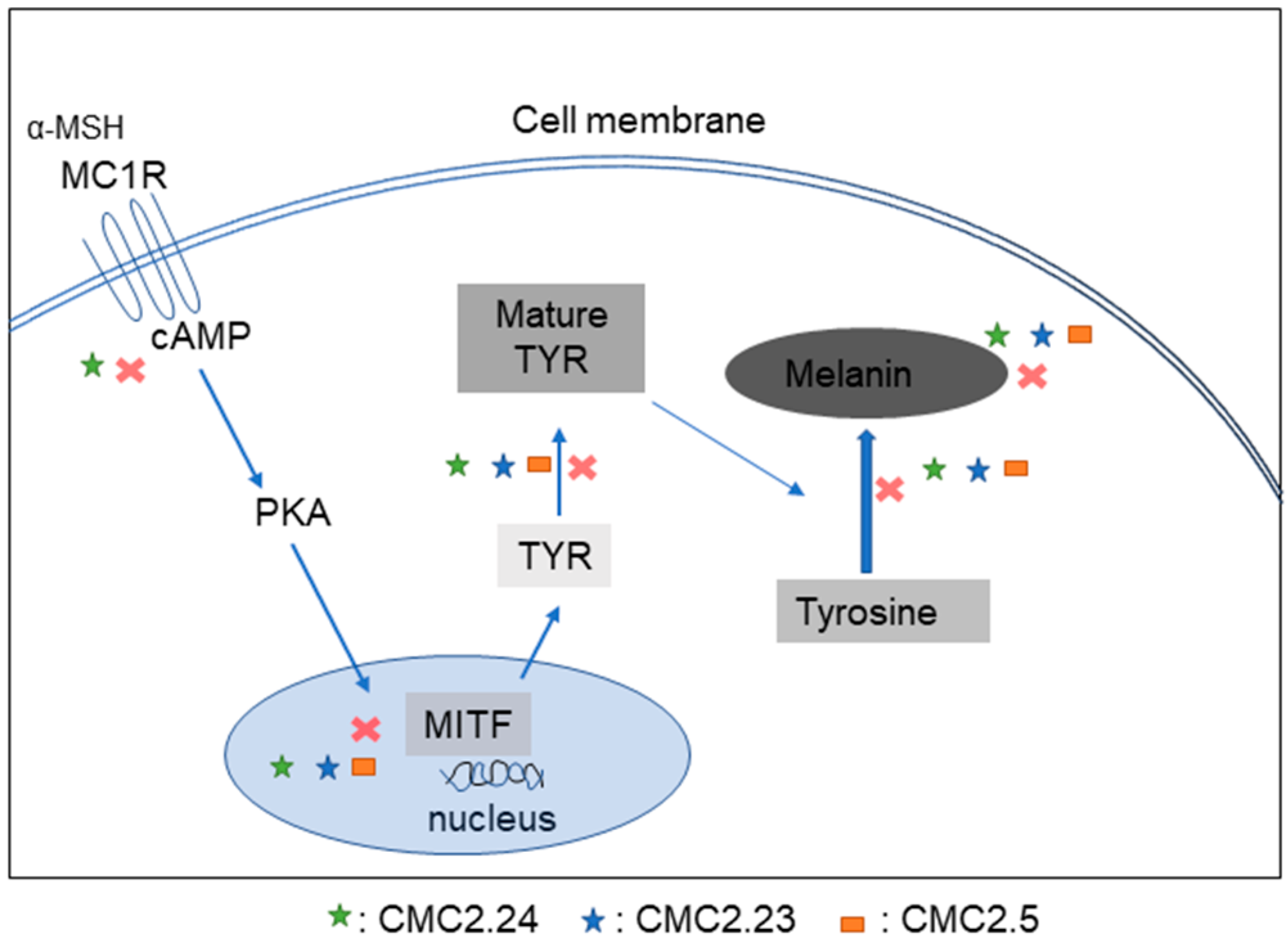
Abstract
Share and Cite
Goenka, S.; Johnson, F.; Simon, S.R. Novel Chemically Modified Curcumin (CMC) Derivatives Inhibit Tyrosinase Activity and Melanin Synthesis in B16F10 Mouse Melanoma Cells. Biomolecules 2021, 11, 674. https://doi.org/10.3390/biom11050674
Goenka S, Johnson F, Simon SR. Novel Chemically Modified Curcumin (CMC) Derivatives Inhibit Tyrosinase Activity and Melanin Synthesis in B16F10 Mouse Melanoma Cells. Biomolecules. 2021; 11(5):674. https://doi.org/10.3390/biom11050674
Chicago/Turabian StyleGoenka, Shilpi, Francis Johnson, and Sanford R. Simon. 2021. "Novel Chemically Modified Curcumin (CMC) Derivatives Inhibit Tyrosinase Activity and Melanin Synthesis in B16F10 Mouse Melanoma Cells" Biomolecules 11, no. 5: 674. https://doi.org/10.3390/biom11050674
APA StyleGoenka, S., Johnson, F., & Simon, S. R. (2021). Novel Chemically Modified Curcumin (CMC) Derivatives Inhibit Tyrosinase Activity and Melanin Synthesis in B16F10 Mouse Melanoma Cells. Biomolecules, 11(5), 674. https://doi.org/10.3390/biom11050674