
Chemoinformatics Studies on a Series of Imidazoles as Cruzain Inhibitors

 , ,
, ,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. QSAR and Molecular Modeling Tools
2.2. Dataset
2.3. Molecular Docking
2.4. QSAR Modeling
2.5. Applicabilty Domain
3. Results and Discussion
3.1. HQSAR and AutoQSAR Models
3.2. CoMFA and CoMSIA Models
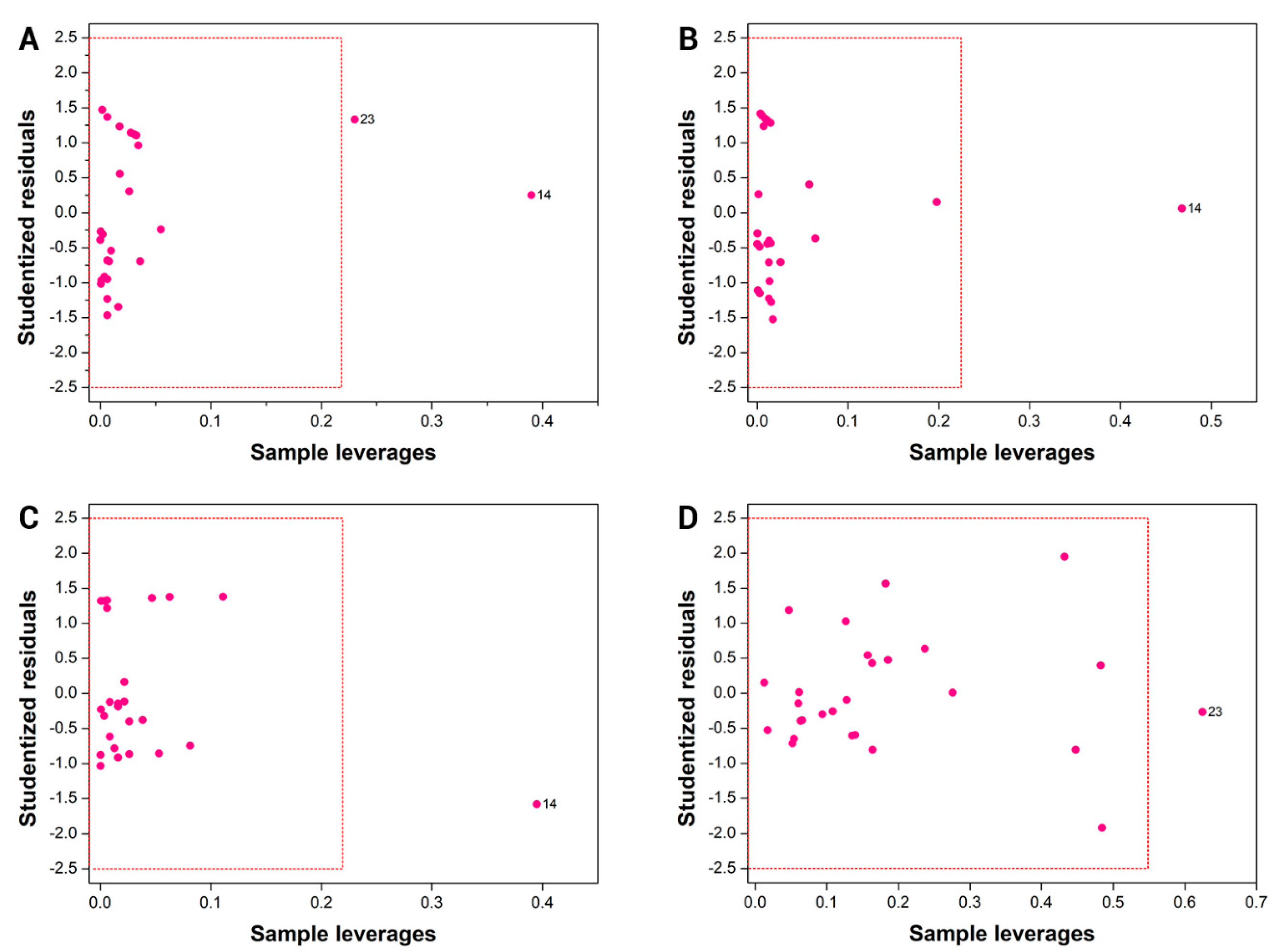
3.3. Applicability Domain
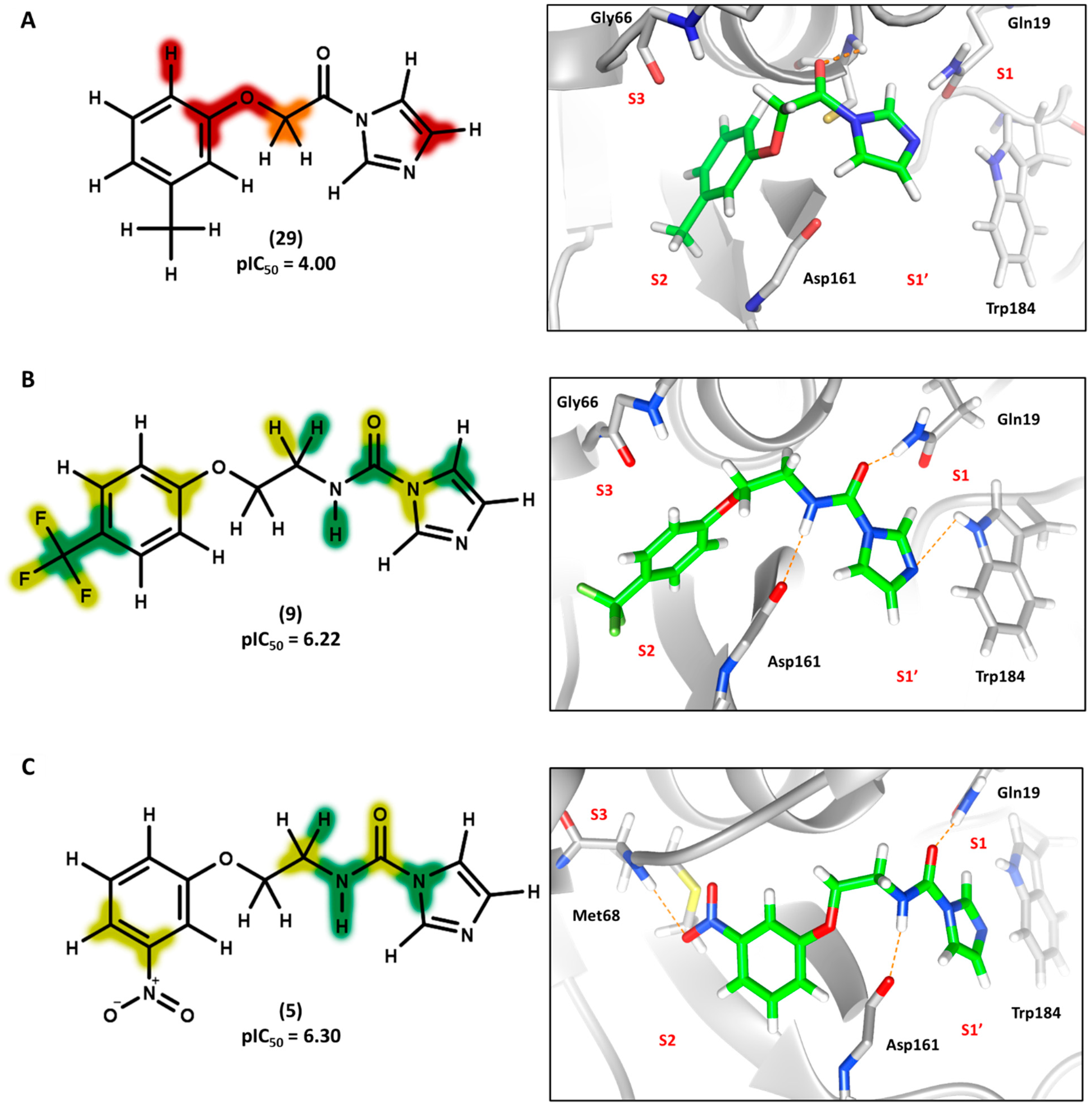
3.4. 2D Contribution Maps
3.5. 3D Contour Maps
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Neglected Tropical Diseases Report by the Director-General. Available online: http://apps.who.int/gb/ebwha/pdf_files/EB146/B146_14-en.pdf?ua=1 (accessed on 30 January 2021).
- World Health Organization. Chagas Disease (American trypanosomiasis). Available online: https://www.who.int/health-topics/chagas-disease#tab=tab_1 (accessed on 30 January 2021).
- Pérez-Molina, J.A.; Molina, I. Chagas disease. Lancet 2018, 391, 82–94. [Google Scholar] [CrossRef]
- Arnal, A.; Waleckx, E.; Rico-Chávez, O.; Herrera, C.; Dumonteil, E. Estimating the current burden of Chagas disease in Mexico: A systematic review and meta-analysis of epidemiological surveys from 2006 to 2017. PLoS Negl. Trop. Dis. 2019, 13, e0006859. [Google Scholar] [CrossRef] [PubMed]
- GBD 2015 DALYs and HALE Collaborators. Global, regional, and national disability-adjusted life-years (DALYs) for 315 diseases and injuries and healthy life expectancy (HALE), 1990–2015: A systematic analysis for the Global Burden of Disease Study. Lancet 2016, 388, 1603–1658. [Google Scholar] [CrossRef]
- Pérez-Molina, J.A.; Crespillo-Andújar, C.; Bosch-Nicolau, P.; Molina, I. Trypanocidal treatment of Chagas disease. Enferm. Infecc. Microbiol. Clin. 2020. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.K.; Kumar, V.; Kaur, K. Imidazole containing natural products as antimicrobial agents: A review. Nat. Prod. J. 2014, 4, 73–81. [Google Scholar] [CrossRef]
- Zhang, L.; Peng, X.M.; Damu, G.L.; Geng, R.X.; Zhou, C.H. Comprehensive review in current developments of imidazole-based medicinal chemistry. Med. Res. Rev. 2014, 34, 340–437. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Ma, Z.; Zhang, D. Synthesis of imidazole-based medicinal molecules utilizing the van Leusen imidazole synthesis. Pharmaceuticals 2020, 13, 37. [Google Scholar] [CrossRef] [PubMed]
- Osei, E.; Kwain, S.; Mawuli, G.T.; Anang, A.K.; Owusu, K.B.; Camas, M.; Camas, A.S.; Ohashi, M.; Alexandru-Crivac, C.N.; Deng, H.; et al. Paenidigyamycin A, potent antiparasitic imidazole alkaloid from the Ghanaian Paenibacillus sp. DE2SH. Mar. Drugs 2018, 17, 9. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; Andricopulo, A.D. Targeting cysteine proteases in trypanosomatid disease drug discovery. Pharmacol. Ther. 2017, 180, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Siqueira-Neto, J.L.; Debnath, A.; McCall, L.I.; Bernatchez, J.A.; Ndao, M.; Reed, S.L.; Rosenthal, P.J. Cysteine proteases in protozoan parasites. PLoS Negl. Trop. Dis. 2018, 12, e0006512. [Google Scholar] [CrossRef] [PubMed]
- de Souza, M.L.; de Oliveira Rezende Junior, C.; Ferreira, R.S.; Espinoza Chávez, R.M.; Ferreira, L.L.G.; Slafer, B.W.; Magalhães, L.G.; Krogh, R.; Oliva, G.; Cruz, F.C.; et al. Discovery of potent, reversible, and competitive cruzain inhibitors with trypanocidal activity: A structure-based drug design approach. J. Chem. Inf. Model. 2020, 60, 1028–1041. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.S.; Dessoy, M.A.; Pauli, I.; Souza, M.L.; Krogh, R.; Sales, A.I.; Oliva, G.; Dias, L.C.; Andricopulo, A.D. Synthesis, biological evaluation, and structure-activity relationships of potent noncovalent and nonpeptidic cruzain inhibitors as anti-Trypanosoma cruzi agents. J. Med. Chem. 2014, 57, 2380–2392. [Google Scholar] [CrossRef] [PubMed]
- Zanatta, N.; Amaral, S.S.; Santos, J.M.; Mello, D.L.; Fernandes, L.D.; Nonacorso, H.G.; Martins, M.A.P.; Andricopulo, A.D.; Borchhardt, D.M. Convergent synthesis and cruzain inhibitory activity of novel 2-(N ‘-benzylidenehydrazino)-4-trifluoromethyl-pyrimidines. Bioorg. Med. Chem. 2008, 16, 10236–10243. [Google Scholar] [CrossRef] [PubMed]
- Chenna, B.C.; Li, L.; Mellott, D.M.; Zhai, X.; Siqueira-Neto, J.L.; Calvet Alvarez, C.; Bernatchez, J.A.; Desormeaux, E.; Alvarez Hernandez, E.; Gomez, J.; et al. Peptidomimetic vinyl heterocyclic inhibitors of cruzain effect antitrypanosomal activity. J. Med. Chem. 2020, 63, 3298–3316. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.L.; Duan, J.; Smith, E.; Von Bargen, C.D.; Sherman, W.; Repasky, M.P. AutoQSAR: An Automated Machine Learning tool for best-practice quantitative structure-activity relationship modeling. Future Med. Chem. 2016, 8, 1825–1839. [Google Scholar] [CrossRef] [PubMed]
- Farutin, V.; Masterson, L.; Andricopulo, A.D.; Cheng, J.M.; Riley, B.; Hakimi, R.; Frazer, J.W.; Cordes, E.H. Structure-activity Relationships for a Class of Inhibitors of Purine Nucleoside Phosphorylase. J. Med. Chem. 1999, 42, 2422–2431. [Google Scholar] [CrossRef] [PubMed]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G.; Abraham, U.; Mietzner, T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem. 1994, 37, 4130–4146. [Google Scholar] [CrossRef]
- Pauli, I.; Ferreira, L.G.; de Souza, M.L.; Oliva, G.; Ferreira, R.S.; Dessoy, M.A.; Slafer, B.W.; Dias, L.C.; Andricopulo, A.D. Molecular modeling and structure-activity relationships for a series of benzimidazole derivatives as cruzain inhibitors. Future Med. Chem. 2017, 9, 641–657. [Google Scholar] [CrossRef] [PubMed]
- Andricopulo, A.D.; Montanari, C.A. Structure-activity relationships for the design of small-molecule inhibitors. Mini-Rev. Med. Chem. 2005, 5, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; Andricopulo, A.D. ADMET modeling approaches in drug discovery. Drug Discov. Today 2019, 24, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Andricopulo, A.D.; Muller, L.A.; Cechinel, V.; Cani, G.S.; Roos, J.F.; Correa, R.; Santos, A.R.S.; Nunes, R.J.; Yunes, R.A. Analgesic activity of cyclic imides: 1,8-naphthalimide and 1,4,5,8-naphthalenediimide derivatives. II Farmaco 2000, 55, 319–321. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK(a) prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.J.; Foloppe, N. Drug-like bioactive structures and conformational coverage with the LigPrep/ConfGen suite: Comparison to programs MOE and catalyst. J. Chem. Inf. Model. 2010, 50, 822–839. [Google Scholar] [CrossRef] [PubMed]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.S.; Simeonov, A.; Jadhav, A.; Eidam, O.; Mott, B.T.; Keiser, M.J.; McKerrow, J.H.; Maloney, D.J.; Irwin, J.J.; Shoichet, B.K. Complementarity between a docking and a high-throughput screen in discovering new cruzain inhibitors. J. Med. Chem. 2010, 53, 4891–4905. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 13, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Dixon, S.L.; Lowrie, J.F.; Sherman, W. Analysis and comparison of 2D fingerprints: Insights into database screening performance using eight fingerprint methods. J. Mol. Graph. Model. 2010, 29, 157–170. [Google Scholar] [CrossRef]
- Bender, A.; Jenkins, J.L.; Scheiber, J.; Sukuru, S.C.; Glick, M.; Davies, J.W. How similar are similarity searching methods? A principal component analysis of molecular descriptor space. J. Chem. Inf. Model. 2009, 49, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Certara. UNITY. Available online: http://www.tripos.com/tripos_resources/fileroot/pdfs/Unity_111408.pdf (accessed on 30 January 2021).
- Willett, P. Similarity-based virtual screening using 2D fingerprints. Drug Discov. Today 2006, 11, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger. Canvas. Available online: https://www.schrodinger.com/canvas (accessed on 30 January 2021).
- Weaver, S.; Gleeson, M.P. The importance of the domain of applicability in QSAR modeling. J. Mol. Graph. Model. 2008, 26, 1315–1326. [Google Scholar] [CrossRef] [PubMed]
- Nunes, C.A.; Freitas, M.P.; Pinheiro, A.C.M.; Bastos, S.C. Chemoface: A novel free user-friendly interface for chemometrics. J. Braz. Chem. Soc. 2012, 23, 2003–2010. [Google Scholar] [CrossRef]
- Gramatica, P. Principles of QSAR models validation: Internal and external. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Saraiva, A.P.B.; Miranda, R.M.; Valente, R.P.P.; Araújo, J.O.; Souza, R.N.B.; Costa, C.H.S.; Oliveira, A.R.S.; Almeida, M.O.; Figueiredo, A.F.; Ferreira, J.E.V.; et al. Molecular description of α-keto-based inhibitors of cruzain with activity against Chagas disease combining 3D-QSAR studies and molecular dynamics. Chem. Biol. Drug Des. 2018, 92, 1475–1487. [Google Scholar] [CrossRef] [PubMed]
- Scotti, M.T.; Scotti, L.; Ishiki, H.M.; Peron, L.M.; de Rezende, L.; do Amaral, A.T. Variable-selection approaches to generate QSAR models for a set of antichagasic semicarbazones and analogues. Chemometr. Intell. Lab. Syst. 2016, 154, 137–149. [Google Scholar] [CrossRef]
- Rosas-Jimenez, J.G.; Garcia-Revilla, M.A.; Madariaga-Mazon, A.; Martinez-Mayorga, K. Predictive Global Models of Cruzain Inhibitors with Large Chemical Coverage. ACS Omega 2021, 6, 6722–6735. [Google Scholar] [CrossRef] [PubMed]
- Kleandrova, V.V.; Speck-Planche, A. The QSAR Paradigm in Fragment-Based Drug Discovery: From the Virtual Generation of Target Inhibitors to Multi-Scale Modeling. Mini Rev. Med. Chem. 2020, 20, 1357–1374. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Structure | pIC50 a |
|---|---|---|
| 1 |  | 6.00 |
| 2 * |  | 6.92 |
| 3 |  | 6.44 |
| 4 * |  | 6.39 |
| 5 |  | 6.30 |
| 6 |  | 6.30 |
| 7 |  | 6.28 |
| 8 |  | 6.24 |
| 9 * |  | 6.22 |
| 10 |  | 6.22 |
| 11 |  | 6.18 |
| 12 * |  | 6.15 |
| 13 |  | 6.12 |
| 14 |  | 6.68 |
| 15 |  | 5.77 |
| 16 * |  | 5.74 |
| 17 |  | 5.74 |
| 18 |  | 5.70 |
| 19 |  | 5.60 |
| 20 |  | 5.55 |
| 21 * |  | 5.52 |
| 22 |  | 5.51 |
| 23 |  | 5.49 |
| 24 |  | 5.49 |
| 25 * |  | 5.32 |
| 26 |  | 5.21 |
| 27 * |  | 5.21 |
| 28 |  | 4.11 |
| 29 |  | 4.00 |
| 30 * |  | 4.00 |
| 31 |  | 4.00 |
| 32 |  | 4.00 |
| 33 |  | 4.00 |
| 34 |  | 4.00 |
| 35 |  | 4.00 |
| 36 * |  | 4.00 |
| 37 |  | 4.00 |
| Model | Fragment Distinction | q2 | r2 | SEE | HL | N |
|---|---|---|---|---|---|---|
| 1 | A/B/C | 0.70 | 0.92 | 0.28 | 199 | 3 |
| 2 | A/B/C/Ch | 0.70 | 0.92 | 0.28 | 199 | 3 |
| 3 | A/B/C/H | 0.72 | 0.90 | 0.33 | 83 | 4 |
| 4 | A/B/C/H/Ch | 0.72 | 0.90 | 0.33 | 83 | 4 |
| Model | Fragment Distinction | Fragment Size | q2 | r2 | r2pred | SEE | HL | N |
|---|---|---|---|---|---|---|---|---|
| 5 | A/B/C | 2–7 | 0.70 | 0.92 | 0.78 | 0.27 | 199 | 3 |
| 6 | A/B/C | 3–7 | 0.71 | 0.92 | 0.80 | 0.28 | 199 | 3 |
| 8 | A/B/C/H | 2–6 | 0.76 | 0.90 | 0.67 | 0.33 | 83 | 4 |
| 7 | A/B/C/Ch | 3–7 | 0.70 | 0.92 | 0.78 | 0.28 | 199 | 3 |
| 9 | A/B/C/H/Ch | 2–6 | 0.76 | 0.90 | 0.67 | 0.33 | 83 | 4 |
| Inhibitor | Experimental | HQSAR | AutoQSAR | CoMFA | CoMSIA | ||||
|---|---|---|---|---|---|---|---|---|---|
| Predicted | Residual 1 | Predicted | Residual 1 | Predicted | Residual 1 | Predicted | Residual 1 | ||
| 1 | 6.00 | 5.34 | 0.66 | 5.49 | 0.51 | 5.78 | 0.22 | 5.85 | 0.15 |
| 2 * | 6.92 | 6.25 | 0.67 | 6.38 | 0.54 | 6.08 | 0.84 | 6.27 | 0.65 |
| 3 | 6.44 | 6.02 | 0.42 | 6.27 | 0.17 | 6.47 | −0.03 | 6.46 | −0.02 |
| 4 * | 6.39 | 5.96 | 0.43 | 5.98 | 0.41 | 6.01 | 0.38 | 6.05 | 0.34 |
| 5 | 6.30 | 6.14 | 0.16 | 6.24 | 0.06 | 6.32 | −0.02 | 6.01 | 0.29 |
| 6 | 6.30 | 6.49 | −0.19 | 6.30 | 0.00 | 6.42 | −0.12 | 6.08 | 0.22 |
| 7 | 6.28 | 5.93 | 0.35 | 5.85 | 0.43 | 6.28 | 0.00 | 6.21 | 0.07 |
| 8 | 6.24 | 5.90 | 0.34 | 6.23 | 0.01 | 6.25 | −0.01 | 6.02 | 0.22 |
| 9 * | 6.22 | 6.60 | −0.38 | 6.11 | 0.11 | 6.04 | 0.18 | 5.93 | 0.29 |
| 10 | 6.22 | 5.99 | 0.23 | 6.04 | 0.18 | 6.19 | 0.03 | 6.16 | 0.06 |
| 11 | 6.18 | 6.07 | 0.11 | 6.36 | −0.18 | 6.17 | 0.01 | 6.09 | 0.09 |
| 12 * | 6.15 | 5.65 | 0.50 | 5.99 | 0.16 | 6.04 | 0.11 | 6.16 | −0.01 |
| 13 | 6.12 | 5.93 | 0.19 | 5.95 | 0.17 | 6.12 | 0.00 | 6.13 | −0.01 |
| 14 | 6.68 | 6.90 | −0.22 | 6.06 | 0.62 | 6.67 | 0.01 | 6.53 | 0.15 |
| 15 | 5.77 | 5.83 | −0.06 | 6.19 | −0.42 | 5.68 | 0.09 | 5.75 | 0.02 |
| 16 * | 5.74 | 5.81 | −0.07 | 6.10 | −0.36 | 6.10 | −0.36 | 6.04 | −0.30 |
| 17 | 5.74 | 5.93 | −0.19 | 6.04 | −0.30 | 5.79 | −0.05 | 5.79 | −0.05 |
| 18 | 5.70 | 5.91 | −0.21 | 5.53 | 0.17 | 5.71 | −0.01 | 5.79 | −0.09 |
| 19 | 5.60 | 5.80 | −0.20 | 5.99 | −0.39 | 5.67 | −0.07 | 5.80 | −0.20 |
| 20 | 5.55 | 5.92 | −0.37 | 5.76 | −0.21 | 5.61 | −0.06 | 6.03 | −0.48 |
| 21 * | 5.52 | 5.82 | −0.30 | 5.85 | −0.33 | 5.55 | −0.03 | 5.87 | −0.35 |
| 22 | 5.51 | 5.93 | −0.42 | 5.78 | −0.27 | 5.56 | −0.05 | 5.85 | −0.34 |
| 23 | 5.49 | 5.63 | −0.14 | 5.14 | 0.35 | 5.54 | −0.05 | 5.66 | −0.17 |
| 24 | 5.49 | 5.44 | 0.05 | 5.52 | −0.03 | 5.39 | 0.10 | 5.62 | −0.13 |
| 25 * | 5.32 | 5.60 | −0.28 | 5.56 | −0.24 | 5.85 | −0.53 | 5.88 | −0.56 |
| 26 | 5.21 | 5.39 | −0.18 | 5.39 | −0.18 | 5.11 | 0.10 | 4.87 | 0.34 |
| 27 * | 5.21 | 4.49 | 0.72 | 4.99 | 0.22 | 4.64 | 0.57 | 4.26 | 0.95 |
| 28 | 4.11 | 4.54 | −0.43 | 4.80 | −0.69 | 4.23 | −0.12 | 4.31 | −0.20 |
| 29 | 4.00 | 4.21 | −0.21 | 4.47 | −0.47 | 3.98 | 0.02 | 3.99 | 0.01 |
| 30 * | 4.00 | 3.90 | 0.10 | 4.07 | −0.07 | 3.91 | 0.09 | 4.18 | −0.18 |
| 31 | 4.00 | 3.89 | 0.11 | 3.99 | 0.01 | 4.03 | −0.03 | 3.81 | 0.19 |
| 32 | 4.00 | 4.08 | −0.08 | 3.95 | 0.05 | 3.98 | 0.02 | 3.95 | 0.05 |
| 33 | 4.00 | 3.85 | 0.15 | 4.17 | −0.17 | 3.94 | 0.06 | 4.06 | −0.06 |
| 34 | 4.00 | 3.95 | 0.05 | 3.79 | 0.21 | 4.04 | −0.04 | 4.23 | −0.23 |
| 35 | 4.00 | 3.98 | 0.02 | 3.79 | 0.21 | 3.96 | 0.04 | 3.82 | 0.18 |
| 36 * | 4.00 | 3.95 | 0.05 | 3.96 | 0.04 | 3.80 | 0.20 | 4.55 | −0.55 |
| 37 | 4.00 | 4.01 | −0.01 | 3.87 | 0.13 | 4.06 | −0.06 | 4.08 | −0.08 |
| Training Set (%) | Score | r2 | SD | q2 | RMSE | N | Fingerprint |
|---|---|---|---|---|---|---|---|
| 70 | 0.85 | 0.85 | 0.40 | 0.82 | 0.37 | 2 | Desc |
| 72 | 0.90 | 0.89 | 0.32 | 0.90 | 0.29 | 2 | Dendritic |
| 74 | 0.82 | 0.82 | 0.44 | 0.78 | 0.39 | 2 | Desc |
| 76 | 0.81 | 0.81 | 0.43 | 0.78 | 0.40 | 2 | Desc |
| 78 | 0.81 | 0.81 | 0.43 | 0.78 | 0.40 | 2 | Desc |
| 80 | 0.82 | 0.82 | 0.43 | 0.91 | 0.26 | 2 | Desc |
| Model | q2 | r2 | r2pred | SEE | N | F | S | E | H | D | A |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CoMFA | 0.72 | 0.99 | 0.81 | 0.08 | 6 | 595.85 | 0.41 | 0.59 | - | - | - |
| CoMSIA | 0.63 | 0.96 | 0.73 | 0.21 | 3 | 179.60 | 0.11 | 0.34 | 0.17 | 0.14 | 0.24 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medeiros, A.R.; Ferreira, L.L.G.; de Souza, M.L.; de Oliveira Rezende Junior, C.; Espinoza-Chávez, R.M.; Dias, L.C.; Andricopulo, A.D. Chemoinformatics Studies on a Series of Imidazoles as Cruzain Inhibitors. Biomolecules 2021, 11, 579. https://doi.org/10.3390/biom11040579
Medeiros AR, Ferreira LLG, de Souza ML, de Oliveira Rezende Junior C, Espinoza-Chávez RM, Dias LC, Andricopulo AD. Chemoinformatics Studies on a Series of Imidazoles as Cruzain Inhibitors. Biomolecules. 2021; 11(4):579. https://doi.org/10.3390/biom11040579
Chicago/Turabian StyleMedeiros, Alex R., Leonardo L. G. Ferreira, Mariana L. de Souza, Celso de Oliveira Rezende Junior, Rocío Marisol Espinoza-Chávez, Luiz Carlos Dias, and Adriano D. Andricopulo. 2021. "Chemoinformatics Studies on a Series of Imidazoles as Cruzain Inhibitors" Biomolecules 11, no. 4: 579. https://doi.org/10.3390/biom11040579
APA StyleMedeiros, A. R., Ferreira, L. L. G., de Souza, M. L., de Oliveira Rezende Junior, C., Espinoza-Chávez, R. M., Dias, L. C., & Andricopulo, A. D. (2021). Chemoinformatics Studies on a Series of Imidazoles as Cruzain Inhibitors. Biomolecules, 11(4), 579. https://doi.org/10.3390/biom11040579