
The Emerging Role of Epigenetic Mechanisms in the Causation of Aberrant MMP Activity during Human Pathologies and the Use of Medicinal Drugs

Abstract
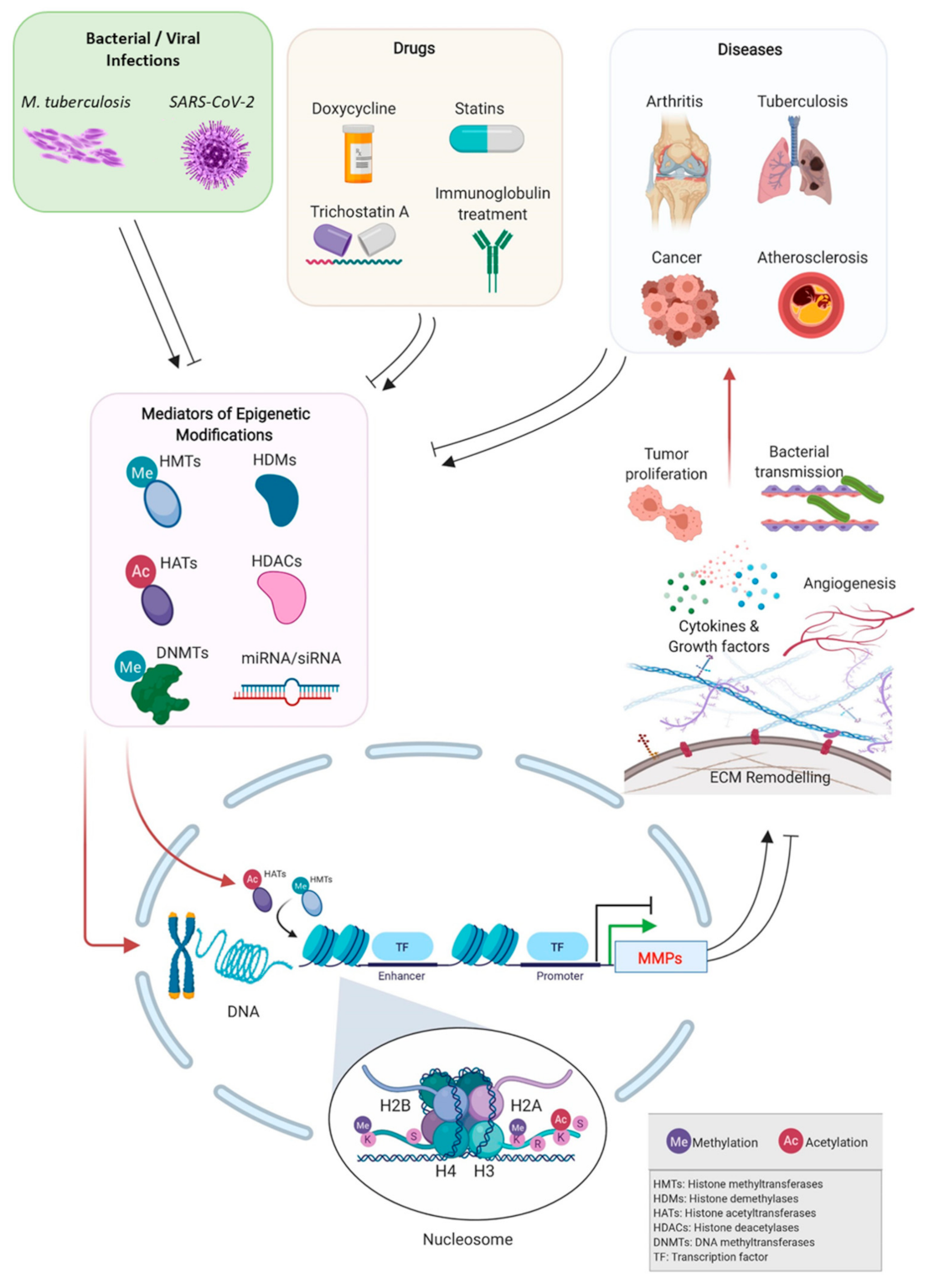
1. Introduction
2. Epigenetic Regulation of MMPs Expression in Physiological Conditions
3. Epigenetic Regulation of MMPs Expression in Pathological Conditions
3.1. Cancer
3.2. Cardiovascular Diseases
3.3. Bone/Cartilage Diseases
3.4. Bacterial Infections—Tuberculosis
3.5. Viral Infections
4. Pharmacologically Induced Epigenetic Control of MMPs
4.1. Epigenetic-Based Therapeutic Control of MMPs and Mediators of Epigenetic Changes
4.2. Statins
4.3. Tetracyclines with MMP Inhibitory Actions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef]
- Cook, R.N.; Sarker, H.; Fernandez-Patron, C. Pathologies of MMP-2 underactivity: A perspective on a neglected condition. Can. J. Physiol. Pharm. 2018, 97, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Felsenfeld, G. A brief history of epigenetics. Cold Spring Harb. Perspect. Biol. 2014, 6, a018200. [Google Scholar] [CrossRef]
- Bednar, J.; Horowitz, R.A.; Grigoryev, S.A.; Carruthers, L.M.; Hansen, J.C.; Koster, A.J.; Woodcock, C.L. Nucleosomes, linker DNA, and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. Proc. Natl. Acad. Sci. USA 1998, 95, 14173–14178. [Google Scholar] [CrossRef] [PubMed]
- Marino-Ramirez, L.; Kann, M.G.; Shoemaker, B.A.; Landsman, D. Histone structure and nucleosome stability. Expert. Rev. Proteom. 2005, 2, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zheng, C.; Thiriet, C.; Hayes, J.J. The core histone N-terminal tail domains negatively regulate binding of transcription factor IIIA to a nucleosome containing a 5S RNA gene via a novel mechanism. Mol. Cell Biol. 2005, 25, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Hayes, J.J.; Pruss, D.; Wolffe, A.P. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell 1993, 72, 73–84. [Google Scholar] [CrossRef]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef]
- Robertson, K.D. DNA methylation and chromatin—Unraveling the tangled web. Oncogene 2002, 21, 5361–5379. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Kooistra, S.M.; Helin, K. Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell Biol. 2012, 13, 297–311. [Google Scholar] [CrossRef]
- Trievel, R.C. Structure and function of histone methyltransferases. Crit. Rev. Eukaryot. Gene Expr. 2004, 14, 147–169. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.K.; Chow, M.Y.; Zhang, Y.; Leung, S.W. siRNA Versus miRNA as Therapeutics for Gene Silencing. Mol. Nucleic Acids 2015, 4, e252. [Google Scholar] [CrossRef]
- Vandooren, J.; Van den Steen, P.E.; Opdenakker, G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): The next decade. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 222–272. [Google Scholar] [CrossRef]
- Massaro, M.; Zampolli, A.; Scoditti, E.; Carluccio, M.A.; Storelli, C.; Distante, A.; De Caterina, R. Statins inhibit cyclooxygenase-2 and matrix metalloproteinase-9 in human endothelial cells: Anti-angiogenic actions possibly contributing to plaque stability. Cardiovasc. Res. 2010, 86, 311–320. [Google Scholar] [CrossRef]
- Luan, Z.; Chase, A.J.; Newby, A.C. Statins inhibit secretion of metalloproteinases-1, -2, -3, and -9 from vascular smooth muscle cells and macrophages. Arter. Thromb. Vasc. Biol. 2003, 23, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.C.; Mamotte, C.D.S. Pleiotropic and Adverse Effects of Statins-Do Epigenetics Play a Role? J. Pharm. Exp. 2017, 362, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Hanemaaijer, R.; Visser, H.; Koolwijk, P.; Sorsa, T.; Salo, T.; Golub, L.M.; van Hinsbergh, V.W. Inhibition of MMP synthesis by doxycycline and chemically modified tetracyclines (CMTs) in human endothelial cells. Adv. Dent. Res. 1998, 12, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Hardy, E.; Hardy-Sosa, A.; Fernandez-Patron, C. MMP-2: Is too low as bad as too high in the cardiovascular system? Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1332–H1340. [Google Scholar] [CrossRef]
- Yan, C.; Boyd, D.D. Regulation of matrix metalloproteinase gene expression. J. Cell Physiol. 2007, 211, 19–26. [Google Scholar] [CrossRef]
- Chernov, A.V.; Strongin, A.Y. Epigenetic regulation of matrix metalloproteinases and their collagen substrates in cancer. Biomol Concepts 2011, 2, 135–147. [Google Scholar] [CrossRef]
- Wang, H.; Ogawa, M.; Wood, J.R.; Bartolomei, M.S.; Sammel, M.D.; Kusanovic, J.P.; Walsh, S.W.; Romero, R.; Strauss, J.F., 3rd. Genetic and epigenetic mechanisms combine to control MMP1 expression and its association with preterm premature rupture of membranes. Hum. Mol. Genet. 2008, 17, 1087–1096. [Google Scholar] [CrossRef]
- Chicoine, E.; Esteve, P.O.; Robledo, O.; Van Themsche, C.; Potworowski, E.F.; St-Pierre, Y. Evidence for the role of promoter methylation in the regulation of MMP-9 gene expression. Biochem. Biophys. Res. Commun. 2002, 297, 765–772. [Google Scholar] [CrossRef]
- Couillard, J.; Demers, M.; Lavoie, G.; St-Pierre, Y. The role of DNA hypomethylation in the control of stromelysin gene expression. Biochem. Biophys. Res. Commun. 2006, 342, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L. Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev. 2002, 12, 142–148. [Google Scholar] [CrossRef]
- Sudarsanam, P.; Winston, F. The Swi/Snf family nucleosome-remodeling complexes and transcriptional control. Trends Genet. 2000, 16, 345–351. [Google Scholar] [CrossRef]
- Yan, C.; Wang, H.; Toh, Y.; Boyd, D.D. Repression of 92-kDa type IV collagenase expression by MTA1 is mediated through direct interactions with the promoter via a mechanism, which is both dependent on and independent of histone deacetylation. J. Biol. Chem. 2003, 278, 2309–2316. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Young, B.D.; Li, S.; Qi, X.; Richardson, J.A.; Olson, E.N. Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell 2006, 126, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Young, D.A.; Lakey, R.L.; Pennington, C.J.; Jones, D.; Kevorkian, L.; Edwards, D.R.; Cawston, T.E.; Clark, I.M. Histone deacetylase inhibitors modulate metalloproteinase gene expression in chondrocytes and block cartilage resorption. Arthritis. Res. 2005, 7, R503–R512. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar]
- Mitsiades, N.; Yu, W.H.; Poulaki, V.; Tsokos, M.; Stamenkovic, I. Matrix metalloproteinase-7-mediated cleavage of Fas ligand protects tumor cells from chemotherapeutic drug cytotoxicity. Cancer Res. 2001, 61, 577–581. [Google Scholar] [PubMed]
- Bergers, G.; Brekken, R.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2000, 2, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Boire, A.; Covic, L.; Agarwal, A.; Jacques, S.; Sherifi, S.; Kuliopulos, A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 2005, 120, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Chernov, A.V.; Sounni, N.E.; Remacle, A.G.; Strongin, A.Y. Epigenetic control of the invasion-promoting MT1-MMP/MMP-2/TIMP-2 axis in cancer cells. J. Biol. Chem. 2009, 284, 12727–12734. [Google Scholar] [CrossRef]
- Sato, N.; Maehara, N.; Su, G.H.; Goggins, M. Effects of 5-aza-2’-deoxycytidine on matrix metalloproteinase expression and pancreatic cancer cell invasiveness. J. Natl. Cancer Inst. 2003, 95, 327–330. [Google Scholar] [CrossRef]
- Klassen, L.M.B.; Chequin, A.; Manica, G.C.M.; Biembengut, I.V.; Toledo, M.B.; Baura, V.A.; de, O.P.F.; Ramos, E.A.S.; Costa, F.F.; de Souza, E.M.; et al. MMP9 gene expression regulation by intragenic epigenetic modifications in breast cancer. Gene 2018, 642, 461–466. [Google Scholar] [CrossRef]
- Brockmeyer, P.; Hemmerlein, B. Epigenetic modification suppresses proliferation, migration and invasion of urothelial cancer cell lines. Oncol. Lett. 2016, 12, 1693–1700. [Google Scholar] [CrossRef][Green Version]
- Wang, S.; Wu, W.; Claret, F.X. Mutual regulation of microRNAs and DNA methylation in human cancers. Epigenetics 2017, 12, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Raghuwanshi, S.K.; Smith, N.; Rivers, E.J.; Thomas, A.J.; Sutton, N.; Hu, Y.; Mukhopadhyay, S.; Chen, X.L.; Leung, T.; Richardson, R.M. G protein-coupled receptor kinase 6 deficiency promotes angiogenesis, tumor progression, and metastasis. J. Immunol. 2013, 190, 5329–5336. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Wu, D.; Chen, J.; Wang, P.; Lv, X.; Huang, J. Hypermethylation of the G protein-coupled receptor kinase 6 (GRK6) promoter inhibits binding of C/EBPalpha, and GRK6 knockdown promotes cell migration and invasion in lung adenocarcinoma cells. FEBS Open Bio 2019, 9, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Cock-Rada, A.M.; Medjkane, S.; Janski, N.; Yousfi, N.; Perichon, M.; Chaussepied, M.; Chluba, J.; Langsley, G.; Weitzman, J.B. SMYD3 promotes cancer invasion by epigenetic upregulation of the metalloproteinase MMP-9. Cancer Res. 2012, 72, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Asuthkar, S.; Velpula, K.K.; Chetty, C.; Gorantla, B.; Rao, J.S. Epigenetic regulation of miRNA-211 by MMP-9 governs glioma cell apoptosis, chemosensitivity and radiosensitivity. Oncotarget 2012, 3, 1439–1454. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Sun, M.; Sader, S. Matrix metalloproteinases in cardiovascular disease. Can. J. Cardiol. 2006, 22 (Suppl. B), 25B–30B. [Google Scholar] [CrossRef]
- Chen, K.C.; Wang, Y.S.; Hu, C.Y.; Chang, W.C.; Liao, Y.C.; Dai, C.Y.; Juo, S.H. OxLDL up-regulates microRNA-29b, leading to epigenetic modifications of MMP-2/MMP-9 genes: A novel mechanism for cardiovascular diseases. FASEB J. 2011, 25, 1718–1728. [Google Scholar] [CrossRef]
- Newby, A.C. Matrix metalloproteinases regulate migration, proliferation, and death of vascular smooth muscle cells by degrading matrix and non-matrix substrates. Cardiovasc. Res. 2006, 69, 614–624. [Google Scholar] [CrossRef]
- Kuo, H.C.; Li, S.C.; Huang, L.H.; Huang, Y.H. Epigenetic hypomethylation and upregulation of matrix metalloproteinase 9 in Kawasaki disease. Oncotarget 2017, 8, 60875–60891. [Google Scholar] [CrossRef][Green Version]
- Hardy, E.; Fernandez-Patron, C. Destroy to Rebuild: The Connection Between Bone Tissue Remodeling and Matrix Metalloproteinases. Front Physiol. 2020, 11, 47. [Google Scholar] [CrossRef]
- Fernandez-Tajes, J.; Soto-Hermida, A.; Vazquez-Mosquera, M.E.; Cortes-Pereira, E.; Mosquera, A.; Fernandez-Moreno, M.; Oreiro, N.; Fernandez-Lopez, C.; Fernandez, J.L.; Rego-Perez, I.; et al. Genome-wide DNA methylation analysis of articular chondrocytes reveals a cluster of osteoarthritic patients. Ann. Rheum. Dis. 2014, 73, 668–677. [Google Scholar] [CrossRef]
- Roach, H.I.; Yamada, N.; Cheung, K.S.; Tilley, S.; Clarke, N.M.; Oreffo, R.O.; Kokubun, S.; Bronner, F. Association between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regions. Arthritis. Rheum. 2005, 52, 3110–3124. [Google Scholar] [CrossRef]
- Song, J.; Jin, E.H.; Kim, D.; Kim, K.Y.; Chun, C.H.; Jin, E.J. MicroRNA-222 regulates MMP-13 via targeting HDAC-4 during osteoarthritis pathogenesis. BBA Clin. 2015, 3, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, N.; Rasheed, Z.; Ramamurthy, S.; Anbazhagan, A.N.; Voss, F.R.; Haqqi, T.M. MicroRNA-27b regulates the expression of matrix metalloproteinase 13 in human osteoarthritis chondrocytes. Arthritis Rheum. 2010, 62, 1361–1371. [Google Scholar] [CrossRef]
- Araki, Y.; Mimura, T. Matrix Metalloproteinase Gene Activation Resulting from Disordred Epigenetic Mechanisms in Rheumatoid Arthritis. Int. J. Mol. Sci. 2017, 18, 905. [Google Scholar] [CrossRef] [PubMed]
- Karouzakis, E.; Gay, R.E.; Michel, B.A.; Gay, S.; Neidhart, M. DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2009, 60, 3613–3622. [Google Scholar] [CrossRef]
- Araki, Y.; Tsuzuki Wada, T.; Aizaki, Y.; Sato, K.; Yokota, K.; Fujimoto, K.; Kim, Y.T.; Oda, H.; Kurokawa, R.; Mimura, T. Histone Methylation and STAT-3 Differentially Regulate Interleukin-6-Induced Matrix Metalloproteinase Gene Activation in Rheumatoid Arthritis Synovial Fibroblasts. Arthritis Rheumatol. 2016, 68, 1111–1123. [Google Scholar] [PubMed]
- Wang, X.; Tang, K.; Wang, Y.; Chen, Y.; Yang, M.; Gu, C.; Wang, J.; Yuan, Y. Elevated microRNA1455p increases matrix metalloproteinase9 by activating the nuclear factorkappaB pathway in rheumatoid arthritis. Mol. Med. Rep. 2019, 20, 2703–2711. [Google Scholar] [PubMed]
- Moores, R.C.; Brilha, S.; Schutgens, F.; Elkington, P.T.; Friedland, J.S. Epigenetic Regulation of Matrix Metalloproteinase-1 and -3 Expression in Mycobacterium tuberculosis Infection. Front. Immunol. 2017, 8, 602. [Google Scholar] [CrossRef]
- Elkington, P.; Shiomi, T.; Breen, R.; Nuttall, R.K.; Ugarte-Gil, C.A.; Walker, N.F.; Saraiva, L.; Pedersen, B.; Mauri, F.; Lipman, M.; et al. MMP-1 drives immunopathology in human tuberculosis and transgenic mice. J. Clin. Investig. 2011, 121, 1827–1833. [Google Scholar] [CrossRef]
- Al Shammari, B.; Shiomi, T.; Tezera, L.; Bielecka, M.K.; Workman, V.; Sathyamoorthy, T.; Mauri, F.; Jayasinghe, S.N.; Robertson, B.D.; D’Armiento, J.; et al. The Extracellular Matrix Regulates Granuloma Necrosis in Tuberculosis. J. Infect. Dis. 2015, 212, 463–473. [Google Scholar] [CrossRef]
- Green, J.A.; Elkington, P.T.; Pennington, C.J.; Roncaroli, F.; Dholakia, S.; Moores, R.C.; Bullen, A.; Porter, J.C.; Agranoff, D.; Edwards, D.R.; et al. Mycobacterium tuberculosis upregulates microglial matrix metalloproteinase-1 and -3 expression and secretion via NF-kappaB- and Activator Protein-1-dependent monocyte networks. J. Immunol. 2010, 184, 6492–6503. [Google Scholar] [CrossRef]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Michael, L.F.; Asahara, H.; Shulman, A.I.; Kraus, W.L.; Montminy, M. The phosphorylation status of a cyclic AMP-responsive activator is modulated via a chromatin-dependent mechanism. Mol. Cell Biol. 2000, 20, 1596–1603. [Google Scholar] [CrossRef] [PubMed]
- Moser, M.A.; Hagelkruys, A.; Seiser, C. Transcription and beyond: The role of mammalian class I lysine deacetylases. Chromosoma 2014, 123, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Barchowsky, A.; Frleta, D.; Vincenti, M.P. Integration of the NF-kappaB and mitogen-activated protein kinase/AP-1 pathways at the collagenase-1 promoter: Divergence of IL-1 and TNF-dependent signal transduction in rabbit primary synovial fibroblasts. Cytokine 2000, 12, 1469–1479. [Google Scholar] [CrossRef]
- Oh, J.; Takahashi, R.; Kondo, S.; Mizoguchi, A.; Adachi, E.; Sasahara, R.M.; Nishimura, S.; Imamura, Y.; Kitayama, H.; Alexander, D.B.; et al. The membrane-anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis. Cell 2001, 107, 789–800. [Google Scholar] [CrossRef]
- Liu, L.T.; Chang, H.C.; Chiang, L.C.; Hung, W.C. Histone deacetylase inhibitor up-regulates RECK to inhibit MMP-2 activation and cancer cell invasion. Cancer Res 2003, 63, 3069–3072. [Google Scholar]
- Garrington, T.P.; Johnson, G.L. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr. Opin. Cell Biol. 1999, 11, 211–218. [Google Scholar] [CrossRef]
- Vincenti, M.P.; Brinckerhoff, C.E. Transcriptional regulation of collagenase (MMP-1, MMP-13) genes in arthritis: Integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthritis Res. 2002, 4, 157–164. [Google Scholar] [CrossRef]
- Ashburner, B.P.; Westerheide, S.D.; Baldwin, A.S., Jr. The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol. Cell Biol. 2001, 21, 7065–7077. [Google Scholar] [CrossRef]
- Mittelstadt, M.L.; Patel, R.C. AP-1 mediated transcriptional repression of matrix metalloproteinase-9 by recruitment of histone deacetylase 1 in response to interferon beta. PLoS ONE 2012, 7, e42152. [Google Scholar] [CrossRef]
- Ram, M.; Sherer, Y.; Shoenfeld, Y. Matrix metalloproteinase-9 and autoimmune diseases. J Clin Immunol 2006, 26, 299–307. [Google Scholar] [CrossRef]
- Galewska, Z.; Romanowicz, L.; Jaworski, S.; Bankowski, E. Gelatinase matrix metalloproteinase (MMP)-2 and MMP-9 of the umbilical cord blood in preeclampsia. Clin. Chem. Lab. Med. 2008, 46, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Elkington, P.T.; O’Kane, C.M.; Friedland, J.S. The paradox of matrix metalloproteinases in infectious disease. Clin. Exp. Immunol. 2005, 142, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Pagenstecher, A.; Stalder, A.K.; Kincaid, C.L.; Volk, B.; Campbell, I.L. Regulation of matrix metalloproteinases and their inhibitor genes in lipopolysaccharide-induced endotoxemia in mice. Am. J. Pathol. 2000, 157, 197–210. [Google Scholar] [CrossRef]
- Aung, H.T.; Schroder, K.; Himes, S.R.; Brion, K.; van Zuylen, W.; Trieu, A.; Suzuki, H.; Hayashizaki, Y.; Hume, D.A.; Sweet, M.J.; et al. LPS regulates proinflammatory gene expression in macrophages by altering histone deacetylase expression. FASEB J. 2006, 20, 1315–1327. [Google Scholar] [CrossRef]
- Seo, Y.L.; Heo, S.; Jang, K.L. Hepatitis C virus core protein overcomes H2O2-induced apoptosis by downregulating p14 expression via DNA methylation. J. Gen. Virol. 2015, 96 Pt 4, 822–832. [Google Scholar] [CrossRef]
- Menachery, V.D.; Eisfeld, A.J.; Schafer, A.; Josset, L.; Sims, A.C.; Proll, S.; Fan, S.; Li, C.; Neumann, G.; Tilton, S.C.; et al. Pathogenic influenza viruses and coronaviruses utilize similar and contrasting approaches to control interferon-stimulated gene responses. mBio 2014, 5, e01174-14. [Google Scholar] [CrossRef]
- Menachery, V.D.; Schafer, A.; Burnum-Johnson, K.E.; Mitchell, H.D.; Eisfeld, A.J.; Walters, K.B.; Nicora, C.D.; Purvine, S.O.; Casey, C.P.; Monroe, M.E.; et al. MERS-CoV and H5N1 influenza virus antagonize antigen presentation by altering the epigenetic landscape. Proc. Natl. Acad. Sci. USA 2018, 115, E1012–E1021. [Google Scholar] [CrossRef] [PubMed]
- Atlante, S.; Mongelli, A.; Barbi, V.; Martelli, F.; Farsetti, A.; Gaetano, C. The epigenetic implication in coronavirus infection and therapy. Clin. Epigenetics 2020, 12, 156. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Sawalha, A.H.; Zhao, M.; Coit, P.; Lu, Q. Epigenetic dysregulation of ACE2 and interferon-regulated genes might suggest increased COVID-19 susceptibility and severity in lupus patients. medRxiv 2020, 108410. [Google Scholar] [CrossRef] [PubMed]
- Corley, M.J.; Pang, A.P.S.; Dody, K.; Mudd, P.A.; Patterson, B.K.; Seethamraju, H.; Bram, Y.; Peluso, M.J.; Torres, L.; Iyer, N.S.; et al. Genome-wide DNA methylation profiling of peripheral blood reveals an epigenetic signature associated with severe COVID-19. J. Leukoc. Biol. 2021, e5HI0720–466R. [Google Scholar] [CrossRef]
- Ueland, T.; Holter, J.C.; Holten, A.R.; Muller, K.E.; Lind, A.; Bekken, G.K.; Dudman, S.; Aukrust, P.; Dyrhol-Riise, A.M.; Heggelund, L. Distinct and early increase in circulating MMP-9 in COVID-19 patients with respiratory failure. J. Infect. 2020, 81, e41–e43. [Google Scholar] [CrossRef]
- Elkington, P.T.; Friedland, J.S. Matrix metalloproteinases in destructive pulmonary pathology. Thorax 2006, 61, 259–266. [Google Scholar] [CrossRef] [PubMed]
- O’Kane, C.M.; McKeown, S.W.; Perkins, G.D.; Bassford, C.R.; Gao, F.; Thickett, D.R.; McAuley, D.F. Salbutamol up-regulates matrix metalloproteinase-9 in the alveolar space in the acute respiratory distress syndrome. Crit. Care Med. 2009, 37, 2242–2249. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.J.; Coleman, A.M.; Gogan, K.M.; Phelan, J.J.; Cilian, O.M.; Dunne, P.J.; Basdeo, S.A.; Keane, J. Inhibiting Histone Deacetylases in Human Macrophages Promotes Glycolysis, IL-1beta, and T Helper Cell Responses to Mycobacterium tuberculosis. Front Immunol. 2020, 11, 1609. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Zhang, H.; McNair, N.N.; Mead, J.R.; Zhu, G. The Existing Drug Vorinostat as a New Lead Against Cryptosporidiosis by Targeting the Parasite Histone Deacetylases. J. Infect. Dis. 2018, 217, 1110–1117. [Google Scholar] [CrossRef]
- Wang, X.; Song, Y.; Jacobi, J.L.; Tuan, R.S. Inhibition of histone deacetylases antagonized FGF2 and IL-1beta effects on MMP expression in human articular chondrocytes. Growth Factors 2009, 27, 40–49. [Google Scholar] [CrossRef]
- Ateia, I.M.; Sutthiboonyapan, P.; Kamarajan, P.; Jin, T.; Godovikova, V.; Kapila, Y.L.; Fenno, J.C. Treponema denticola increases MMP-2 expression and activation in the periodontium via reversible DNA and histone modifications. Cell Microbiol. 2018, 20, e12815. [Google Scholar] [CrossRef]
- Adhyaru, B.B.; Jacobson, T.A. Safety and efficacy of statin therapy. Nat. Rev. Cardiol. 2018, 15, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, S.; Hayashi, H.; Kinoshita, K.; Abe, M.; Kuroki, H.; Tokunaga, R.; Tomiyasu, S.; Tanaka, H.; Sugita, H.; Arita, T.; et al. Statins inhibit tumor progression via an enhancer of zeste homolog 2-mediated epigenetic alteration in colorectal cancer. Int. J. Cancer 2014, 135, 2528–2536. [Google Scholar] [CrossRef] [PubMed]
- Karlic, H.; Thaler, R.; Gerner, C.; Grunt, T.; Proestling, K.; Haider, F.; Varga, F. Inhibition of the mevalonate pathway affects epigenetic regulation in cancer cells. Cancer Genet. 2015, 208, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Feig, J.E.; Shang, Y.; Rotllan, N.; Vengrenyuk, Y.; Wu, C.; Shamir, R.; Torra, I.P.; Fernandez-Hernando, C.; Fisher, E.A.; Garabedian, M.J. Statins promote the regression of atherosclerosis via activation of the CCR7-dependent emigration pathway in macrophages. PLoS ONE 2011, 6, e28534. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Lin, J.H.; Chou, C.W.; Chang, Y.F.; Yeh, S.H.; Chen, C.C. Statins increase p21 through inhibition of histone deacetylase activity and release of promoter-associated HDAC1/2. Cancer Res. 2008, 68, 2375–2383. [Google Scholar] [CrossRef]
- Marks, P.A. Histone deacetylase inhibitors: A chemical genetics approach to understanding cellular functions. Biochim Biophys. Acta 2010, 1799, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Xu, W.S. Histone deacetylase inhibitors: Potential in cancer therapy. J. Cell Biochem. 2009, 107, 600–608. [Google Scholar] [CrossRef]
- Li, H.; Rauch, T.; Chen, Z.X.; Szabo, P.E.; Riggs, A.D.; Pfeifer, G.P. The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J. Biol. Chem. 2006, 281, 19489–19500. [Google Scholar] [CrossRef]
- Kamio, K.; Liu, X.D.; Sugiura, H.; Togo, S.; Kawasaki, S.; Wang, X.; Ahn, Y.; Hogaboam, C.; Rennard, S.I. Statins inhibit matrix metalloproteinase release from human lung fibroblasts. Eur. Respir. J. 2010, 35, 637–646. [Google Scholar] [CrossRef]
- Ikeda, U.; Shimpo, M.; Ohki, R.; Inaba, H.; Takahashi, M.; Yamamoto, K.; Shimada, K. Fluvastatin inhibits matrix metalloproteinase-1 expression in human vascular endothelial cells. Hypertension 2000, 36, 325–329. [Google Scholar] [CrossRef]
- Bellosta, S.; Via, D.; Canavesi, M.; Pfister, P.; Fumagalli, R.; Paoletti, R.; Bernini, F. HMG-CoA reductase inhibitors reduce MMP-9 secretion by macrophages. Arter. Thromb. Vasc. Biol. 1998, 18, 1671–1678. [Google Scholar] [CrossRef]
- Kim, S.E.; Thanh Thuy, T.T.; Lee, J.H.; Ro, J.Y.; Bae, Y.A.; Kong, Y.; Ahn, J.Y.; Lee, D.S.; Oh, Y.M.; Lee, S.D.; et al. Simvastatin inhibits induction of matrix metalloproteinase-9 in rat alveolar macrophages exposed to cigarette smoke extract. Exp. Mol. Med. 2009, 41, 277–287. [Google Scholar] [CrossRef]
- Golub, L.M.; Ramamurthy, N.S.; McNamara, T.F.; Greenwald, R.A.; Rifkin, B.R. Tetracyclines inhibit connective tissue breakdown: New therapeutic implications for an old family of drugs. Crit. Rev. Oral Biol. Med. 1991, 2, 297–321. [Google Scholar] [CrossRef]
- Jonat, C.; Chung, F.Z.; Baragi, V.M. Transcriptional downregulation of stromelysin by tetracycline. J. Cell Biochem. 1996, 60, 341–347. [Google Scholar] [CrossRef]
- Liu, J.; Xiong, W.; Baca-Regen, L.; Nagase, H.; Baxter, B.T. Mechanism of inhibition of matrix metalloproteinase-2 expression by doxycycline in human aortic smooth muscle cells. J. Vasc. Surg. 2003, 38, 1376–1383. [Google Scholar] [CrossRef]
- Uitto, V.J.; Firth, J.D.; Nip, L.; Golub, L.M. Doxycycline and chemically modified tetracyclines inhibit gelatinase A (MMP-2) gene expression in human skin keratinocytes. Ann. N. Y. Acad. Sci. 1994, 732, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Zucker, S.; Cao, J.; Chen, W.T. Critical appraisal of the use of matrix metalloproteinase inhibitors in cancer treatment. Oncogene 2000, 19, 6642–6650. [Google Scholar] [CrossRef] [PubMed]
- Cerisano, G.; Buonamici, P.; Valenti, R.; Sciagra, R.; Raspanti, S.; Santini, A.; Carrabba, N.; Dovellini, E.V.; Romito, R.; Pupi, A.; et al. Early short-term doxycycline therapy in patients with acute myocardial infarction and left ventricular dysfunction to prevent the ominous progression to adverse remodelling: The TIPTOP trial. Eur. Heart J. 2014, 35, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Becker, E.; Bengs, S.; Aluri, S.; Opitz, L.; Atrott, K.; Stanzel, C.; Castro, P.A.R.; Rogler, G.; Frey-Wagner, I. Doxycycline, metronidazole and isotretinoin: Do they modify microRNA/mRNA expression profiles and function in murine T-cells? Sci. Rep. 2016, 6, 37082. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.S.; Maguire, E.M.; Bai, Y.S.; Huang, L.; Liu, Y.; Xu, L.; Fauzi, I.; Zhang, S.Q.; Xiao, Q.; Ma, N.F. A Novel Regulatory Axis, CHD1L-MicroRNA 486-Matrix Metalloproteinase 2, Controls Spermatogonial Stem Cell Properties. Mol. Cell Biol. 2019, 39, e00357-18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fu, J.; Zhang, Z.; Qin, H. miR-486-5p regulates the migration and invasion of colorectal cancer cells through targeting PIK3R1. Oncol. Lett. 2018, 15, 7243–7248. [Google Scholar] [CrossRef]
- Hu, H.; Xu, H.; Lu, F.; Zhang, J.; Xu, L.; Xu, S.; Jiang, H.; Zeng, Q.; Chen, E.; He, Z. Exosome-Derived miR-486-5p Regulates Cell Cycle, Proliferation and Metastasis in Lung Adenocarcinoma via Targeting NEK2. Front. Bioeng. Biotechnol. 2020, 8, 259. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Target Gene | Epigenetic Control Mechanism | Reference |
|---|---|---|
| MMP1 |
| [23,54] [29,30,57] [55] |
| MMP2 |
| [35,36] [22,45,109,110] [41] |
| MMP3 |
| [25,49,50] [55] [57] |
| MMP7 |
| [41] |
| MMP9 |
| [24,47,49,50] [28] [22,43,110,111] [37,42] [55] |
| MMP10 |
| [25] [29,30] |
| MMP13 |
| [49,50] [22,51,52] [55] [29,30,51] |
| MMP14 |
| [35,36,54] [22] |
| MMP16 |
| [22] |
| TIMP2 |
| [35] |
| TIMP3 |
| [22] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarker, H.; Haimour, A.; Toor, R.; Fernandez-Patron, C. The Emerging Role of Epigenetic Mechanisms in the Causation of Aberrant MMP Activity during Human Pathologies and the Use of Medicinal Drugs. Biomolecules 2021, 11, 578. https://doi.org/10.3390/biom11040578
Sarker H, Haimour A, Toor R, Fernandez-Patron C. The Emerging Role of Epigenetic Mechanisms in the Causation of Aberrant MMP Activity during Human Pathologies and the Use of Medicinal Drugs. Biomolecules. 2021; 11(4):578. https://doi.org/10.3390/biom11040578
Chicago/Turabian StyleSarker, Hassan, Ayman Haimour, Ravneet Toor, and Carlos Fernandez-Patron. 2021. "The Emerging Role of Epigenetic Mechanisms in the Causation of Aberrant MMP Activity during Human Pathologies and the Use of Medicinal Drugs" Biomolecules 11, no. 4: 578. https://doi.org/10.3390/biom11040578
APA StyleSarker, H., Haimour, A., Toor, R., & Fernandez-Patron, C. (2021). The Emerging Role of Epigenetic Mechanisms in the Causation of Aberrant MMP Activity during Human Pathologies and the Use of Medicinal Drugs. Biomolecules, 11(4), 578. https://doi.org/10.3390/biom11040578