Mechanisms of Primary Membranous Nephropathy

Abstract
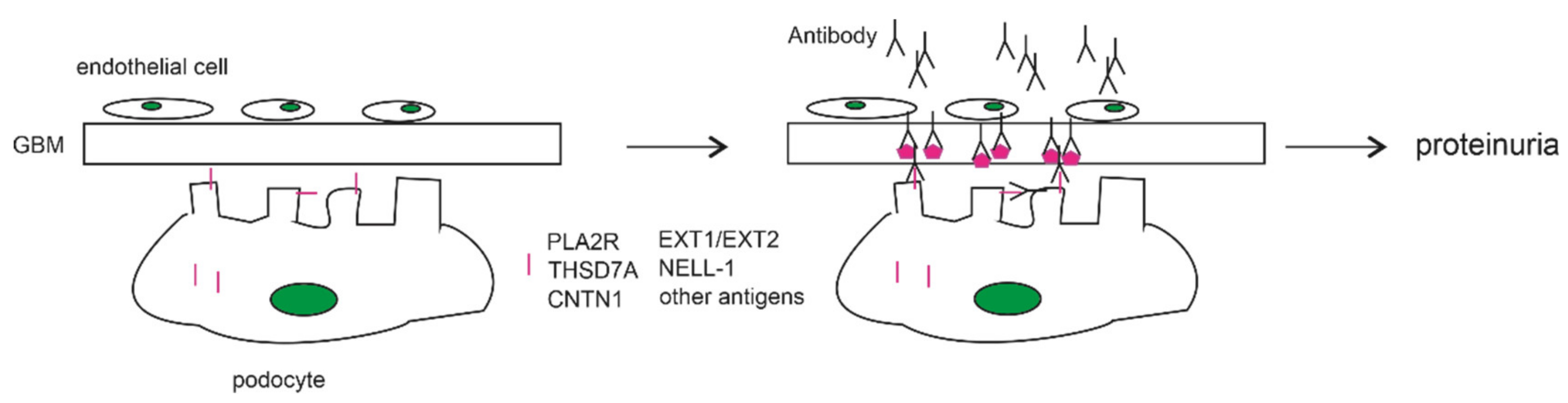
1. Introduction
2. Contributions of Autoimmunity to PLA2R in MN Pathogenesis
2.1. The Association of Anti-PLA2R Antibodies with Primary MN
2.2. The Anti-PLA2R Antibody as a Cause of PMN
3. THSD7A as a Podocyte Antigen of Membranous Nephropathy
4. Mechanisms Underlying PLA2R- and THSD7A-Contributed MN Pathogenesis
4.1. Genetic Factors
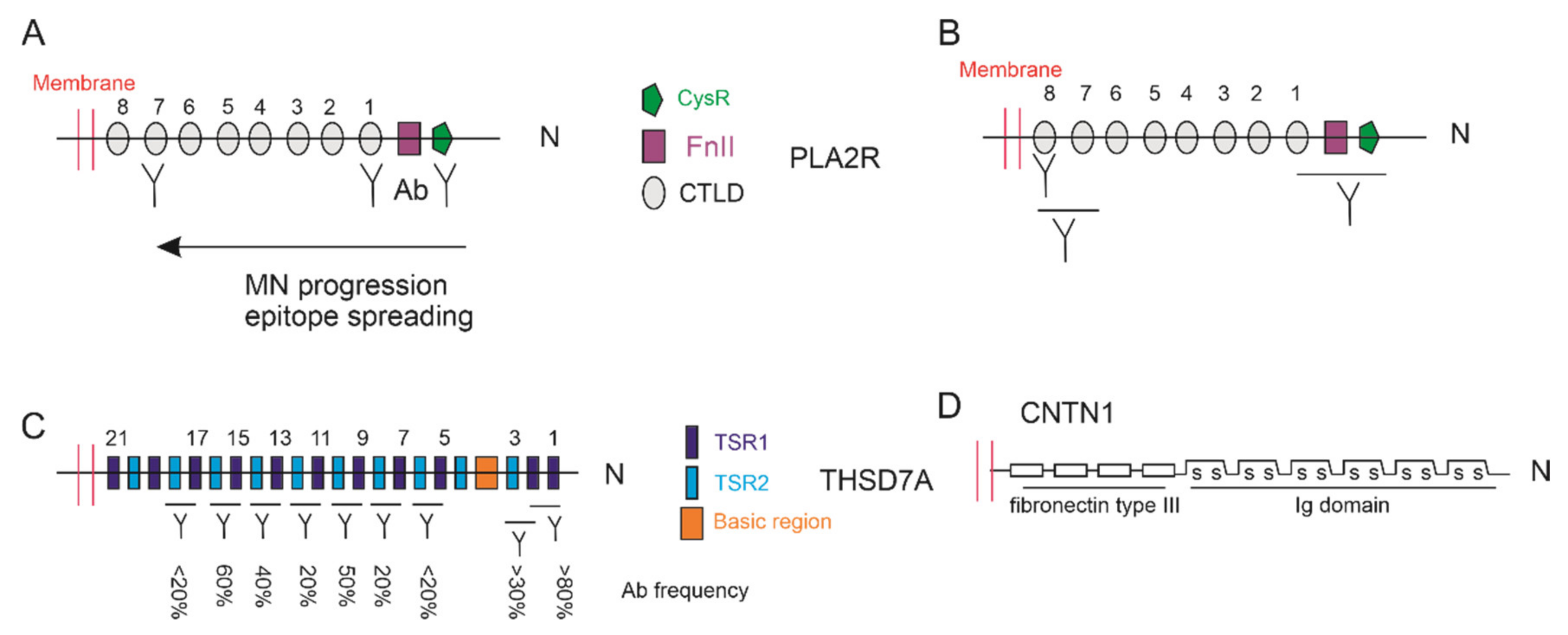
4.2. Epitopes of PLA2R and THSD7A
4.3. Complement Activation in PLA2R- and THSD7A-Contributed MN
4.4. Physiological Impact of PLA2R and THSD7A on MN Pathogenesis
5. Other MN-Associated Antibodies Targeting Podocyte Antigens
5.1. Exostosin 1 and Exostosin 2
5.2. Neural Cell Adhesion Molecule 1 (NCAM-1)
5.3. Neural Epidermal Growth Factor-like 1 (NELL-1)
5.4. Contactin 1 (CNTN1)
5.5. Semaphorin 3B, High Temperature Recombinant Protein A1 (HTRA1), and Protocadherin 7 (PCDH7)
6. Other Aspects
7. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- D’Arienzo, A.; Andreani, L.; Sacchetti, F.; Colangeli, S.; Capanna, R. Hereditary multiple exostoses: Current insights. Orthop. Res. Rev. 2019, 11, 199–211. [Google Scholar] [CrossRef]
- Ponticelli, C.; Glassock, R.J. Glomerular diseases: Membranous nephropathy—A modern view. Clin. J. Am. Soc. Nephrol. 2014, 9, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Sinico, R.A.; Mezzina, N.; Trezzi, B.; Ghiggeri, G.M.; Radice, A. Immunology of membranous nephropathy: From animal models to humans. Clin. Exp. Immunol. 2016, 183, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Simon, P.; Ramee, M.P.; Autuly, V.; Laruelle, E.; Charasse, C.; Cam, G.; Ang, K.S. Epidemiology of primary glomerular diseases in a french region. Variations according to period and age. Kidney Int. 1994, 46, 1192–1198. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, P.; Agodoa, L.; Gellert, R.; Stewart, J.H.; Buccianti, G.; Lowenfels, A.B.; Wolfe, R.A.; Jones, E.; Disney, A.P.; Briggs, D.; et al. Distribution of primary renal diseases leading to end-stage renal failure in the United States, Europe, and Australia/New Zealand: Results from an international comparative study. Am. J. Kidney Dis. 2000, 35, 157–165. [Google Scholar] [CrossRef]
- Gupta, S.; Pepper, R.J.; Ashman, N.; Walsh, S.B. Nephrotic syndrome: Oedema formation and its treatment with diuretics. Front. Physiol. 2018, 9, 1868. [Google Scholar] [CrossRef]
- Vaziri, N.D. Disorders of lipid metabolism in nephrotic syndrome: Mechanisms and consequences. Kidney Int. 2016, 90, 41–52. [Google Scholar] [CrossRef]
- Van de Logt, A.E.; Fresquet, M.; Wetzels, J.F.; Brenchley, P. The anti-pla2r antibody in membranous nephropathy: What we know and what remains a decade after its discovery. Kidney Int. 2019, 96, 1292–1302. [Google Scholar] [CrossRef]
- Heymann, W.; Hackel, D.B.; Harwood, S.; Wilson, S.G.; Hunter, J.L. Production of nephrotic syndrome in rats by Freund’s adjuvants and rat kidney suspensions. Proc. Soc. Exp. Biol. Med. 1959, 100, 660–664. [Google Scholar] [CrossRef]
- Kerjaschki, D.; Farquhar, M.G. The pathogenic antigen of heymann nephritis is a membrane glycoprotein of the renal proximal tubule brush border. Proc. Natl. Acad. Sci. USA 1982, 79, 5557–5561. [Google Scholar] [CrossRef]
- Beck, L.H., Jr.; Bonegio, R.G.; Lambeau, G.; Beck, D.M.; Powell, D.W.; Cummins, T.D.; Klein, J.B.; Salant, D.J. M-type phospholipase a2 receptor as target antigen in idiopathic membranous nephropathy. N. Engl. J. Med. 2009, 361, 11–21. [Google Scholar] [CrossRef]
- Tomas, N.M.; Beck, L.H., Jr.; Meyer-Schwesinger, C.; Seitz-Polski, B.; Ma, H.; Zahner, G.; Dolla, G.; Hoxha, E.; Helmchen, U.; Dabert-Gay, A.S.; et al. Thrombospondin type-1 domain-containing 7a in idiopathic membranous nephropathy. N. Engl. J. Med. 2014, 371, 2277–2287. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Li, J.; He, F.; Lv, Y.; Liu, W.; Wu, P.; Huang, J.; Wei, S.; Gao, H. The diagnosis accuracy of pla2r-ab in the diagnosis of idiopathic membranous nephropathy: A meta-analysis. PLoS ONE 2014, 9, e104936. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, S.; Akiyama, M.; Imai, E.; Ozaki, T.; Matsuo, S.; Maruyama, S. Prevalence of anti-phospholipase a2 receptor antibodies in Japanese patients with membranous nephropathy. Clin. Exp. Nephrol. 2015, 19, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Hofstra, J.M.; Wetzels, J.F. Anti-pla(2)r antibodies in membranous nephropathy: Ready for routine clinical practice? Neth. J. Med. 2012, 70, 109–113. [Google Scholar] [PubMed]
- Iwakura, T.; Ohashi, N.; Kato, A.; Baba, S.; Yasuda, H. Prevalence of enhanced granular expression of thrombospondin type-1 domain-containing 7a in the glomeruli of Japanese patients with idiopathic membranous nephropathy. PLoS ONE 2015, 10, e0138841. [Google Scholar] [CrossRef]
- Akiyama, S.; Imai, E.; Maruyama, S. Immunology of membranous nephropathy. F1000Research 2019, 8, f1000. [Google Scholar] [CrossRef]
- Meyer-Schwesinger, C.; Tomas, N.M.; Dehde, S.; Seifert, L.; Hermans-Borgmeyer, I.; Wiech, T.; Koch-Nolte, F.; Huber, T.B.; Zahner, G. A novel mouse model of phospholipase a2 receptor 1-associated membranous nephropathy mimics podocyte injury in patients. Kidney Int. 2020, 97, 913–919. [Google Scholar] [CrossRef]
- Tomas, N.M.; Hoxha, E.; Reinicke, A.T.; Fester, L.; Helmchen, U.; Gerth, J.; Bachmann, F.; Budde, K.; Koch-Nolte, F.; Zahner, G.; et al. Autoantibodies against thrombospondin type 1 domain-containing 7a induce membranous nephropathy. J. Clin. Investig. 2016, 126, 2519–2532. [Google Scholar] [CrossRef]
- Rojas-Rivera, J.E.; Carriazo, S.; Ortiz, A. Treatment of idiopathic membranous nephropathy in adults: Kdigo 2012, cyclophosphamide and cyclosporine a are out, rituximab is the new normal. Clin. Kidney J. 2019, 12, 629–638. [Google Scholar] [CrossRef]
- Fervenza, F.C.; Appel, G.B.; Barbour, S.J.; Rovin, B.H.; Lafayette, R.A.; Aslam, N.; Jefferson, J.A.; Gipson, P.E.; Rizk, D.V.; Sedor, J.R.; et al. Rituximab or cyclosporine in the treatment of membranous nephropathy. N. Engl. J. Med. 2019, 381, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Stahl, R.A.; Reinhard, L.; Hoxha, E. Characterization of autoantibodies in primary membranous nephropathy and their clinical significance. Expert Rev. Clin. Immunol. 2019, 15, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Ronco, P.; Debiec, H. Molecular pathogenesis of membranous nephropathy. Annu. Rev. Pathol. 2020, 15, 287–313. [Google Scholar] [CrossRef]
- Fogo, A.B.; Lusco, M.A.; Najafian, B.; Alpers, C.E. Ajkd atlas of renal pathology: Membranous nephropathy. Am. J. Kidney Dis. 2015, 66, e15–e17. [Google Scholar] [CrossRef]
- VanBeek, C.; Haas, M. Anti-pla2r-associated membranous nephropathy: A review with emphasis on diagnostic testing methods. Clin. Nephrol. 2015, 84, 1–9. [Google Scholar] [CrossRef]
- Li, X.; Wei, D.; Zhou, Z.; Wang, B.; Xu, Y.; Pan, J.; Yang, C.; Lu, J.; Qiu, Y. Anti-pla2r antibodies in Chinese patients with membranous nephropathy. Med. Sci. Monit. 2016, 22, 1630–1636. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Luo, W.; Gong, S.; Ding, X. Detection and clinical significance of glomerular m-type phospholipase a2 receptor in patients with idiopathic membranous nephropathy. Intern. Med. J. 2016, 46, 1318–1322. [Google Scholar] [CrossRef] [PubMed]
- Radice, A.; Pieruzzi, F.; Trezzi, B.; Ghiggeri, G.; Napodano, P.; d’Amico, M.; Stellato, T.; Brugnano, R.; Ravera, F.; Rolla, D.; et al. Diagnostic specificity of autoantibodies to m-type phospholipase a2 receptor (pla2r) in differentiating idiopathic membranous nephropathy (imn) from secondary forms and other glomerular diseases. J. Nephrol. 2018, 31, 271–278. [Google Scholar] [CrossRef]
- Guo, N.; Cao, Y.; Dai, H.; Yuan, L.; Shi, L.; Zhang, Y. Anti-phospholipase a2 receptor (anti-pla2r) antibody in diagnosis and treatment of idiopathic membranous nephropathy: A single-center observational study in China. Med. Sci. Monit. 2019, 25, 9364–9368. [Google Scholar] [CrossRef]
- Pang, L.; Zhang, A.M.; Li, H.X.; Du, J.L.; Jiao, L.L.; Duan, N.; Liu, Y.; Yu, D. Serum anti-pla2r antibody and glomerular pla2r deposition in Chinese patients with membranous nephropathy: A cross-sectional study. Medicine 2017, 96, e7218. [Google Scholar] [CrossRef]
- Qin, H.Z.; Zhang, M.C.; Le, W.-B.; Ren, Q.; Chen, D.C.; Zeng, C.H.; Liu, L.; Zuo, K.; Xu, F.; Liu, Z.H. Combined assessment of phospholipase a2 receptor autoantibodies and glomerular deposits in membranous nephropathy. J. Am. Soc. Nephrol. 2016, 27, 3195–3203. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Zhang, M.F.; Cui, Z.; Wang, J.; Wang, M.; Zhang, Y.M.; Wang, F.; Wang, X.; Meng, L.Q.; Cheng, X.Y.; et al. Antibodies against m-type phospholipase a2 receptor may predict treatment response and outcome in membranous nephropathy. Am. J. Nephrol. 2018, 48, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Han, W.W.; Tang, L.J.; Kong, X.L.; Yang, H.; Xu, D.M. Clinical significance of autoantibodies in the assessment and treatment of idiopathic membranous nephropathy. Exp. Ther. Med. 2019, 17, 1825–1830. [Google Scholar] [CrossRef]
- Ghiggeri, G.M.; Seitz-Polski, B.; Justino, J.; Zaghrini, C.; Payre, C.; Brglez, V.; Dolla, G.; Sinico, A.; Scolari, F.; Vaglio, A.; et al. Multi-autoantibody signature and clinical outcome in membranous nephropathy. Clin. J. Am. Soc. Nephrol. 2020, 15, 1762–1776. [Google Scholar] [CrossRef]
- Dong, D.; Fan, T.T.; Wang, Y.Y.; Zhang, L.; Song, L.; Zhang, L. Relationship between renal tissues phospholipase a2 receptor and its serum antibody and clinical condition and prognosis of idiopathic membranous nephropathy: A meta-analysis. BMC Nephrol. 2019, 20, 444. [Google Scholar] [CrossRef]
- Rao, S.J.; Shen, Q.; Wang, H.M.; Tang, S.; Wang, X.Y. The association of anti-pla2r with clinical manifestations and outcomes in idiopathic membranous nephropathy: A meta-analysis. Int. Urol. Nephrol. 2020, 52, 2123–2133. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhao, Y. Prognostic value of phospholipase a2 receptor in primary membranous nephropathy: A systematic review and meta-analysis. Int. Urol. Nephrol. 2019, 51, 1581–1596. [Google Scholar] [CrossRef]
- Liang, Y.; Wan, J.; Chen, Y.; Pan, Y. Serum anti-phospholipase a2 receptor (pla2r) antibody detected at diagnosis as a predictor for clinical remission in patients with primary membranous nephropathy: A meta-analysis. BMC Nephrol. 2019, 20, 360. [Google Scholar] [CrossRef]
- Zhang, Q.; Huang, B.; Liu, X.; Liu, B.; Zhang, Y.; Zhang, Z.; Hua, J.; Fan, Y.; Hu, L.; Meng, M.; et al. Ultrasensitive quantitation of anti-phospholipase a2 receptor antibody as a diagnostic and prognostic indicator of idiopathic membranous nephropathy. Sci. Rep. 2017, 7, 12049. [Google Scholar] [CrossRef]
- Liu, Y.; Li, X.; Ma, C.; Wang, P.; Liu, J.; Su, H.; Zhuo, H.; Kong, X.; Xu, D.; Xu, D. Serum anti-pla2r antibody as a diagnostic biomarker of idiopathic membranous nephropathy: The optimal cut-off value for Chinese patients. Clin. Chim. Acta 2018, 476, 9–14. [Google Scholar] [CrossRef]
- Tampoia, M.; Migliucci, F.; Villani, C.; Abbracciavento, L.; Rossini, M.; Fumarulo, R.; Gesualdo, L.; Montinaro, V. Definition of a new cut-off for the anti-phospholipase a2 receptor (pla2r) autoantibody immunoassay in patients affected by idiopathic membranous nephropathy. J. Nephrol. 2018, 31, 899–905. [Google Scholar] [CrossRef]
- Dahnrich, C.; Saschenbrecker, S.; Gunnarsson, I.; Schlumberger, W.; Ronco, P.; Debiec, H. Development of a standardized chemiluminescence immunoassay for the detection of autoantibodies against human m-type phospholipase a2 receptor in primary membranous nephropathy. Kidney Int. Rep. 2020, 5, 182–188. [Google Scholar] [CrossRef]
- Li, C.; Li, P.; Guo, W.; Chen, L.; Li, J.; Wang, R.; Chen, B. The optimal anti-pla2r cutoff for the diagnosis of idiopathic membranous nephropathy: A single-center retrospective study. Korean J. Intern. Med. 2020. [Google Scholar] [CrossRef]
- Grupper, A.; Cornell, L.D.; Fervenza, F.C.; Beck, L.H., Jr.; Lorenz, E.; Cosio, F.G. Recurrent membranous nephropathy after kidney transplantation: Treatment and long-term implications. Transplantation 2016, 100, 2710–2716. [Google Scholar] [CrossRef]
- Gupta, G.; Fattah, H.; Ayalon, R.; Kidd, J.; Gehr, T.; Quintana, L.F.; Kimball, P.; Sadruddin, S.; Massey, H.D.; Kumar, D.; et al. Pre-transplant phospholipase a2 receptor autoantibody concentration is associated with clinically significant recurrence of membranous nephropathy post-kidney transplantation. Clin. Transplant. 2016, 30, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Hoxha, E.; Harendza, S.; Pinnschmidt, H.; Panzer, U.; Stahl, R.A. Pla2r antibody levels and clinical outcome in patients with membranous nephropathy and non-nephrotic range proteinuria under treatment with inhibitors of the renin-angiotensin system. PLoS ONE 2014, 9, e110681. [Google Scholar] [CrossRef]
- Burbelo, P.D.; Joshi, M.; Chaturvedi, A.; Little, D.J.; Thurlow, J.S.; Waldman, M.; Olson, S.W. Detection of pla2r autoantibodies before the diagnosis of membranous nephropathy. J. Am. Soc. Nephrol. 2020, 31, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Borza, D.B.; Zhang, J.J.; Beck, L.H., Jr.; Meyer-Schwesinger, C.; Luo, W. Mouse models of membranous nephropathy: The road less travelled by. Am. J. Clin. Exp. Immunol. 2013, 2, 135–145. [Google Scholar]
- Hamilton, P.; Kanigicherla, D.; Hanumapura, P.; Walz, L.; Kramer, D.; Fischer, M.; Brenchley, P.; Mitra, S. Peptide gam immunoadsorption therapy in primary membranous nephropathy (prism): Phase ii trial investigating the safety and feasibility of peptide gam immunoadsorption in anti-pla2 r positive primary membranous nephropathy. J. Clin. Apher. 2018, 33, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.G.; Larsen, C.P. Tissue staining for thsd7a in glomeruli correlates with serum antibodies in primary membranous nephropathy: A clinicopathological study. Mod. Pathol. 2018, 31, 616–622. [Google Scholar] [CrossRef]
- Hoxha, E.; Beck, L.H., Jr.; Wiech, T.; Tomas, N.M.; Probst, C.; Mindorf, S.; Meyer-Schwesinger, C.; Zahner, G.; Stahl, P.R.; Schopper, R.; et al. An indirect immunofluorescence method facilitates detection of thrombospondin type 1 domain-containing 7a-specific antibodies in membranous nephropathy. J. Am. Soc. Nephrol. 2017, 28, 520–531. [Google Scholar] [CrossRef]
- Zaghrini, C.; Seitz-Polski, B.; Justino, J.; Dolla, G.; Payre, C.; Jourde-Chiche, N.; van de Logt, A.E.; Booth, C.; Rigby, E.; Lonnbro-Widgren, J.; et al. Novel elisa for thrombospondin type 1 domain-containing 7a autoantibodies in membranous nephropathy. Kidney Int. 2019, 95, 666–679. [Google Scholar] [CrossRef]
- Zhang, D.; Zou, J.; Zhang, C.; Zhang, W.; Lin, F.; Jiang, G. Clinical and histological features of phospholipase a2 receptor-associated and thrombospondin type-i domain-containing 7a-associated idiopathic membranous nephropathy: A single center retrospective study from China. Med. Sci. Monit. 2018, 24, 5076–5083. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cui, Z.; Lu, J.; Probst, C.; Zhang, Y.M.; Wang, X.; Qu, Z.; Wang, F.; Meng, L.Q.; Cheng, X.Y.; et al. Circulating antibodies against thrombospondin type-i domain-containing 7a in Chinese patients with idiopathic membranous nephropathy. Clin. J. Am. Soc. Nephrol. 2017, 12, 1642–1651. [Google Scholar] [CrossRef]
- Larsen, C.P.; Cossey, L.N.; Beck, L.H. Thsd7a staining of membranous glomerulopathy in clinical practice reveals cases with dual autoantibody positivity. Mod. Pathol. 2016, 29, 421–426. [Google Scholar] [CrossRef]
- Ren, S.; Wu, C.; Zhang, Y.; Wang, A.Y.; Li, G.; Wang, L.; Hong, D. An update on clinical significance of use of thsd7a in diagnosing idiopathic membranous nephropathy: A systematic review and meta-analysis of thsd7a in imn. Ren. Fail. 2018, 40, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Beck, L.H., Jr. Pla2r and thsd7a: Disparate paths to the same disease? J. Am. Soc. Nephrol. 2017, 28, 2579–2589. [Google Scholar] [CrossRef]
- Liu, Y.; Zheng, S.; Ma, C.; Lian, Y.; Zheng, X.; Guan, P.; Wang, B.; Gong, X.; Gao, F.; Liang, L.; et al. Meta-analysis of the diagnostic efficiency of thsd7a-ab for the diagnosis of idiopathic membranous nephropathy. Glob. Chall. 2020, 4, 1900099. [Google Scholar] [CrossRef]
- Weinmann-Menke, J.; Holtz, S.; Sollinger, D.; Dorken, M.; Boedecker, S.; Schamberger, B.; Pfister, F.; Amann, K.; Lutz, J. Treatment of membranous nephropathy in patients with thsd7a antibodies using immunoadsorption. Am. J. Kidney Dis. 2019, 74, 849–852. [Google Scholar] [CrossRef] [PubMed]
- Tomas, N.M.; Meyer-Schwesinger, C.; von Spiegel, H.; Kotb, A.M.; Zahner, G.; Hoxha, E.; Helmchen, U.; Endlich, N.; Koch-Nolte, F.; Stahl, R.A.K. A heterologous model of thrombospondin type 1 domain-containing 7a-associated membranous nephropathy. J. Am. Soc. Nephrol. 2017, 28, 3262–3277. [Google Scholar] [CrossRef] [PubMed]
- Boerries, M.; Grahammer, F.; Eiselein, S.; Buck, M.; Meyer, C.; Goedel, M.; Bechtel, W.; Zschiedrich, S.; Pfeifer, D.; Laloe, D.; et al. Molecular fingerprinting of the podocyte reveals novel gene and protein regulatory networks. Kidney Int. 2013, 83, 1052–1064. [Google Scholar] [CrossRef]
- Zhang, C.; Leng, L.; Li, Z.; Zhao, Y.; Jiao, J. Identification of biomarkers and drug repurposing candidates based on an immune-, inflammation- and membranous glomerulonephritis-associated triplets network for membranous glomerulonephritis. BMC Med. Genom. 2020, 13, 5. [Google Scholar] [CrossRef] [PubMed]
- Stanescu, H.C.; Arcos-Burgos, M.; Medlar, A.; Bockenhauer, D.; Kottgen, A.; Dragomirescu, L.; Voinescu, C.; Patel, N.; Pearce, K.; Hubank, M.; et al. Risk hla-dqa1 and pla(2)r1 alleles in idiopathic membranous nephropathy. N. Engl. J. Med. 2011, 364, 616–626. [Google Scholar] [CrossRef]
- Lv, J.; Hou, W.; Zhou, X.; Liu, G.; Zhou, F.; Zhao, N.; Hou, P.; Zhao, M.; Zhang, H. Interaction between pla2r1 and hla-dqa1 variants associates with anti-pla2r antibodies and membranous nephropathy. J. Am. Soc. Nephrol. 2013, 24, 1323–1329. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Kumar, V.; Kumar, A.; Yadav, A.K.; Nada, R.; Kumar, H.; Kumar, V.; Rathi, M.; Kohli, H.S.; Gupta, K.L.; et al. Pla2r antibodies, glomerular pla2r deposits and variations in pla2r1 and hla-dqa1 genes in primary membranous nephropathy in south asians. Nephrol. Dial. Transplant. 2016, 31, 1486–1493. [Google Scholar] [CrossRef] [PubMed]
- Dhaouadi, T.; Abdellatif, J.; Trabelsi, R.; Gaied, H.; Chamkhi, S.; Sfar, I.; Goucha, R.; Ben Hamida, F.; Ben Abdallah, T.; Gorgi, Y. Pla2r antibody, pla2r rs4664308 polymorphism and pla2r mrna levels in tunisian patients with primary membranous nephritis. PLoS ONE 2020, 15, e0240025. [Google Scholar] [CrossRef]
- Sekula, P.; Li, Y.; Stanescu, H.C.; Wuttke, M.; Ekici, A.B.; Bockenhauer, D.; Walz, G.; Powis, S.H.; Kielstein, J.T.; Brenchley, P.; et al. Genetic risk variants for membranous nephropathy: Extension of and association with other chronic kidney disease aetiologies. Nephrol. Dial. Transplant. 2017, 32, 325–332. [Google Scholar] [CrossRef]
- Cui, Z.; Xie, L.J.; Chen, F.J.; Pei, Z.Y.; Zhang, L.J.; Qu, Z.; Huang, J.; Gu, Q.H.; Zhang, Y.M.; Wang, X.; et al. Mhc class ii risk alleles and amino acid residues in idiopathic membranous nephropathy. J. Am. Soc. Nephrol. 2017, 28, 1651–1664. [Google Scholar] [CrossRef]
- Wang, H.Y.; Cui, Z.; Xie, L.J.; Zhang, L.J.; Pei, Z.Y.; Chen, F.J.; Qu, Z.; Huang, J.; Zhang, Y.M.; Wang, X.; et al. Hla class ii alleles differing by a single amino acid associate with clinical phenotype and outcome in patients with primary membranous nephropathy. Kidney Int. 2018, 94, 974–982. [Google Scholar] [CrossRef]
- Le, W.B.; Shi, J.S.; Zhang, T.; Liu, L.; Qin, H.Z.; Liang, S.; Zhang, Y.W.; Zheng, C.X.; Jiang, S.; Qin, W.S.; et al. Hla-drb1*15:01 and hla-drb3*02:02 in pla2r-related membranous nephropathy. J. Am. Soc. Nephrol. 2017, 28, 1642–1650. [Google Scholar] [CrossRef]
- Tao, T.; Wang, J.; Lei, S.; Hu, Z. Identical twins with idiopathic membranous nephropathy. J. Nephrol. 2020. [Google Scholar] [CrossRef]
- Cantarelli, C.; Jarque, M.; Angeletti, A.; Manrique, J.; Hartzell, S.; O’Donnell, T.; Merritt, E.; Laserson, U.; Perin, L.; Donadei, C.; et al. A comprehensive phenotypic and functional immune analysis unravels circulating anti-phospholipase a2 receptor antibody secreting cells in membranous nephropathy patients. Kidney Int. Rep. 2020, 5, 1764–1776. [Google Scholar] [CrossRef]
- Tozzoli, R. Receptor autoimmunity: Diagnostic and therapeutic implications. Auto Immun. Highlights 2020, 11, 1. [Google Scholar] [CrossRef]
- Fresquet, M.; Jowitt, T.A.; Gummadova, J.; Collins, R.; O’Cualain, R.; McKenzie, E.A.; Lennon, R.; Brenchley, P.E. Identification of a major epitope recognized by pla2r autoantibodies in primary membranous nephropathy. J. Am. Soc. Nephrol. 2015, 26, 302–313. [Google Scholar] [CrossRef]
- Seitz-Polski, B.; Dolla, G.; Payre, C.; Girard, C.A.; Polidori, J.; Zorzi, K.; Birgy-Barelli, E.; Jullien, P.; Courivaud, C.; Krummel, T.; et al. Epitope spreading of autoantibody response to pla2r associates with poor prognosis in membranous nephropathy. J. Am. Soc. Nephrol. 2016, 27, 1517–1533. [Google Scholar] [CrossRef] [PubMed]
- Beck, L.H., Jr.; Salant, D.J. Refining our understanding of the pla2r-antibody response in primary membranous nephropathy: Looking forward, looking back. J. Am. Soc. Nephrol. 2020, 31, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Seitz-Polski, B.; Debiec, H.; Rousseau, A.; Dahan, K.; Zaghrini, C.; Payre, C.; Esnault, V.L.M.; Lambeau, G.; Ronco, P. Phospholipase a2 receptor 1 epitope spreading at baseline predicts reduced likelihood of remission of membranous nephropathy. J. Am. Soc. Nephrol. 2018, 29, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Kao, L.; Lam, V.; Waldman, M.; Glassock, R.J.; Zhu, Q. Identification of the immunodominant epitope region in phospholipase a2 receptor-mediating autoantibody binding in idiopathic membranous nephropathy. J. Am. Soc. Nephrol. 2015, 26, 291–301. [Google Scholar] [CrossRef]
- Reinhard, L.; Zahner, G.; Menzel, S.; Koch-Nolte, F.; Stahl, R.A.K.; Hoxha, E. Clinical relevance of domain-specific phospholipase a2 receptor 1 antibody levels in patients with membranous nephropathy. J. Am. Soc. Nephrol. 2020, 31, 197–207. [Google Scholar] [CrossRef]
- Seifert, L.; Hoxha, E.; Eichhoff, A.M.; Zahner, G.; Dehde, S.; Reinhard, L.; Koch-Nolte, F.; Stahl, R.A.K.; Tomas, N.M. The most n-terminal region of thsd7a is the predominant target for autoimmunity in thsd7a-associated membranous nephropathy. J. Am. Soc. Nephrol. 2018, 29, 1536–1548. [Google Scholar] [CrossRef]
- Fresquet, M.; Rhoden, S.J.; Jowitt, T.A.; McKenzie, E.A.; Roberts, I.; Lennon, R.; Brenchley, P.E. Autoantigens pla2r and thsd7a in membranous nephropathy share a common epitope motif in the n-terminal domain. J. Autoimmun. 2020, 106, 102308. [Google Scholar] [CrossRef]
- Doi, T.; Mayumi, M.; Kanatsu, K.; Suehiro, F.; Hamashima, Y. Distribution of igg subclasses in membranous nephropathy. Clin. Exp. Immunol. 1984, 58, 57–62. [Google Scholar]
- Kon, S.P.; Coupes, B.; Short, C.D.; Solomon, L.R.; Raftery, M.J.; Mallick, N.P.; Brenchley, P.E. Urinary c5b-9 excretion and clinical course in idiopathic human membranous nephropathy. Kidney Int. 1995, 48, 1953–1958. [Google Scholar] [CrossRef]
- Papagianni, A.A.; Alexopoulos, E.; Leontsini, M.; Papadimitriou, M. C5b-9 and adhesion molecules in human idiopathic membranous nephropathy. Nephrol. Dial. Transplant. 2002, 17, 57–63. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Beck, L.H., Jr.; Salant, D.J. Membranous nephropathy: From models to man. J. Clin. Investig. 2014, 124, 2307–2314. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Sandor, D.G.; Beck, L.H., Jr. The role of complement in membranous nephropathy. Semin. Nephrol. 2013, 33, 531–542. [Google Scholar] [CrossRef]
- Salant, D.J.; Belok, S.; Madaio, M.P.; Couser, W.G. A new role for complement in experimental membranous nephropathy in rats. J. Clin. Investig. 1980, 66, 1339–1350. [Google Scholar] [CrossRef]
- Cybulsky, A.V.; Rennke, H.G.; Feintzeig, I.D.; Salant, D.J. Complement-induced glomerular epithelial cell injury. Role of the membrane attack complex in rat membranous nephropathy. J. Clin. Investig. 1986, 77, 1096–1107. [Google Scholar] [CrossRef] [PubMed]
- Baker, P.J.; Ochi, R.F.; Schulze, M.; Johnson, R.J.; Campbell, C.; Couser, W.G. Depletion of c6 prevents development of proteinuria in experimental membranous nephropathy in rats. Am. J. Pathol. 1989, 135, 185–194. [Google Scholar] [PubMed]
- Luo, W.; Olaru, F.; Miner, J.H.; Beck, L.H., Jr.; van der Vlag, J.; Thurman, J.M.; Borza, D.B. Alternative pathway is essential for glomerular complement activation and proteinuria in a mouse model of membranous nephropathy. Front. Immunol. 2018, 9, 1433. [Google Scholar] [CrossRef]
- Kawata, N.; Kang, D.; Aiuchi, T.; Obama, T.; Yoshitake, O.; Shibata, T.; Takimoto, M.; Itabe, H.; Honda, K. Proteomics of human glomerulonephritis by laser microdissection and liquid chromatography-tandem mass spectrometry. Nephrology 2020, 25, 351–359. [Google Scholar] [CrossRef]
- Ravindran, A.; Madden, B.; Charlesworth, M.C.; Sharma, R.; Sethi, A.; Debiec, H.; Cattran, D.; Fervenza, F.C.; Smith, R.J.; Ronco, P.; et al. Proteomic analysis of complement proteins in membranous nephropathy. Kidney Int. Rep. 2020, 5, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Debiec, H.; Hanoy, M.; Francois, A.; Guerrot, D.; Ferlicot, S.; Johanet, C.; Aucouturier, P.; Godin, M.; Ronco, P. Recurrent membranous nephropathy in an allograft caused by igg3kappa targeting the pla2 receptor. J. Am. Soc. Nephrol. 2012, 23, 1949–1954. [Google Scholar] [CrossRef] [PubMed]
- Haddad, G.; Lorenzen, J.M.; Ma, H.; de Haan, N.; Seeger, H.; Zaghrini, C.; Brandt, S.; Kolling, M.; Wegmann, U.; Kiss, B.; et al. Altered glycosylation of igg4 promotes lectin complement pathway activation in anti-pla2r1-associated membranous nephropathy. J. Clin. Investig. 2021, 131, e140453. [Google Scholar] [CrossRef]
- Zhang, M.F.; Huang, J.; Zhang, Y.M.; Qu, Z.; Wang, X.; Wang, F.; Meng, L.Q.; Cheng, X.Y.; Cui, Z.; Liu, G.; et al. Complement activation products in the circulation and urine of primary membranous nephropathy. BMC Nephrol. 2019, 20, 313. [Google Scholar] [CrossRef]
- Bally, S.; Debiec, H.; Ponard, D.; Dijoud, F.; Rendu, J.; Faure, J.; Ronco, P.; Dumestre-Perard, C. Phospholipase a2 receptor-related membranous nephropathy and mannan-binding lectin deficiency. J. Am. Soc. Nephrol. 2016, 27, 3539–3544. [Google Scholar] [CrossRef] [PubMed]
- Seikrit, C.; Ronco, P.; Debiec, H. Factor h autoantibodies and membranous nephropathy. N. Engl. J. Med. 2018, 379, 2479–2481. [Google Scholar] [CrossRef] [PubMed]
- Schiller, B.; He, C.; Salant, D.J.; Lim, A.; Alexander, J.J.; Quigg, R.J. Inhibition of complement regulation is key to the pathogenesis of active heymann nephritis. J. Exp. Med. 1998, 188, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Makou, E.; Herbert, A.P.; Barlow, P.N. Functional anatomy of complement factor h. Biochemistry 2013, 52, 3949–3962. [Google Scholar] [CrossRef]
- Herwig, J.; Skuza, S.; Sachs, W.; Sachs, M.; Failla, A.V.; Rune, G.; Meyer, T.N.; Fester, L.; Meyer-Schwesinger, C. Thrombospondin type 1 domain-containing 7a localizes to the slit diaphragm and stabilizes membrane dynamics of fully differentiated podocytes. J. Am. Soc. Nephrol. 2019, 30, 824–839. [Google Scholar] [CrossRef]
- Godel, M.; Grahammer, F.; Huber, T.B. Thrombospondin type-1 domain-containing 7a in idiopathic membranous nephropathy. N. Engl. J. Med. 2015, 372, 1073. [Google Scholar]
- Fresquet, M.; Jowitt, T.A.; McKenzie, E.A.; Ball, M.D.; Randles, M.J.; Lennon, R.; Brenchley, P.E. Pla2r binds to the annexin a2-s100a10 complex in human podocytes. Sci. Rep. 2017, 7, 6876. [Google Scholar] [CrossRef]
- Caldwell, P.R.; Brentjens, J.R.; Camussi, G.; Andres, G. In vivo interaction of antibodies with cell surface proteins used as antigens. Tissue Cell 1986, 18, 809–816. [Google Scholar] [CrossRef]
- Ancian, P.; Lambeau, G.; Mattei, M.G.; Lazdunski, M. The human 180-kda receptor for secretory phospholipases a2. Molecular cloning, identification of a secreted soluble form, expression, and chromosomal localization. J. Biol. Chem. 1995, 270, 8963–8970. [Google Scholar] [CrossRef]
- Kuo, M.W.; Wang, C.H.; Wu, H.C.; Chang, S.J.; Chuang, Y.J. Soluble thsd7a is an n-glycoprotein that promotes endothelial cell migration and tube formation in angiogenesis. PLoS ONE 2011, 6, e29000. [Google Scholar] [CrossRef]
- Sethi, S.; Madden, B.J.; Debiec, H.; Charlesworth, M.C.; Gross, L.; Ravindran, A.; Hummel, A.M.; Specks, U.; Fervenza, F.C.; Ronco, P. Exostosin 1/exostosin 2-associated membranous nephropathy. J. Am. Soc. Nephrol. 2019, 30, 1123–1136. [Google Scholar] [CrossRef]
- Busse-Wicher, M.; Wicher, K.B.; Kusche-Gullberg, M. The exostosin family: Proteins with many functions. Matrix Biol. 2014, 35, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Ludecke, H.J.; Lindow, S.; Horton, W.A.; Lee, B.; Wagner, M.J.; Horsthemke, B.; Wells, D.E. Cloning of the putative tumour suppressor gene for hereditary multiple exostoses (ext1). Nat. Genet. 1995, 11, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Busse, M.; Kusche-Gullberg, M. In vitro polymerization of heparan sulfate backbone by the ext proteins. J. Biol. Chem. 2003, 278, 41333–41337. [Google Scholar] [CrossRef] [PubMed]
- Busse, M.; Feta, A.; Presto, J.; Wilen, M.; Gronning, M.; Kjellen, L.; Kusche-Gullberg, M. Contribution of ext1, ext2, and extl3 to heparan sulfate chain elongation. J. Biol. Chem. 2007, 282, 32802–32810. [Google Scholar] [CrossRef]
- Caza, T.N.; Hassen, S.I.; Kuperman, M.; Sharma, S.G.; Dvanajscak, Z.; Arthur, J.; Edmondson, R.; Storey, A.; Herzog, C.; Kenan, D.J.; et al. Neural cell adhesion molecule 1 is a novel autoantigen in membranous lupus nephritis. Kidney Int. 2020. [Google Scholar] [CrossRef]
- Garcia-Vives, E.; Sole, C.; Moline, T.; Alvarez-Rios, A.M.; Vidal, M.; Agraz, I.; Ordi-Ros, J.; Cortes-Hernandez, J. Antibodies to m-type phospholipase a2 receptor (pla2r) in membranous lupus nephritis. Lupus 2019, 28, 396–405. [Google Scholar] [CrossRef]
- Markovic-Lipkovski, J.; Zivotic, M.; Muller, C.A.; Tampe, B.; Cirovic, S.; Vjestica, J.; Tomanovic, N.; Zeisberg, M.; Muller, G.A. Variable expression of neural cell adhesion molecule isoforms in renal tissue: Possible role in incipient renal fibrosis. PLoS ONE 2015, 10, e0137028. [Google Scholar] [CrossRef]
- Sethi, S.; Debiec, H.; Madden, B.; Charlesworth, M.C.; Morelle, J.; Gross, L.; Ravindran, A.; Buob, D.; Jadoul, M.; Fervenza, F.C.; et al. Neural epidermal growth factor-like 1 protein (nell-1) associated membranous nephropathy. Kidney Int. 2020, 97, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Matsuhashi, S.; Noji, S.; Koyama, E.; Myokai, F.; Ohuchi, H.; Taniguchi, S.; Hori, K. New gene, nel, encoding a m(r) 93 k protein with egf-like repeats is strongly expressed in neural tissues of early stage chick embryos. Dev. Dyn. 1995, 203, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.K.; Katagiri, T.; Suzuki, M.; Shimizu, F.; Fujiwara, T.; Kanemoto, N.; Nakamura, Y.; Hirai, Y.; Maekawa, H.; Takahashi, E. Cloning and characterization of two novel human cdnas (nell1 and nell2) encoding proteins with six egf-like repeats. Genomics 1996, 38, 273–276. [Google Scholar] [CrossRef]
- Zhang, X.; Zara, J.; Siu, R.K.; Ting, K.; Soo, C. The role of nell-1, a growth factor associated with craniosynostosis, in promoting bone regeneration. J. Dent. Res. 2010, 89, 865–878. [Google Scholar] [CrossRef]
- Li, C.; Zhang, X.; Zheng, Z.; Nguyen, A.; Ting, K.; Soo, C. Nell-1 is a key functional modulator in osteochondrogenesis and beyond. J. Dent. Res. 2019, 98, 1458–1468. [Google Scholar] [CrossRef]
- Kun, E.; Tsang, Y.T.M.; Ng, C.W.; Gershenson, D.M.; Wong, K.K. Mek inhibitor resistance mechanisms and recent developments in combination trials. Cancer Treat. Rev. 2020, 92, 102137. [Google Scholar] [CrossRef] [PubMed]
- Caspi, M.; Wittenstein, A.; Kazelnik, M.; Shor-Nareznoy, Y.; Rosin-Arbesfeld, R. Therapeutic targeting of the oncogenic wnt signaling pathway for treating colorectal cancer and other colonic disorders. Adv. Drug Deliv. Rev. 2020, 169, 118–136. [Google Scholar] [CrossRef]
- Kuroda, S.; Oyasu, M.; Kawakami, M.; Kanayama, N.; Tanizawa, K.; Saito, N.; Abe, T.; Matsuhashi, S.; Ting, K. Biochemical characterization and expression analysis of neural thrombospondin-1-like proteins nell1 and nell2. Biochem. Biophys. Res. Commun. 1999, 265, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Imai, A.; Iijima, M.; Yoshimoto, N.; Maturana, A.D.; Kuroda, S.; Niimi, T. Mapping the heparin-binding site of the osteoinductive protein nell1 by site-directed mutagenesis. FEBS Lett. 2015, 589, 4026–4032. [Google Scholar] [CrossRef] [PubMed]
- Ting, K.; Vastardis, H.; Mulliken, J.B.; Soo, C.; Tieu, A.; Do, H.; Kwong, E.; Bertolami, C.N.; Kawamoto, H.; Kuroda, S.; et al. Human nell-1 expressed in unilateral coronal synostosis. J. Bone Miner. Res. 1999, 14, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Carpenter, D.; Bokui, N.; Soo, C.; Miao, S.; Truong, T.; Wu, B.; Chen, I.; Vastardis, H.; Tanizawa, K.; et al. Overexpression of nell-1, a craniosynostosis-associated gene, induces apoptosis in osteoblasts during craniofacial development. J. Bone Miner. Res. 2003, 18, 2126–2134. [Google Scholar] [CrossRef]
- Zhang, X.; Kuroda, S.; Carpenter, D.; Nishimura, I.; Soo, C.; Moats, R.; Iida, K.; Wisner, E.; Hu, F.Y.; Miao, S.; et al. Craniosynostosis in transgenic mice overexpressing nell-1. J. Clin. Investig. 2002, 110, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.; Raskind, W.; Blanton, S.H.; Pauli, R.M.; Gregg, R.G.; Francomano, C.A.; Puffenberger, E.; Conrad, E.U.; Schmale, G.; Schellenberg, G.; et al. Genetic heterogeneity in families with hereditary multiple exostoses. Am. J. Hum. Genet. 1993, 53, 71–79. [Google Scholar]
- Ahmad, S.B.; Appel, G.B. Antigens, antibodies, and membranous nephropathy: A decade of progress. Kidney Int. 2020, 97, 29–31. [Google Scholar] [CrossRef]
- Bizzoca, A.; Corsi, P.; Gennarini, G. The mouse f3/contactin glycoprotein: Structural features, functional properties and developmental significance of its regulated expression. Cell Adhes. Migr. 2009, 3, 53–63. [Google Scholar] [CrossRef]
- Boyle, M.E.; Berglund, E.O.; Murai, K.K.; Weber, L.; Peles, E.; Ranscht, B. Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron 2001, 30, 385–397. [Google Scholar] [CrossRef]
- Bhat, M.A.; Rios, J.C.; Lu, Y.; Garcia-Fresco, G.P.; Ching, W.; St Martin, M.; Li, J.; Einheber, S.; Chesler, M.; Rosenbluth, J.; et al. Axon-glia interactions and the domain organization of myelinated axons requires neurexin iv/caspr/paranodin. Neuron 2001, 30, 369–383. [Google Scholar] [CrossRef]
- Querol, L.; Nogales-Gadea, G.; Rojas-Garcia, R.; Martinez-Hernandez, E.; Diaz-Manera, J.; Suarez-Calvet, X.; Navas, M.; Araque, J.; Gallardo, E.; Illa, I. Antibodies to contactin-1 in chronic inflammatory demyelinating polyneuropathy. Ann. Neurol. 2013, 73, 370–380. [Google Scholar] [CrossRef]
- Ng, J.K.; Malotka, J.; Kawakami, N.; Derfuss, T.; Khademi, M.; Olsson, T.; Linington, C.; Odaka, M.; Tackenberg, B.; Pruss, H.; et al. Neurofascin as a target for autoantibodies in peripheral neuropathies. Neurology 2012, 79, 2241–2248. [Google Scholar] [CrossRef]
- Doppler, K.; Appeltshauser, L.; Villmann, C.; Martin, C.; Peles, E.; Kramer, H.H.; Haarmann, A.; Buttmann, M.; Sommer, C. Auto-antibodies to contactin-associated protein 1 (caspr) in two patients with painful inflammatory neuropathy. Brain 2016, 139, 2617–2630. [Google Scholar] [CrossRef]
- Delmont, E.; Manso, C.; Querol, L.; Cortese, A.; Berardinelli, A.; Lozza, A.; Belghazi, M.; Malissart, P.; Labauge, P.; Taieb, G.; et al. Autoantibodies to nodal isoforms of neurofascin in chronic inflammatory demyelinating polyneuropathy. Brain 2017, 140, 1851–1858. [Google Scholar] [CrossRef] [PubMed]
- Joint Task Force of the EFNS and the PNS. European federation of neurological societies/peripheral nerve society guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: Report of a joint task force of the European federation of neurological societies and the peripheral nerve society-first revision. J. Peripher. Nerv. Syst. 2010, 15, 1–9. [Google Scholar]
- Koike, H.; Katsuno, M. Pathophysiology of chronic inflammatory demyelinating polyneuropathy: Insights into classification and therapeutic strategy. Neurol. Ther. 2020, 9, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Delmont, E.; Brodovitch, A.; Kouton, L.; Allou, T.; Beltran, S.; Brisset, M.; Camdessanche, J.P.; Cauquil, C.; Cirion, J.; Dubard, T.; et al. Antibodies against the node of ranvier: A real-life evaluation of incidence, clinical features and response to treatment based on a prospective analysis of 1500 sera. J. Neurol. 2020, 267, 3664–3672. [Google Scholar] [CrossRef] [PubMed]
- Cortese, A.; Lombardi, R.; Briani, C.; Callegari, I.; Benedetti, L.; Manganelli, F.; Luigetti, M.; Ferrari, S.; Clerici, A.M.; Marfia, G.A.; et al. Antibodies to neurofascin, contactin-1, and contactin-associated protein 1 in cidp: Clinical relevance of igg isotype. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e639. [Google Scholar] [CrossRef]
- Miura, Y.; Devaux, J.J.; Fukami, Y.; Manso, C.; Belghazi, M.; Wong, A.H.; Yuki, N.; CNTN1-CIDP Study Group. Contactin 1 igg4 associates to chronic inflammatory demyelinating polyneuropathy with sensory ataxia. Brain 2015, 138, 1484–1491. [Google Scholar] [CrossRef]
- Doppler, K.; Appeltshauser, L.; Wilhelmi, K.; Villmann, C.; Dib-Hajj, S.D.; Waxman, S.G.; Maurer, M.; Weishaupt, A.; Sommer, C. Destruction of paranodal architecture in inflammatory neuropathy with anti-contactin-1 autoantibodies. J. Neurol. Neurosurg. Psychiatry 2015, 86, 720–728. [Google Scholar] [CrossRef]
- Witte, A.S.; Burke, J.F. Membranous glomerulonephritis associated with chronic progressive demyelinating neuropathy. Neurology 1987, 37, 342–345. [Google Scholar] [CrossRef]
- Kohli, A.; Tandon, P.; Kher, V. Chronic inflammatory demyelinating polyradiculoneuropathy with membranous glomerulonephritis: Report of one case. Clin. Neurol. Neurosurg. 1992, 94, 31–33. [Google Scholar] [CrossRef]
- Panjwani, M.; Truong, L.D.; Eknoyan, G. Membranous glomerulonephritis associated with inflammatory demyelinating peripheral neuropathies. Am. J. Kidney Dis. 1996, 27, 279–283. [Google Scholar] [CrossRef]
- Kanemoto, K.; Nakahara, C.; Saitoh, H.; Fukushima, T.; Kashiwagi, R.; Takahashi, M.; Iwasaki, N.; Kamoda, T.; Ohkoshi, N.; Nagata, M.; et al. Renal glucosuria and membranous glomerulonephritis in chronic inflammatory demyelinating polyradiculoneuropathy: Cidp. Nihon Jinzo Gakkai Shi 1999, 41, 511–516. [Google Scholar]
- Mobbs, R.J.; Tuck, R.R.; Hurley, B. Chronic inflammatory demyelinating polyneuropathy associated with membranous glomerulonephritis: Case report. J. Clin. Neurosci. 2000, 7, 454–455. [Google Scholar] [CrossRef][Green Version]
- Wu, A.D.; Russell, J.A.; Bouthout, B.A. Chronic inflammatory demyelinating polyneuropathy and membranous glomerulonephropathy: Report of two cases. J. Clin. Neuromuscul. Dis. 2001, 3, 70–74. [Google Scholar] [CrossRef]
- Emsley, H.C.; Molloy, J. Inflammatory demyelinating polyradiculoneuropathy associated with membranous glomerulonephritis and thrombocytopaenia. Clin. Neurol. Neurosurg. 2002, 105, 23–26. [Google Scholar] [CrossRef]
- Chen, K.H.; Chang, C.T.; Hung, C.C. Glomerulonephritis associated with chronic inflammatory demyelinating polyneuropathy. Ren. Fail. 2006, 28, 255–259. [Google Scholar] [CrossRef][Green Version]
- Smyth, S.; Menkes, D.L. Coincident membranous glomerulonephritis and chronic inflammatory demyelinating polyradiculoneuropathy: Questioning the autoimmunity hypothesis. Muscle Nerve 2008, 37, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.H.; Kokubun, N.; Fukami, Y.; Miyaji, K.; Yuki, N. Chronic inflammatory demyelinating polyneuropathy with membranous nephropathy. J. Peripher. Nerv. Syst. 2015, 20, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Ogata, H.; Yamasaki, R.; Sasaguri, T.; Ko, S.; Yamashita, K.; Xu, Z.; Matsushita, T.; Tateishi, T.; Akiyama, S.; et al. Chronic inflammatory demyelinating polyneuropathy with concurrent membranous nephropathy: An anti-paranode and podocyte protein antibody study and literature survey. Front. Neurol. 2018, 9, 997. [Google Scholar] [CrossRef] [PubMed]
- Reid, R.A.; Bronson, D.D.; Young, K.M.; Hemperly, J.J. Identification and characterization of the human cell adhesion molecule contactin. Mol. Brain Res. 1994, 21, 1–8. [Google Scholar] [CrossRef]
- Nazarali, S.; Mathey, E.K.; Tang, D.; Margetts, P.J.; Baker, S.K. Chronic inflammatory demyelinating polyneuropathy and concurrent membranous nephropathy. Can. J. Neurol. Sci. 2020, 47, 585–587. [Google Scholar] [CrossRef] [PubMed]
- Sistani, L.; Rodriguez, P.Q.; Hultenby, K.; Uhlen, M.; Betsholtz, C.; Jalanko, H.; Tryggvason, K.; Wernerson, A.; Patrakka, J. Neuronal proteins are novel components of podocyte major processes and their expression in glomerular crescents supports their role in crescent formation. Kidney Int. 2013, 83, 63–71. [Google Scholar] [CrossRef]
- Sethi, S.; Debiec, H.; Madden, B.; Vivarelli, M.; Charlesworth, M.C.; Ravindran, A.; Gross, L.; Ulinski, T.; Buob, D.; Tran, C.L.; et al. Semaphorin 3b-associated membranous nephropathy is a distinct type of disease predominantly present in pediatric patients. Kidney Int. 2020, 98, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Guan, F.; Villegas, G.; Teichman, J.; Mundel, P.; Tufro, A. Autocrine class 3 semaphorin system regulates slit diaphragm proteins and podocyte survival. Kidney Int. 2006, 69, 1564–1569. [Google Scholar] [CrossRef]
- Al-Rabadi, L.; Caza, T.; Avillach, C.; Rodan, A.R.; Williams, B.; Abraham, J.; Revelo Penafiel, M.P.; Andeen, N.K.; Kawalit, I.; Clayton, F.; et al. High temperature recombinant protein a1 (htra1): A novel antigen in membranous nephropathy. In Proceedings of the 2020 ASN Kidney Week, Simulive, 22–25 October 2020; pp. 25–26. [Google Scholar]
- Sethi, S.; Madden, B.J.; Gross, L.; Negron, V.C.; Charlesworth, C.; Debiec, H.; Ronco, P.M.; Fervenza, F.C. Protocadherin 7-associated membranous nephropathy. In Proceedings of the 2020 ASN Kidney Week, Simulive, 22–25 October 2020; p. 26. [Google Scholar]
- Sethi, S. New ’antigens’ in membranous nephropathy. J. Am. Soc. Nephrol. 2021, 32, 268–278. [Google Scholar] [CrossRef]
- Kanigicherla, D.; Gummadova, J.; McKenzie, E.A.; Roberts, S.A.; Harris, S.; Nikam, M.; Poulton, K.; McWilliam, L.; Short, C.D.; Venning, M.; et al. Anti-pla2r antibodies measured by elisa predict long-term outcome in a prevalent population of patients with idiopathic membranous nephropathy. Kidney Int. 2013, 83, 940–948. [Google Scholar] [CrossRef]
- Kaya, B.; Paydas, S.; Balal, M.; Eren Erdogan, K.; Gonlusen, G. Renal expression of pla2r, thsd7a, and igg4 in patients with membranous nephropathy and correlation with clinical findings. Int. J. Clin. Pract. 2020, e13855. [Google Scholar]
- Von Haxthausen, F.; Reinhard, L.; Pinnschmidt, H.O.; Rink, M.; Soave, A.; Hoxha, E.; Stahl, R.A.K. Antigen-specific igg subclasses in primary and malignancy-associated membranous nephropathy. Front. Immunol. 2018, 9, 3035. [Google Scholar] [CrossRef]
- Matsumoto, A.; Matsui, I.; Namba, T.; Sakaguchi, Y.; Mizuno, H.; Shirayama, Y.; Shimada, K.; Hashimoto, N.; Doi, Y.; Yamaguchi, S.; et al. Vegf-a links angiolymphoid hyperplasia with eosinophilia (alhe) to thsd7a membranous nephropathy: A report of 2 cases. Am. J. Kidney Dis. 2019, 73, 880–885. [Google Scholar] [CrossRef]
- Xian, L.; Dong, D.; Luo, J.; Zhuo, L.; Li, K.; Zhang, P.; Wang, W.; Xu, Y.; Xu, G.; Wang, L.; et al. Expression of thsd7a in neoplasm tissues and its relationship with proteinuria. BMC Nephrol. 2019, 20, 332. [Google Scholar] [CrossRef]
- Reinhard, L.; Stahl, R.A.K.; Hoxha, E. Is primary membranous nephropathy a complement mediated disease? Mol. Immunol. 2020, 128, 195–204. [Google Scholar] [CrossRef]
- Schubart, A.; Anderson, K.; Mainolfi, N.; Sellner, H.; Ehara, T.; Adams, C.M.; Mac Sweeney, A.; Liao, S.M.; Crowley, M.; Littlewood-Evans, A.; et al. Small-molecule factor b inhibitor for the treatment of complement-mediated diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 7926–7931. [Google Scholar] [CrossRef]
- Mainolfi, N.; Ehara, T.; Karki, R.G.; Anderson, K.; Mac Sweeney, A.; Liao, S.M.; Argikar, U.A.; Jendza, K.; Zhang, C.; Powers, J.; et al. Discovery of 4-((2s,4s)-4-ethoxy-1-((5-methoxy-7-methyl-1h-indol-4-yl)methyl)piperidin-2-yl)be nzoic acid (lnp023), a factor b inhibitor specifically designed to be applicable to treating a diverse array of complement mediated diseases. J. Med. Chem. 2020, 63, 5697–5722. [Google Scholar] [CrossRef]
- Kaplon, H.; Muralidharan, M.; Schneider, Z.; Reichert, J.M. Antibodies to watch in 2020. MAbs 2020, 12, 1703531. [Google Scholar] [CrossRef]
- Ahmadi, A.; Moghadasali, R.; Ezzatizadeh, V.; Taghizadeh, Z.; Nassiri, S.M.; Asghari-Vostikolaee, M.H.; Alikhani, M.; Hadi, F.; Rahbarghazi, R.; Yazdi, R.S.; et al. Transplantation of mouse induced pluripotent stem cell-derived podocytes in a mouse model of membranous nephropathy attenuates proteinuria. Sci. Rep. 2019, 9, 15467. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Ojo, D.; Kapoor, A.; Lin, X.; Pinthus, J.H.; Aziz, T.; Bismar, T.A.; Wei, F.; Wong, N.; de Melo, J.; et al. Neural cell adhesion protein cntn1 promotes the metastatic progression of prostate cancer. Cancer Res. 2016, 76, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Wong, N.; Hung, C.; Chen, W.X.; Tang, D. Contactin-1 reduces e-cadherin expression via activating akt in lung cancer. PLoS ONE 2013, 8, e65463. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Li, T.; Kapoor, A.; Major, P.; Tang, D. Contactin 1: An important and emerging oncogenic protein promoting cancer progression and metastasis. Genes 2020, 11, 874. [Google Scholar] [CrossRef]
- Liang, Y.; Ma, C.; Li, F.; Nie, G.; Zhang, H. The role of contactin 1 in cancers: What we know so far. Front. Oncol. 2020, 10, 574208. [Google Scholar] [CrossRef]
- Abrahamson, D.R.; Steenhard, B.M.; Stroganova, L.; Zelenchuk, A.; St John, P.L.; Petroff, M.G.; Patarroyo, M.; Borza, D.B. Maternal alloimmune igg causes anti-glomerular basement membrane disease in perinatal transgenic mice that express human laminin alpha5. Kidney Int. 2019, 96, 1320–1331. [Google Scholar] [CrossRef]
- Vivarelli, M.; Emma, F. A new mouse model of anti-gbm disease sheds light on maternal transfer of alloantibodies in glomerular disease. Kidney Int. 2019, 96, 1272–1274. [Google Scholar] [CrossRef]
- Wang, J.; Wang, M.; Cui, Z.; Zhao, M.H. Epitope mapping of human alpha3(iv)nc1-induced membranous nephropathy in mice. Am. J. Nephrol. 2020, 51, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Glassock, R.J. Membranous nephropathy due to anti-gbm antibodies of mice and men. Am. J. Nephrol. 2020, 51, 96–98. [Google Scholar] [CrossRef]
- Xu, X.; Wang, G.; Chen, N.; Lu, T.; Nie, S.; Xu, G.; Zhang, P.; Luo, Y.; Wang, Y.; Wang, X.; et al. Long-term exposure to air pollution and increased risk of membranous nephropathy in china. J. Am. Soc. Nephrol. 2016, 27, 3739–3746. [Google Scholar] [CrossRef]
- Wang, Z.; Wen, L.; Dou, Y.; Zhao, Z. Human anti-thrombospondin type 1 domain-containing 7a antibodies induce membranous nephropathy through activation of lectin complement pathway. Biosci. Rep. 2018, 38, BSR20180131. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Liang, L.; Liu, L.; Tang, X.; Tang, L.; Chen, P.; Chen, J.; Wang, Z.; Cao, W.; et al. Effect of glomerular mannose-binding lectin deposition on the prognosis of idiopathic membranous nephropathy. Kidney Blood Press. Res. 2020, 45, 713–726. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| PMN Cases | Control Cases 1 | Sensitivity | Specificity | Refs |
|---|---|---|---|---|
| 69 | 386 | 71% | 100% | [39] |
| 57 | 84 | 82.5% | 75% | [40] |
| 67 | 236 | 88.1% | 96% | [41] |
| 155 | 154 | 83.9% | 99.4% | [42] |
| 374 | 296 | 80.8% | 98% | [43] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, Y.; Xu, H.; Tang, D. Mechanisms of Primary Membranous Nephropathy. Biomolecules 2021, 11, 513. https://doi.org/10.3390/biom11040513
Gu Y, Xu H, Tang D. Mechanisms of Primary Membranous Nephropathy. Biomolecules. 2021; 11(4):513. https://doi.org/10.3390/biom11040513
Chicago/Turabian StyleGu, Yan, Hui Xu, and Damu Tang. 2021. "Mechanisms of Primary Membranous Nephropathy" Biomolecules 11, no. 4: 513. https://doi.org/10.3390/biom11040513
APA StyleGu, Y., Xu, H., & Tang, D. (2021). Mechanisms of Primary Membranous Nephropathy. Biomolecules, 11(4), 513. https://doi.org/10.3390/biom11040513