
The Cys Sense: Thiol Redox Switches Mediate Life Cycles of Cellular Proteins


Abstract
:1. Introduction
2. Cellular Oxidants: Origin, Targets and Benefits
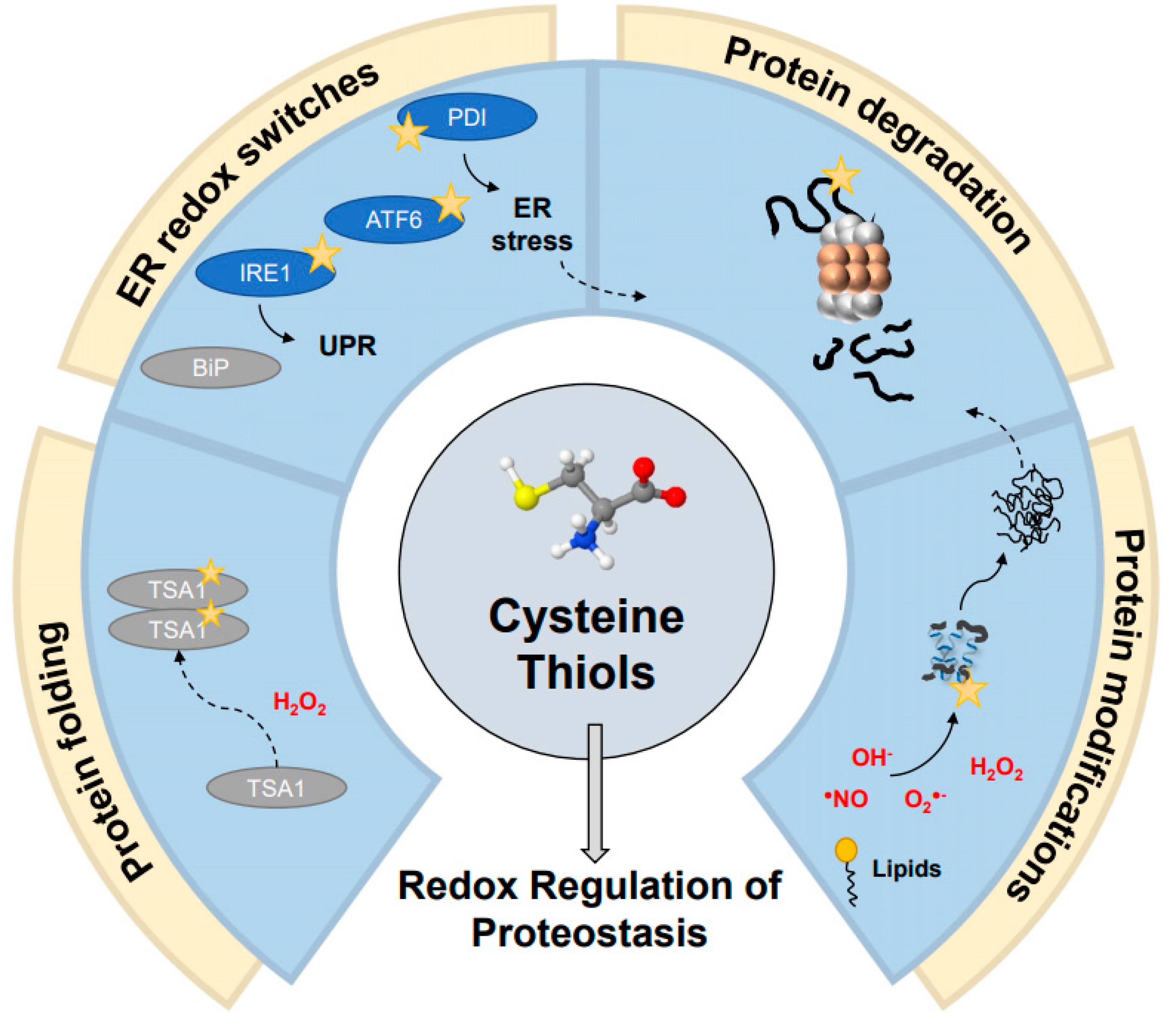
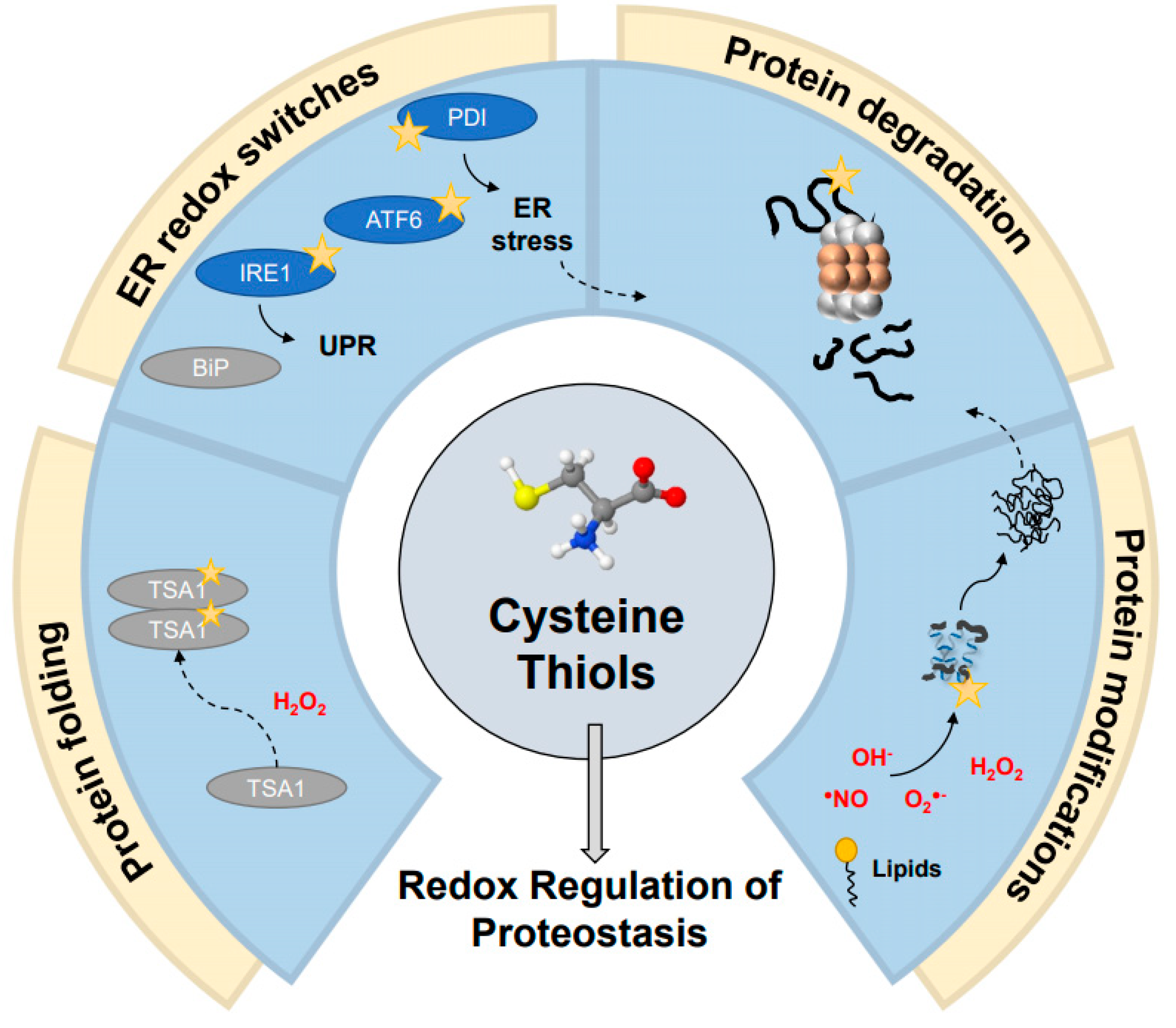
3. Cysteine Thiols: The Central Components of Redox-Regulation of Proteostasis
4. Integrative Approaches for Discovering New Redox Switches in PQC
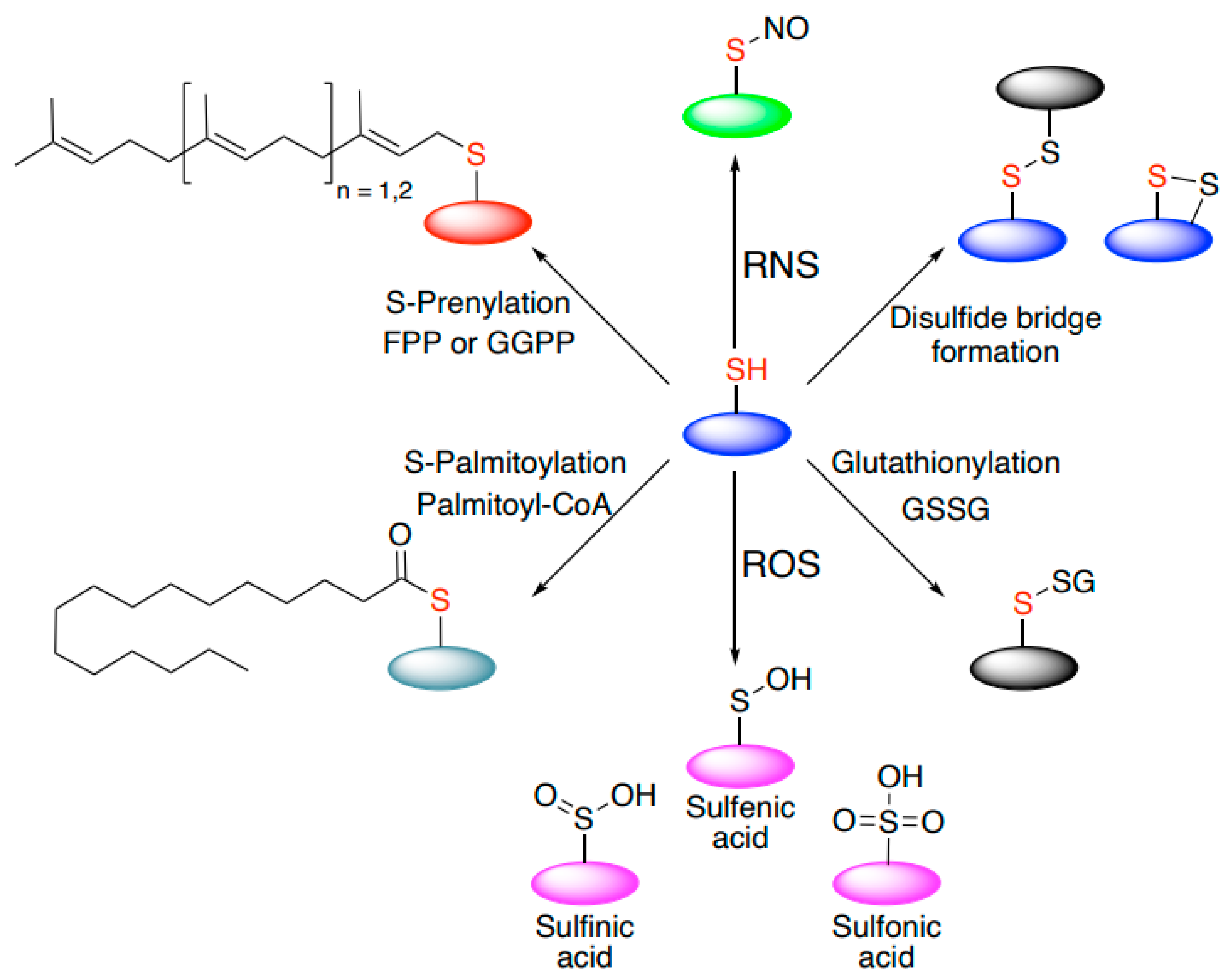
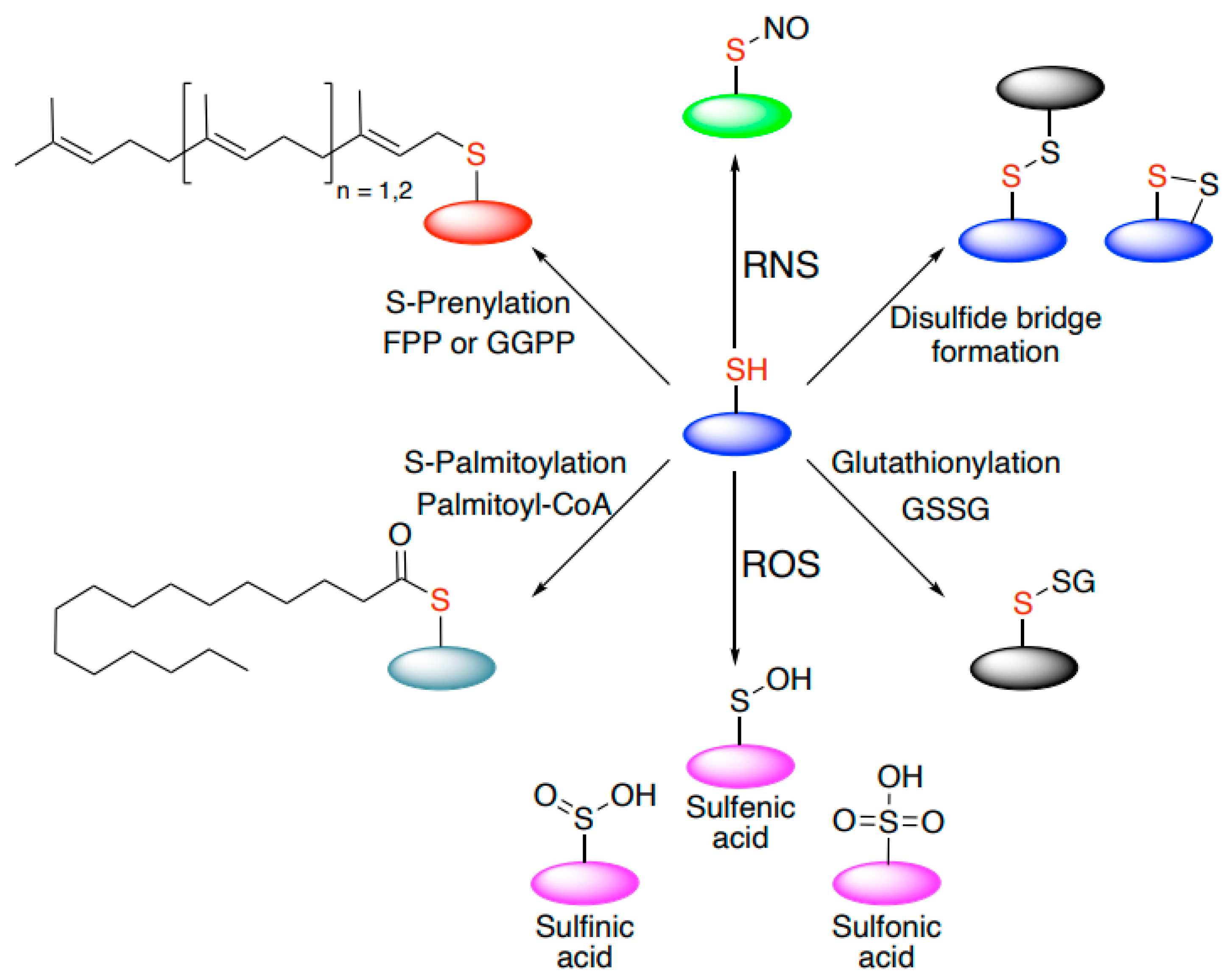
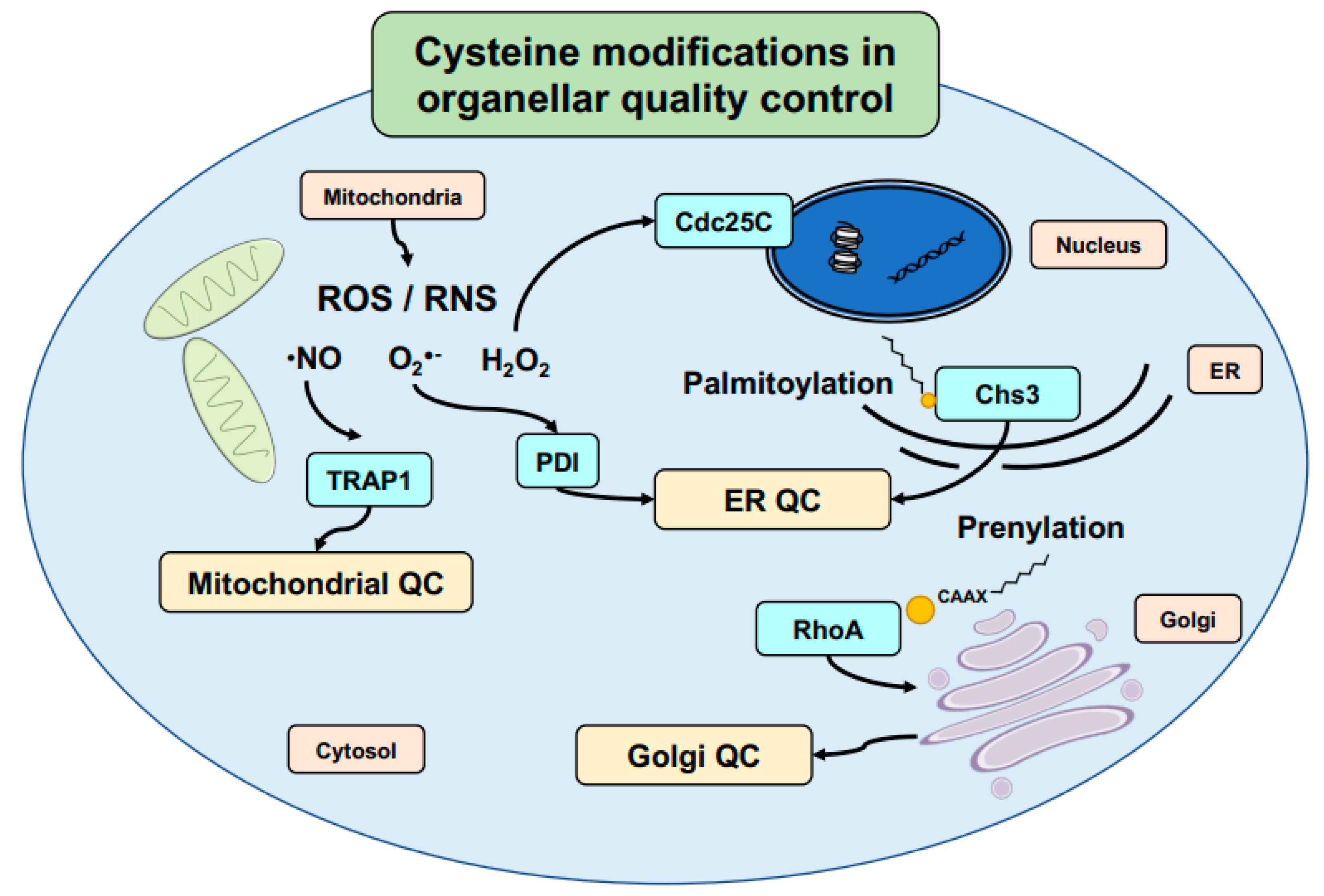
5. Cysteine-Mediated Modifications: An Efficient Mechanism to Regulate Signal Transduction and Protein Localization in Cells
6. Thiol Editing in the ER Is Mediated by Molecular Redox Switches
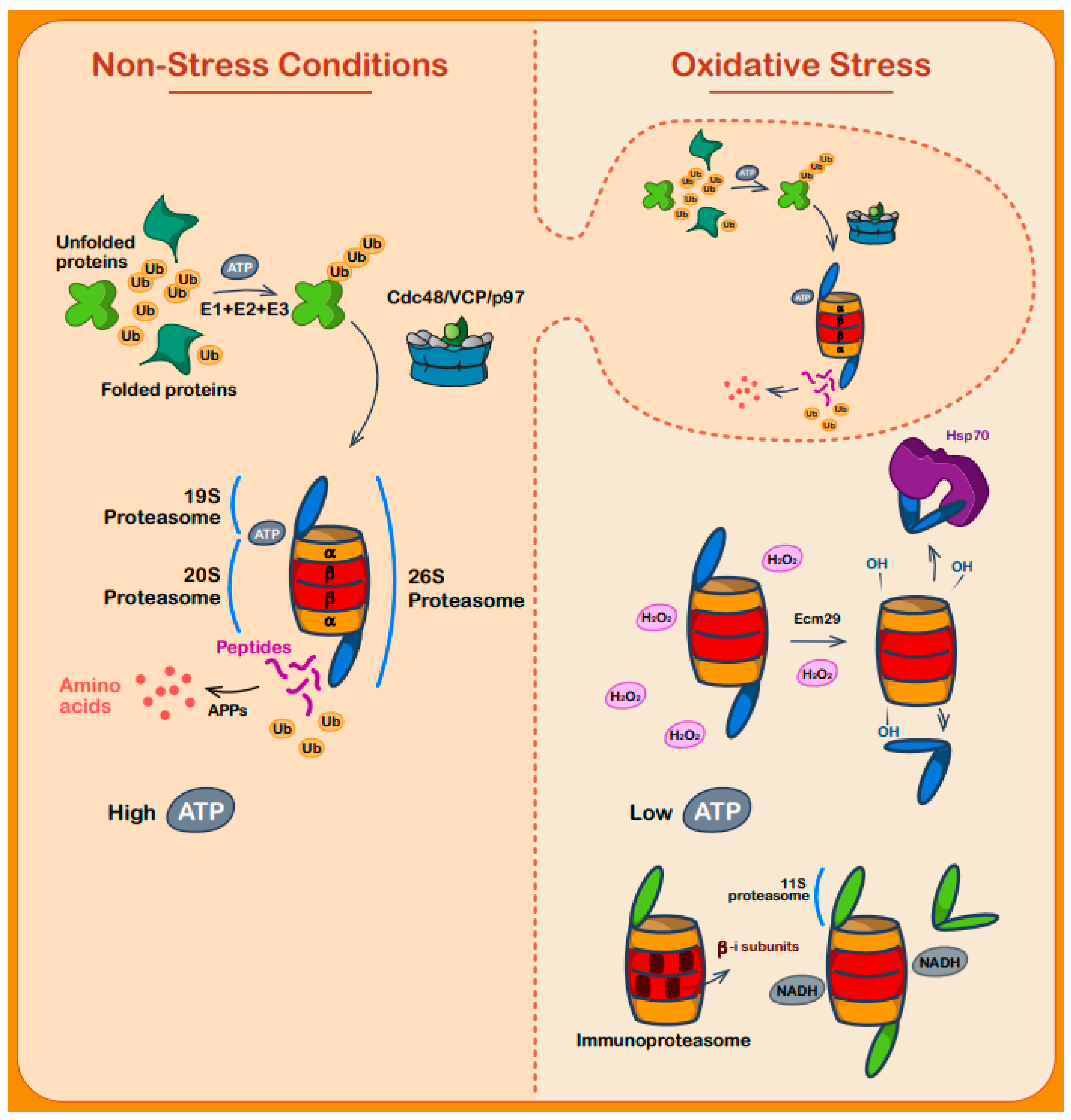
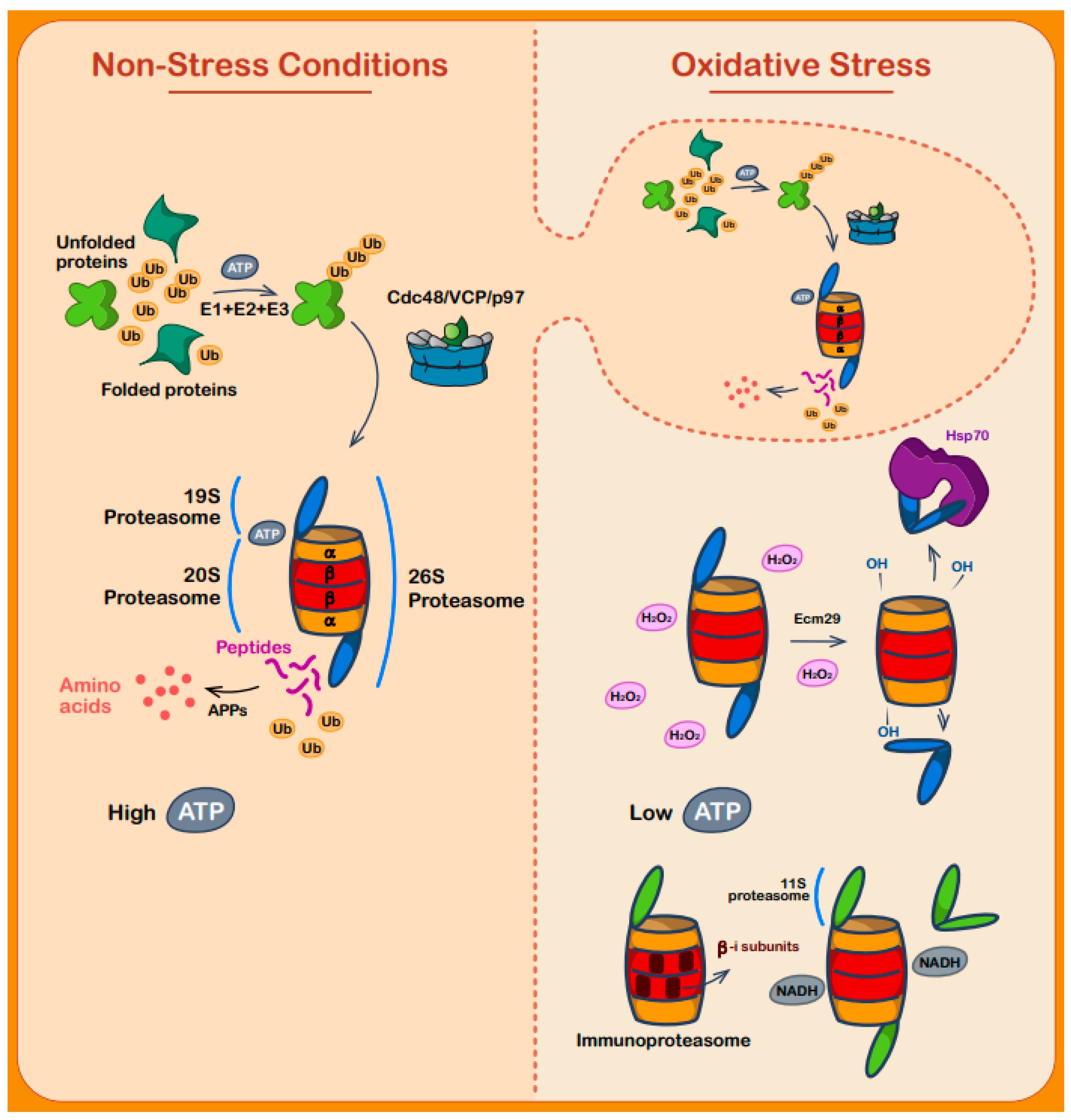
7. Regulation of Protein Degradation during Oxidative Stress
8. Protein Degradation by Redox Sensitive Proteins
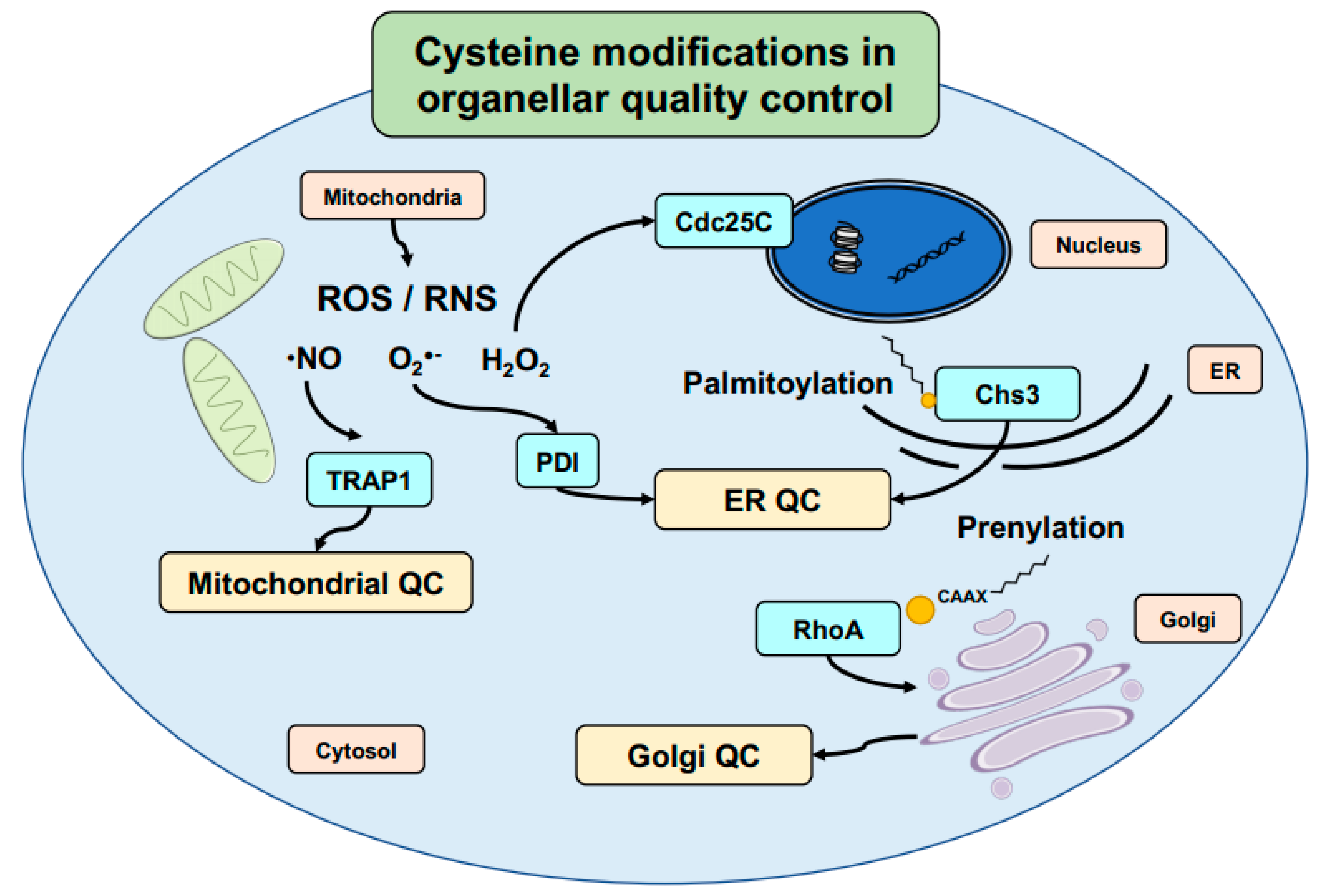
9. Aging, Subcellular Localization, and Cysteine Oxidation
10. Cell Cycle and Redox Status Are Highly Connected
11. Conclusions and Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brandman, O.; Stewart-Ornstein, J.; Wong, D.; Larson, A.; Williams, C.C.; Li, G.-W.; Zhou, S.; King, D.; Shen, P.S.; Weibezahn, J.; et al. A Ribosome-Bound Quality Control Complex Triggers Degradation of Nascent Peptides and Signals Translation Stress. Cell 2012, 151, 1042–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandman, O.; Hegde, R.S. Ribosome-associated protein quality control. Nat. Struct. Mol. Biol. 2016, 23, 7–15. [Google Scholar] [CrossRef]
- Santiago, A.M.; Gonçalves, D.L.; Morano, K.A. Mechanisms of sensing and response to proteotoxic stress. Exp. Cell Res. 2020, 395, 112240. [Google Scholar] [CrossRef]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Ulrich Hartl, F. Molecular Chaperone Functions in Protein Folding and Proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef]
- Enam, C.; Geffen, Y.; Ravid, T.; Gardner, R.G. Protein Quality Control Degradation in the Nucleus. Annu. Rev. Biochem. 2018, 87, 725–749. [Google Scholar] [CrossRef]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial proteins: From biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Moehle, E.A.; Shen, K.; Dillin, A. Mitochondrial proteostasis in the context of cellular and organismal health and aging. J. Biol. Chem. 2019, 294, 5396–5407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef]
- Bardwell, J.C.A.; Jakob, U. Conditional disorder in chaperone action. Trends Biochem. Sci. 2012, 37, 517–525. [Google Scholar] [CrossRef] [Green Version]
- Suss, O.; Reichmann, D. Protein plasticity underlines activation and function of ATP-independent chaperones. Front. Mol. Biosci. 2015, 2. [Google Scholar] [CrossRef] [Green Version]
- Mayer, M.P.; Gierasch, L.M. Recent advances in the structural and mechanistic aspects of Hsp70 molecular chaperones. J. Biol. Chem. 2019, 294, 2085–2097. [Google Scholar] [CrossRef] [Green Version]
- Laskowska, E.; Kuczyńska-Wiśnik, D.; Lipińska, B. Proteomic analysis of protein homeostasis and aggregation. J. Proteom. 2018, 198, 98–112. [Google Scholar] [CrossRef]
- Reichmann, D.; Voth, W.; Jakob, U. Maintaining a Healthy Proteome during Oxidative Stress. Mol. Cell 2018, 69, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Morimoto, R.I. The Biology of Proteostasis in Aging and Disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [Green Version]
- Saarikangas, J.; Barral, Y. Protein aggregation as a mechanism of adaptive cellular responses. Curr. Genet. 2016, 62, 711–724. [Google Scholar] [CrossRef]
- Korovila, I.; Hugo, M.; Castro, J.P.; Weber, D.; Höhn, A.; Grune, T.; Jung, T. Proteostasis, oxidative stress and aging. Redox Biol. 2017, 13, 550–567. [Google Scholar] [CrossRef] [PubMed]
- Sitia, R.; Braakman, I. Quality control in the endoplasmic reticulum protein factory. Nature 2003, 426, 891–894. [Google Scholar] [CrossRef]
- Tsai, F.T.F.; Jeng, W.; Lee, S.; Sung, N.; Lee, J. Molecular chaperones: Guardians of the proteome in normal and disease states. F1000Research 2015, 4, 1448. [Google Scholar]
- Brandvold, K.R.; Morimoto, R.I. The Chemical Biology of Molecular Chaperones—Implications for Modulation of Proteostasis. J. Mol. Biol. 2015, 427, 2931–2947. [Google Scholar] [CrossRef] [Green Version]
- Jakob, U.; Muse, W.; Eser, M.; Bardwell, J.C.A. Chaperone activity with a redox switch. Cell 1999, 96, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Tapley, T.L.; Körnera, J.L.; Barge, M.T.; Hupfeld, J.; Schauerte, J.A.; Gafni, A.; Jakob, U.; Bardwell, J.C.A. Structural plasticity of an acid-activated chaperone allows promiscuous substrate binding. Proc. Natl. Acad. Sci. USA 2009, 106, 5557–5562. [Google Scholar] [CrossRef] [Green Version]
- Dahl, J.U.; Koldewey, P.; Salmon, L.; Horowitz, S.; Bardwell, J.C.A.; Jakob, U. HdeB functions as an acid-protective chaperone in bacteria. J. Biol. Chem. 2015, 290, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Haslbeck, M.; Weinkauf, S.; Buchner, J. Small heat shock proteins: Simplicity meets complexity. J. Biol. Chem. 2019, 294, 2121–2132. [Google Scholar] [CrossRef] [Green Version]
- Alderson, T.R.; Ying, J.; Bax, A.; Benesch, J.L.P.; Baldwin, A.J. Conditional Disorder in Small Heat-shock Proteins. J. Mol. Biol. 2020, 432, 3033–3049. [Google Scholar] [CrossRef]
- Richter, K.; Haslbeck, M.; Buchner, J. The Heat Shock Response: Life on the Verge of Death. Mol. Cell 2010, 40, 253–266. [Google Scholar] [CrossRef]
- Mogk, A.; Kummer, E.; Bukau, B. Cooperation of Hsp70 and Hsp100 chaperone machines in protein disaggregation. Front. Mol. Biosci. 2015, 2, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebeaud, M.; Mallik, S.; Goloubinoff, P.; Tawfik, D. On the evolution of chaperones and co-chaperones and the expansion of proteomes across the Tree of Life. bioRxiv 2020. [Google Scholar] [CrossRef]
- Powers, E.T.; Balch, W.E. Diversity in the origins of proteostasis networks-a driver for protein function in evolution. Nat. Rev. Mol. Cell Biol. 2013, 14, 237–248. [Google Scholar] [CrossRef]
- Russell, R.; Karzai, A.W.; Mehl, A.F.; McMacken, R. DnaJ dramatically stimulates ATP hydrolysis by DnaK: Insight into targeting of Hsp70 proteins to polypeptide substrates. Biochemistry 1999, 38, 4165–4176. [Google Scholar] [CrossRef]
- Xu, H. Cochaperones enable Hsp70 to use ATP energy to stabilize native proteins out of the folding equilibrium. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Dean, M.E.; Johnson, J.L. Human Hsp90 cochaperones: Perspectives on tissue-specific expression and identification of cochaperones with similar in vivo functions. Cell Stress Chaperones 2020, 26. [Google Scholar] [CrossRef]
- Cyr, D.M. Cooperation of the molecular chaperone Ydj1 with specific Hsp70 homologs to suppress protein aggregation. FEBS Lett. 1995, 359, 129–132. [Google Scholar] [CrossRef] [Green Version]
- Luke, M.M.; Sutton, A.; Arndt, K.T. Characterization of SIS1, a Saccharomyces cerevisiae homologue of bacterial dnaJ proteins. J. Cell Biol. 1991, 114, 623–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.H.; Kukushkin, Y.; Gupta, R.; Chen, T.; Konagai, A.; Hipp, M.S.; Hayer-Hartl, M.; Hartl, F.U. PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell 2013, 154, 134–145. [Google Scholar] [CrossRef] [Green Version]
- Faust, O.; Abayev-Avraham, M.; Wentink, A.S.; Maurer, M.; Nillegoda, N.B.; London, N.; Bukau, B.; Rosenzweig, R. HSP40 proteins use class-specific regulation to drive HSP70 functional diversity. Nature 2020, 587, 489–494. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Andreasson, C.; Barducci, A.; Cheetham, M.E.; Cyr, D.; Emanuelsson, C.; Genevaux, P.; Gestwicki, J.E.; Goloubinoff, P.; Huerta-Cepas, J.; et al. Function, evolution, and structure of J-domain proteins. Cell Stress Chaperones 2019, 24, 7–15. [Google Scholar] [CrossRef]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 2019, 20, 665–680. [Google Scholar] [CrossRef]
- Kirstein, J.; Arnsburg, K.; Scior, A.; Szlachcic, A.; Guilbride, D.L.; Morimoto, R.I.; Bukau, B.; Nillegoda, N.B. In vivo properties of the disaggregase function of J-proteins and Hsc70 in Caenorhabditis elegans stress and aging. Aging Cell 2017, 16, 1414–1424. [Google Scholar] [CrossRef]
- Feleciano, D.R.; Arnsburg, K.; Kirstein, J. Interplay between redox and protein homeostasis. Worm 2016, 5, e1170273. [Google Scholar] [CrossRef] [Green Version]
- Ushioda, R.; Hoseki, J.; Araki, K.; Jansen, G.; Thomas, D.Y.; Nagata, K. ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science 2008, 321, 569–572. [Google Scholar] [CrossRef]
- Abildgaard, A.B.; Gersing, S.K.; Larsen-Ledet, S.; Nielsen, S.V.; Stein, A.; Lindorff-Larsen, K.; Hartmann-Petersen, R. Co-chaperones in targeting and delivery of misfolded proteins to the 26s proteasome. Biomolecules 2020, 10, 1141. [Google Scholar] [CrossRef]
- Shiber, A.; Ravid, T. Chaperoning proteins for destruction: Diverse roles of Hsp70 chaperones and their co-chaperones in targeting misfolded proteins to the proteasome. Biomolecules 2014, 4, 704–724. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, B.S.; Belenghi, B.; Levine, A. Oxidative stress increased respiration and generation of reactive oxygen species, resulting in ATP depletion, opening of mitochondrial permeability transition, and programmed cell death. Plant Physiol. 2002, 128, 1271–1281. [Google Scholar] [CrossRef] [Green Version]
- Colussi, C.; Albertini, M.C.; Coppola, S.; Rovidati, S.; Galli, F.; Ghibelli, L. H2O2-induced block of glycolysis as an active ADP-ribosylation reaction protecting cells from apoptosis. FASEB J. 2000, 14, 2266–2276. [Google Scholar] [CrossRef] [Green Version]
- Schraufstatter, I.U.; Hyslop, P.A.; Hinshaw, D.B.; Spragg, R.G.; Sklar, L.A.; Cochrane, C.G. Hydrogen peroxide-induced injury of cells and its prevention by inhibitors of poly(ADP-ribose) polymerase. Proc. Natl. Acad. Sci. USA 1986, 83, 4908–4912. [Google Scholar] [CrossRef] [Green Version]
- Goemans, C.V.; Vertommen, D.; Agrebi, R.; Collet, J.F. CnoX Is a Chaperedoxin: A Holdase that Protects Its Substrates from Irreversible Oxidation. Mol. Cell 2018, 70, 614–627.e7. [Google Scholar] [CrossRef] [Green Version]
- Voth, W.; Schick, M.; Gates, S.; Li, S.; Vilardi, F.; Gostimskaya, I.; Southworth, D.R.; Schwappach, B.; Jakob, U. The protein targeting factor Get3 functions as ATP-Independent chaperone under oxidative stress conditions. Mol. Cell 2014, 56, 116–127. [Google Scholar] [CrossRef] [Green Version]
- Jensen, P.K. Antimycin-insensitive oxidation of succinate and reduced nicotinamide-adenine dinucleotide in electron-transport particles I. pH dependency and hydrogen peroxide formation. BBA Enzymol. Biol. Oxid. 1966, 122, 157–166. [Google Scholar] [CrossRef]
- Loschen, G.; Azzi, A.; Richter, C.; Flohé, L. Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Lett. 1974, 42, 68–72. [Google Scholar] [CrossRef] [Green Version]
- Loschen, G.; Azzi, A.; Flohé, L. Mitochondrial H2O2 formation: Relationship with energy conservation. FEBS Lett. 1973, 33, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef]
- Fridovich, I. Superoxide dismutases. Annu. Rev. Biochem. 1975, 44, 147–159. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef]
- Holmström, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Yoboue, E.D.; Sitia, R.; Simmen, T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 2018, 9, 331. [Google Scholar] [CrossRef] [Green Version]
- Suh, Y.-A.; Arnold, R.S.; Lassegue, B.; Shi, J.; Xu, X.; Sorescu, D.; Chung, A.B.; Griendling, K.K.; Lambeth, J.D. Cell transformation by the superoxide-generating oxidase Mox1. Nature 1999, 401, 79–82. [Google Scholar] [CrossRef]
- Takeya, R.; Sumimoto, H. Regulation of novel superoxide-producing NAD(P)H oxidases. Antioxid. Redox Signal. 2006, 8, 1523–1532. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Maker, H.S.; Weiss, C.; Silides, D.J.; Cohen, G. Coupling of Dopamine Oxidation (Monoamine Oxidase Activity) to Glutathione Oxidation Via the Generation of Hydrogen Peroxide in Rat Brain Homogenates. J. Neurochem. 1981, 36, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Shao, L.; Spitz, D.R. Reactive oxygen species in normal and tumor stem cells. In Advances in Cancer Research; Academic Press Inc.: Cambridge, MA, USA, 2014; Volume 122, pp. 1–67. [Google Scholar]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef]
- Fourquet, S.; Guerois, R.; Biard, D.; Toledano, M.B. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. J. Biol. Chem. 2010, 285, 8463–8471. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, E.M.; McGinity, C.; Wink, D.A.; McVicar, D.W. Nitric oxide in macrophage immunometabolism: Hiding in plain sight. Metabolites 2020, 10, 429. [Google Scholar] [CrossRef]
- Nathan, C.; Ding, A. Snapshot: Reactive oxygen intermediates (ROI). Cell 2010, 140, 951–951.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Loake, G.J.; Chu, C. Cross-talk of nitric oxide and reactive oxygen species in plant programed cell death. Front. Plant Sci. 2013, 4, 314. [Google Scholar] [CrossRef] [Green Version]
- Apel, K.; Hirt, H. Reactive oxygen species: Metabolism, oxidative stress, and signal transduction. Annu. Rev. Plant Biol. 2004, 55, 373–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Feldman, N.B.; Lutsenko, S.V. ROS and RNS signalling: Adaptive redox switches through oxidative/nitrosative protein modifications. Free Radic. Res. 2018, 52, 507–543. [Google Scholar] [CrossRef]
- Babior, B.M.; Kipnes, R.S.; Curnutte, J.T. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J. Clin. Investig. 1973, 52, 741–744. [Google Scholar] [CrossRef]
- Rice, M.E. H2O2: A dynamic neuromodulator. Neuroscientist 2011, 17, 389–406. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T. Oxidant signals and oxidative stress. Curr. Opin. Cell Biol. 2003, 15, 247–254. [Google Scholar] [CrossRef]
- Koshland, D.E. The molecule of the year. Science 1992, 258, 1861. [Google Scholar] [CrossRef] [Green Version]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Del Valle, N.R.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [Green Version]
- Benhar, M. Roles of mammalian glutathione peroxidase and thioredoxin reductase enzymes in the cellular response to nitrosative stress. Free Radic. Biol. Med. 2018, 127, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Madeo, F.; Fröhlich, E.; Ligr, M.; Grey, M.; Sigrist, S.J.; Wolf, D.H.; Fröhlich, K.U. Oxygen stress: A regulator of apoptosis in yeast. J. Cell Biol. 1999, 145, 757–767. [Google Scholar] [CrossRef]
- Squier, T.C. Oxidative stress and protein aggregation during biological aging. Exp. Gerontol. 2001, 36, 1539–1550. [Google Scholar] [CrossRef]
- Fernando, R.; Drescher, C.; Nowotny, K.; Grune, T.; Castro, J.P. Impaired proteostasis during skeletal muscle aging. Free Radic. Biol. Med. 2019, 132, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Soares, T.R.; Reis, S.D.; Pinho, B.R.; Duchen, M.R.; Oliveira, J.M.A. Targeting the proteostasis network in Huntington’s disease. Ageing Res. Rev. 2019, 49, 92–103. [Google Scholar] [CrossRef]
- Mesika, R.; Reichmann, D. When safeguarding goes wrong: Impact of oxidative stress on protein homeostasis in health and neurodegenerative disorders. In Advances in Protein Chemistry and Structural Biology; Academic Press Inc.: Cambridge, MA, USA, 2019; Volume 114, pp. 221–264. ISBN 9780128155578. [Google Scholar]
- Butterfield, D.A. Amyloid beta-peptide (1-42)-induced oxidative stress and neurotoxicity: Implications for neurodegeneration in Alzheimer’s disease brain. A review. Free Radic. Res. 2002, 36, 1307–1313. [Google Scholar] [CrossRef]
- Jenner, P. Oxidative stress in Parkinson’s disease. Ann. Neurol. 2003, 53, S26–S38. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.V.; Hora, S.; Pal, A.; Jha, S.; Taneja, R. Stressing the (Epi)Genome: Dealing with Reactive Oxygen Species in Cancer. Antioxid. Redox Signal. 2017, 29, 1273–1292. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. The Free Radical Theory of Aging. Antioxid. Redox Signal. 2003, 5, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.-W.; Chen, Y.-C.; Hsieh, W.-L.; Chiou, S.-H.; Kao, C.-L. Ageing and neurodegenerative diseases. Ageing Res. Rev. 2010, 9, S36–S46. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, B.; Chang, C. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat. Chem. Biol. 2011, 7, 504–511. [Google Scholar] [CrossRef] [Green Version]
- Paiva, C.N.; Bozza, M.T. Are Reactive Oxygen Species Always Detrimental to Pathogens? Antioxid. Redox Signal. 2014, 20, 1000–1037. [Google Scholar] [CrossRef] [Green Version]
- Brandes, N.; Schmitt, S.; Jakob, U. Thiol-Based Redox Switches in Eukaryotic Proteins. Antioxid. Redox Signal. 2009, 11, 997–1014. [Google Scholar] [CrossRef]
- Rimon, O.; Suss, O.; Goldenberg, M.; Fassler, R.; Yogev, O.; Amartely, H.; Propper, G.; Friedler, A.; Reichmann, D. A role of metastable regions and their connectivity in the inactivation of a redox-regulated chaperone and its inter-chaperone crosstalk. Antioxid. Redox Signal. 2017, 27, 1252–1267. [Google Scholar] [CrossRef]
- Topf, U.; Suppanz, I.; Samluk, L.; Wrobel, L.; Böser, A.; Sakowska, P.; Knapp, B.; Pietrzyk, M.K.; Chacinska, A.; Warscheid, B. Quantitative proteomics identifies redox switches for global translation modulation by mitochondrially produced reactive oxygen species. Nat. Commun. 2018, 9, 324. [Google Scholar] [CrossRef]
- Knuesting, J.; Scheibe, R. Small Molecules Govern Thiol Redox Switches. Trends Plant Sci. 2018, 23, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A. Antioxidant Function of Thioredoxin and Glutaredoxin Systems. Antioxid. Redox Signal. 2000, 2, 811–820. [Google Scholar] [CrossRef]
- Rhee, S.G.; Kil, I.S. Multiple Functions and Regulation of Mammalian Peroxiredoxins. Annu. Rev. Biochem. 2017, 86, 749–775. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Benhar, M. Oxidants, Antioxidants and Thiol Redox Switches in the Control of Regulated Cell Death Pathways. Antioxidants 2020, 9, 309. [Google Scholar] [CrossRef] [Green Version]
- Liedgens, L.; Zimmermann, J.; Wäschenbach, L.; Geissel, F.; Laporte, H.; Gohlke, H.; Morgan, B.; Deponte, M. Quantitative assessment of the determinant structural differences between redox-active and inactive glutaredoxins. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Go, Y.M.; Roede, J.R.; Walker, D.I.; Duong, D.M.; Seyfried, N.T.; Orr, M.; Liang, Y.; Pennell, K.D.; Jones, D.P. Selective targeting of the cysteine proteome by thioredoxin and glutathione redox systems. Mol. Cell. Proteom. 2013, 12, 3285–3296. [Google Scholar] [CrossRef] [Green Version]
- Gould, N.S.; Evans, P.; Martínez-Acedo, P.; Marino, S.M.; Gladyshev, V.N.; Carroll, K.S.; Ischiropoulos, H. Site-Specific Proteomic Mapping Identifies Selectively Modified Regulatory Cysteine Residues in Functionally Distinct Protein Networks. Chem. Biol. 2015, 22, 965–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fomenko, D.E.; Marino, S.M.; Gladyshev, V.N. Functional diversity of cysteine residues in proteins and unique features of catalytic redox-active cysteines in thiol oxidoreductases. Mol. Cells 2008, 26, 228–235. [Google Scholar]
- Brandes, N.; Reichmann, D.; Tienson, H.; Leichert, L.I.; Jakob, U. Using quantitative redox proteomics to dissect the yeast redoxome. J. Biol. Chem. 2011, 286, 41893–41903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genest, O.; Wickner, S.; Doyle, S.M. Hsp90 and Hsp70 chaperones: Collaborators in protein remodeling. J. Biol. Chem. 2019, 294, 2109–2120. [Google Scholar] [CrossRef] [Green Version]
- Ulrich, K.; Schwappach, B.; Jakob, U. Thiol-based switching mechanisms of stress-sensing chaperones. Biol. Chem. 2020. [Google Scholar] [CrossRef]
- Wood, Z.A.; Schröder, E.; Harris, J.R.; Poole, L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003, 28, 32–40. [Google Scholar] [CrossRef]
- Jang, H.H.; Lee, K.O.; Chi, Y.H.; Jung, B.G.; Park, S.K.; Park, J.H.; Lee, J.R.; Lee, S.S.; Moon, J.C.; Yun, J.W.; et al. Two enzymes in one: Two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell 2004, 117, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Macdiarmid, C.W.; Taggart, J.; Kerdsomboon, K.; Kubisiak, M.; Panascharoen, S.; Schelble, K.; Eide, D.J. Peroxiredoxin chaperone activity is critical for protein homeostasis in zinc-deficient yeast. J. Biol. Chem. 2013, 288, 31313–31327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanzén, S.; Vielfort, K.; Yang, J.; Roger, F.; Andersson, V.; Zamarbide-Forés, S.; Andersson, R.; Malm, L.; Palais, G.; Biteau, B.; et al. Lifespan Control by Redox-Dependent Recruitment of Chaperones to Misfolded Proteins. Cell 2016, 166, 140–151. [Google Scholar] [CrossRef] [Green Version]
- Biteau, B.; Labarre, J.; Toledano, M.B. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature 2003, 425, 980–984. [Google Scholar] [CrossRef]
- Sunico, C.R.; Sultan, A.; Nakamura, T.; Dolatabadi, N.; Parker, J.; Shan, B.; Han, X.; Yates, J.R.; Masliah, E.; Ambasudhan, R.; et al. Role of sulfiredoxin as a peroxiredoxin-2 denitrosylase in human iPSC-derived dopaminergic neurons. Proc. Natl. Acad. Sci. USA 2016, 113, E7564–E7571. [Google Scholar] [CrossRef] [Green Version]
- Detienne, G.; De Haes, W.; Mergan, L.; Edwards, S.L.; Temmerman, L.; Van Bael, S. Beyond ROS clearance: Peroxiredoxins in stress signaling and aging. Ageing Res. Rev. 2018, 44, 33–48. [Google Scholar] [CrossRef]
- Kamariah, N.; Eisenhaber, B.; Eisenhaber, F.; Grüber, G. Molecular mechanism of the Escherichia coli AhpC in the function of a chaperone under heat-shock conditions. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, F.; Castro, H.; Cruz, T.; Tse, E.; Koldewey, P.; Southworth, D.R.; Tomás, A.M.; Jakob, U. Mitochondrial peroxiredoxin functions as crucial chaperone reservoir in Leishmania infantum. Proc. Natl. Acad. Sci. USA 2015, 112, E616–E624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saccoccia, F.; Di Micco, P.; Boumis, G.; Brunori, M.; Koutris, I.; Miele, A.E.; Morea, V.; Sriratana, P.; Williams, D.L.; Bellelli, A.; et al. Moonlighting by different stressors: Crystal structure of the chaperone species of a 2-Cys peroxiredoxin. Structure 2012, 20, 429–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zivanovic, J.; Kouroussis, E.; Kohl, J.B.; Adhikari, B.; Bursac, B.; Schott-Roux, S.; Petrovic, D.; Miljkovic, J.L.; Thomas-Lopez, D.; Jung, Y.; et al. Selective Persulfide Detection Reveals Evolutionarily Conserved Antiaging Effects of S-Sulfhydration. Cell Metab. 2019, 30, 1152–1170.e13. [Google Scholar] [CrossRef]
- Gu, F.; Duc, T.N.; Stuible, M.; Dubé, N.; Tremblay, M.L.; Chevet, E. Protein-tyrosine phosphatase 1B potentiates IRE1 signaling during endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 49689–49693. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Muramatsu, A.; Saito, R.; Iso, T.; Shibata, T.; Kuwata, K.; Kawaguchi, S.; Iwawaki, T.; Adachi, S.; Suda, H.; et al. Molecular Mechanism of Cellular Oxidative Stress Sensing by Keap1. Cell Rep. 2019, 28, 746–758.e4. [Google Scholar] [CrossRef] [Green Version]
- Dóka, É.; Ida, T.; Dagnell, M.; Abiko, Y.; Luong, N.C.; Balog, N.; Takata, T.; Espinosa, B.; Nishimura, A.; Cheng, Q.; et al. Control of protein function through oxidation and reduction of persulfidated states. Sci. Adv. 2020, 6, eaax8358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharapov, M.G.; Novoselov, V.I.; Penkov, N.V.; Fesenko, E.E.; Vedunova, M.V.; Bruskov, V.I.; Gudkov, S.V. Protective and adaptogenic role of peroxiredoxin 2 (Prx2) in neutralization of oxidative stress induced by ionizing radiation. Free Radic. Biol. Med. 2019, 134, 76–86. [Google Scholar] [CrossRef]
- Ding, C.; Fan, X.; Wu, G. Peroxiredoxin 1—An antioxidant enzyme in cancer. J. Cell. Mol. Med. 2017, 21, 193–202. [Google Scholar] [CrossRef]
- McCaffrey, K.; Braakman, I. Protein quality control at the endoplasmic reticulum. Essays Biochem. 2016, 60, 227–235. [Google Scholar] [CrossRef]
- Aramin, S.; Fassler, R.; Chikne, V.; Goldenberg, M.; Arian, T.; Kolet Eliaz, L.; Rimon, O.; Ram, O.; Michaeli, S.; Reichmann, D. TrypOx, a Novel Eukaryotic Homolog of the Redox-Regulated Chaperone Hsp33 in Trypanosoma brucei. Front. Microbiol. 2020, 11, 1844. [Google Scholar] [CrossRef]
- Ilbert, M.; Horst, J.; Ahrens, S.; Winter, J.; Graf, P.C.F.; Lilie, H.; Jakob, U. The redox-switch domain of Hsp33 functions as dual stress sensor. Nat. Struct. Mol. Biol. 2007, 14, 556–563. [Google Scholar] [CrossRef] [Green Version]
- Winter, J.; Ilbert, M.; Graf, P.C.F.; Özcelik, D.; Jakob, U. Bleach Activates a Redox-Regulated Chaperone by Oxidative Protein Unfolding. Cell 2008, 135, 691–701. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.L.; Bohovych, I.; Wong, E.O.Y.; Lambert, J.P.; Gingras, A.C.; Khalimonchuk, O.; Cowen, L.E.; Leach, M.D. Ydj1 governs fungal morphogenesis and stress response, and facilitates mitochondrial protein import via Mas1 and Mas2. Microb. Cell 2017, 4, 342–361. [Google Scholar] [CrossRef]
- Reichmann, D.; Xu, Y.; Cremers, C.M.; Ilbert, M.; Mittelman, R.; Fitzgerald, M.C.; Jakob, U. Order out of disorder: Working cycle of an intrinsically unfolded chaperone. Cell 2012, 148, 947–957. [Google Scholar] [CrossRef] [Green Version]
- Groitl, B.; Horowitz, S.; Makepeace, K.A.T.; Petrotchenko, E.V.; Borchers, C.H.; Reichmann, D.; Bardwell, J.C.A.; Jakob, U. Protein unfolding as a switch from self-recognition to high-affinity client binding. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef]
- Cremers, C.M.; Reichmann, D.; Hausmann, J.; Ilbert, M.; Jakob, U. Unfolding of metastable linker region is at the core of Hsp33 activation as a redox-regulated chaperone. J. Biol. Chem. 2010, 285, 11243–11251. [Google Scholar] [CrossRef] [Green Version]
- Goemans, C.V.; Beaufay, F.; Arts, I.S.; Agrebi, R.; Vertommen, D.; Collet, J.F. The Chaperone and Redox Properties of CnoX Chaperedoxins Are Tailored to the Proteostatic Needs of Bacterial Species. MBio 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdős, G.; Mészáros, B.; Reichmann, D.; Dosztányi, Z. Large-Scale Analysis of Redox-Sensitive Conditionally Disordered Protein Regions Reveals Their Widespread Nature and Key Roles in High-Level Eukaryotic Processes. Proteomics 2019, 19, 1800070. [Google Scholar] [CrossRef]
- Leichert, L.I.; Gehrke, F.; Gudiseva, H.V.; Blackwell, T.; Ilbert, M.; Walker, A.K.; Strahler, J.R.; Andrews, P.C.; Jakob, U. Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 8197–8202. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Jedrychowski, M.P.; Schweppe, D.K.; Huttlin, E.L.; Yu, Q.; Heppner, D.E.; Li, J.; Long, J.; Mills, E.L.; Szpyt, J.; et al. A Quantitative Tissue-Specific Landscape of Protein Redox Regulation during Aging. Cell 2020, 180, 968–983.e24. [Google Scholar] [CrossRef]
- McDonagh, B.; Sakellariou, G.K.; Smith, N.T.; Brownridge, P.; Jackson, M.J. Differential cysteine labeling and global label-free proteomics reveals an altered metabolic state in skeletal muscle aging. J. Proteome Res. 2014, 13, 5008–5021. [Google Scholar] [CrossRef]
- Rosenwasser, S.; Van Creveld, S.G.; Schatz, D.; Malitsky, S.; Tzfadia, O.; Aharoni, A.; Levin, Y.; Gabashvili, A.; Feldmesser, E.; Vardi, A. Mapping the diatom redox-sensitive proteome provides insight into response to nitrogen stress in the marine environment. Proc. Natl. Acad. Sci. USA 2014, 111, 2740–2745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Carroll, K.S.; Liebler, D.C. The expanding landscape of the thiol redox proteome. Mol. Cell. Proteom. 2016, 15, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Carroll, K.S. Activity-Based Sensing for Site-Specific Proteomic Analysis of Cysteine Oxidation. Acc. Chem. Res. 2020, 53, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.H.; Li, L.; Parisien, M.; Wu, J.; Bederman, I.; Gao, Z.; Krokowski, D.; Chirieleison, S.M.; Abbott, D.; Wang, B.; et al. Discovery of a redox thiol switch: Implications for cellular energy metabolism. Mol. Cell. Proteom. 2020, 19, 852–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerashchenko, M.V.; Lobanov, A.V.; Gladyshev, V.N. Genome-wide ribosome profiling reveals complex translational regulation in response to oxidative stress. Proc. Natl. Acad. Sci. USA 2012, 109, 17394–17399. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.M.; Kim, H.G.; Son, C.G. Tissue-specific profiling of oxidative stress-associated transcriptome in a healthy mouse model. Int. J. Mol. Sci. 2018, 19, 3174. [Google Scholar] [CrossRef] [Green Version]
- Radzinski, M.; Fassler, R.; Yogev, O.; Breuer, W.; Shai, N.; Gutin, J.; Ilyas, S.; Geffen, Y.; Tsytkin-Kirschenzweig, S.; Nahmias, Y.; et al. Temporal profiling of redox-dependent heterogeneity in single cells. Elife 2018, 7, e37623. [Google Scholar] [CrossRef]
- Ayer, A.; Fellermeier, S.; Fife, C.; Li, S.S.; Smits, G.; Meyer, A.J.; Dawes, I.W.; Perrone, G.G. A Genome-Wide Screen in Yeast Identifies Specific Oxidative Stress Genes Required for the Maintenance of Sub-Cellular Redox Homeostasis. PLoS ONE 2012, 7, e44278. [Google Scholar] [CrossRef]
- Kim, H.H.-J.; Ha, S.; Lee, H.Y.; Lee, K.-J.K. ROSics: Chemistry and proteomics of cysteine modifications in redox biology. Mass Spectrom. Rev. 2015, 34, 184–208. [Google Scholar] [CrossRef] [Green Version]
- Rizza, S.; Montagna, C.; Cardaci, S.; Maiani, E.; Di Giacomo, G.; Sanchez-Quiles, V.; Blagoev, B.; Rasola, A.; De Zio, D.; Stamler, J.S.; et al. S-nitrosylation of the mitochondrial chaperone TRAP1 sensitizes hepatocellular carcinoma cells to inhibitors of succinate dehydrogenase. Cancer Res. 2016, 76, 4170–4182. [Google Scholar] [CrossRef] [Green Version]
- Uehara, T.; Nakamura, T.; Yao, D.; Shi, Z.Q.; Gu, Z.; Ma, Y.; Masliah, E.; Nomura, Y.; Lipton, S.A. S-Nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 2006, 441, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.K.Y.; Davey, M.; Sun, B.; Roth, A.F.; Davis, N.G.; Conibear, E. Palmitoylation by the DHHC protein Pfa4 regulates the ER exit of Chs3. J. Cell Biol. 2006, 174, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Savitsky, P.A.; Finkel, T. Redox regulation of Cdc25C. J. Biol. Chem. 2002, 277, 20535–20540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, H.; Toma-Fukai, S.; Kontani, K.; Katada, T.; Shimizu, T.; Goody, R.S. GEF mechanism revealed by the structure of SmgGDS-558 and farnesylated RhoA complex and its implication for a chaperone mechanism. Proc. Natl. Acad. Sci. USA 2018, 115, 9563–9568. [Google Scholar] [CrossRef] [Green Version]
- Resh, M.D. Trafficking and signaling by fatty-acylated and prenylated proteins. Nat. Chem. Biol. 2006, 2, 584–590. [Google Scholar] [CrossRef]
- Abrami, L.; Atrice Kunz, B.; Iacovache, I.; Gisou Van Der Goot, F. Palmitoylation and Ubiquitination Regulate Exit of the Wnt Signaling Protein LRP6 from the Endoplasmic Reticulum. Proc. Natl. Acad. Sci. USA 2008, 105, 5384–5389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diez-Ardanuy, C.; Greaves, J.; Munro, K.R.; Tomkinson, N.C.O.; Chamberlain, L.H. A cluster of palmitoylated cysteines are essential for aggregation of cysteine-string protein mutants that cause neuronal ceroid lipofuscinosis. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Ernst, A.M.; Syed, S.A.; Zaki, O.; Bottanelli, F.; Zheng, H.; Hacke, M.; Xi, Z.; Rivera-Molina, F.; Graham, M.; Rebane, A.A.; et al. S-Palmitoylation Sorts Membrane Cargo for Anterograde Transport in the Golgi. Dev. Cell 2018, 47, 479–493.e7. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Brodsky, J.L. Protein quality control in the secretory pathway. J. Cell Biol. 2019, 218, 3171–3187. [Google Scholar] [CrossRef] [Green Version]
- Berndt, N.; Hamilton, A.D.; Sebti, S.M. Targeting protein prenylation for cancer therapy. Nat. Rev. Cancer 2011, 11, 775–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrera, M.; Boronat, S.; Marte, L.; Vega, M.; Pérez, P.; Ayté, J.; Hidalgo, E. Chaperone-Facilitated Aggregation of Thermo-Sensitive Proteins Shields Them from Degradation during Heat Stress. Cell Rep. 2020, 30, 2430–2443.e4. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Lipton, S.A. Nitric oxide-dependent protein post-translational modifications impair mitochondrial function and metabolism to contribute to neurodegenerative diseases. Antioxid. Redox Signal. 2020, 32, 817–833. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Ruiz, A.; Lamas, S. S-nitrosylation: A potential new paradigm in signal transduction. Cardiovasc. Res. 2004, 62, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Ogura, J.; Ruddock, L.W.; Mano, N. Cysteine 343 in the substrate binding domain is the primary S-Nitrosylated site in protein disulfide isomerase. Free Radic. Biol. Med. 2020, 160, 103–110. [Google Scholar] [CrossRef]
- Wang, K.; Liu, J.Q.; Zhong, T.; Liu, X.L.; Zeng, Y.; Qiao, X.; Xie, T.; Chen, Y.; Gao, Y.Y.; Tang, B.; et al. Phase Separation and Cytotoxicity of Tau are Modulated by Protein Disulfide Isomerase and S-nitrosylation of this Molecular Chaperone. J. Mol. Biol. 2020, 432, 2141–2163. [Google Scholar] [CrossRef] [PubMed]
- Astier, J.; Besson-Bard, A.; Lamotte, O.; Bertoldo, J.; Bourque, S.; Terenzi, H.; Wendehenne, D. Nitric oxide inhibits the ATPase activity of the chaperone-like AAA + ATPase CDC48, a target for S-nitrosylation in cryptogein signalling in tobacco cells. Biochem. J. 2012, 447, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, M.; Takata, T.; Kimura, Y.; Manno, A.; Murakami, K.; Koike, M.; Ohizumi, H.; Hori, S.; Kakizuka, A. ATPase activity of p97/valosin-containing protein is regulated by oxidative modification of the evolutionally conserved cysteine 522 residue in walker a motif. J. Biol. Chem. 2005. [Google Scholar] [CrossRef] [Green Version]
- Fang, C.; Zhang, X.; Zhang, L.; Gao, X.; Yang, P.; Lu, H. Identification of Palmitoylated Transitional Endoplasmic Reticulum ATPase by Proteomic Technique and Pan Antipalmitoylation Antibody. J. Proteome Res. 2016, 15, 956–962. [Google Scholar] [CrossRef]
- Kleizen, B.; Braakman, I. Protein folding and quality control in the endoplasmic reticulum. Curr. Opin. Cell Biol. 2004, 16, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevier, C.S.; Kaiser, C.A. Ero1 and redox homeostasis in the endoplasmic reticulum. Biochim. Biophys. Acta Mol. Cell Res. 2008, 1783, 549–556. [Google Scholar] [CrossRef] [Green Version]
- Ellgaard, L.; Ruddock, L.W. The human protein disulphide isomerase family: Substrate interactions and functional properties. EMBO Rep. 2005, 6, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.K.; Farg, M.A.; Bye, C.R.; McLean, C.A.; Horne, M.K.; Atkin, J.D. Protein disulphide isomerase protects against protein aggregation and is S-nitrosylated in amyotrophic lateral sclerosis. Brain 2010, 133, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Eletto, D.; Chevet, E.; Argon, Y.; Appenzeller-Herzog, C. Redox controls UPR to control redox. J. Cell Sci. 2014, 127, 3649–3658. [Google Scholar] [CrossRef] [Green Version]
- Chadwick, S.R.; Fazio, E.N.; Etedali-Zadeh, P.; Genereaux, J.; Duennwald, M.L.; Lajoie, P. A functional unfolded protein response is required for chronological aging in Saccharomyces cerevisiae. Curr. Genet. 2019, 66, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Teuber, V.; Albert-Gasco, H.; Auyeung, V.C.; Papa, F.R.; Mallucci, G.R.; Hetz, C. Small Molecules to Improve ER Proteostasis in Disease. Trends Pharmacol. Sci. 2019, 40, 684–695. [Google Scholar] [CrossRef]
- Hourihan, J.M.; Moronetti Mazzeo, L.E.; Fernández-Cárdenas, L.P.; Blackwell, T.K. Cysteine Sulfenylation Directs IRE-1 to Activate the SKN-1/Nrf2 Antioxidant Response. Mol. Cell 2016, 63, 553–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra-Moreno, A.; Ang, J.; Welsch, H.; Jochem, M.; Hanna, J. Regulation of the unfolded protein response in yeast by oxidative stress. FEBS Lett. 2019, 593, 1080–1088. [Google Scholar] [CrossRef]
- Eletto, D.; Eletto, D.; Dersh, D.; Gidalevitz, T.; Argon, Y. Protein Disulfide Isomerase A6 Controls the Decay of IRE1α Signaling via Disulfide-Dependent Association. Mol. Cell 2014, 53, 562–576. [Google Scholar] [CrossRef] [Green Version]
- Deshmukh, F.K.; Yaffe, D.; Olshina, M.A.; Ben-Nissan, G.; Sharon, M. The contribution of the 20s proteasome to proteostasis. Biomolecules 2019, 9, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, G.M.; Netto, L.E.S.; Simões, V.; Santos, L.F.A.; Gozzo, F.C.; Demasi, M.A.A.; Oliveira, C.L.P.; Bicev, R.N.; Klitzke, C.F.; Sogayar, M.C.; et al. Redox control of 20S proteasome gating. Antioxid. Redox Signal. 2012, 16, 1183–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiken, C.T.; Kaake, R.M.; Wang, X.; Huang, L. Oxidative Stress-Mediated Regulation of Proteasome Complexes. Mol. Cell. Proteom. 2011. [Google Scholar] [CrossRef] [Green Version]
- Shringarpure, R.; Grune, T.; Mehlhase, J.; Davies, K.J.A. Ubiquitin conjugation is not required for the degradation of oxidized proteins by proteasome. J. Biol. Chem. 2003, 278, 311–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, K.J.A. Degradation of oxidized proteins by the 20S proteasome. In Proceedings of the Biochimie; Elsevier Masson SAS: Amsterdam, The Netherlands, 2001; Volume 83, pp. 301–310. [Google Scholar]
- Yu, Z.; Yu, Y.; Wang, F.; Myasnikov, A.G.; Coffino, P.; Cheng, Y. Allosteric coupling between α-rings of the 20S proteasome. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, A.; Bertolotti, A. Regulation of proteasome assembly and activity in health and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 697–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peth, A.; Uchiki, T.; Goldberg, A.L. ATP-Dependent steps in the binding of ubiquitin conjugates to the 26s proteasome that commit to degradation. Mol. Cell 2010, 40, 671–681. [Google Scholar] [CrossRef] [Green Version]
- Aufderheide, A.; Beck, F.; Stengel, F.; Hartwig, M.; Schweitzer, A.; Pfeifer, G.; Goldberg, A.L.; Sakata, E.; Baumeister, W.; Förster, F. Structural characterization of the interaction of Ubp6 with the 26S proteasome. Proc. Natl. Acad. Sci. USA 2015, 112, 8626–8631. [Google Scholar] [CrossRef] [Green Version]
- Finley, D.; Chen, X.; Walters, K.J. Gates, Channels, and Switches: Elements of the Proteasome Machine. Trends Biochem. Sci. 2016, 41, 77–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husnjak, K.; Elsasser, S.; Zhang, N.; Chen, X.; Randles, L.; Shi, Y.; Hofmann, K.; Walters, K.J.; Finley, D.; Dikic, I. Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 2008, 453, 481–488. [Google Scholar] [CrossRef] [Green Version]
- Richly, H.; Rape, M.; Braun, S.; Rumpf, S.; Hoege, C.; Jentsch, S. A Series of Ubiquitin Binding Factors Connects CDC48/p97 to Substrate Multiubiquitylation and Proteasomal Targeting. Cell 2005, 120, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, H.X.; Chen, W.; Hong, S.T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benaroudj, N.; Zwickl, P.; Seemüller, E.; Baumeister, W.; Goldberg, A.L. ATP hydrolysis by the proteasome regulatory complex PAN serves multiple functions in protein degradation. Mol. Cell 2003, 11, 69–78. [Google Scholar] [CrossRef]
- Groll, M.; Ditzel, L.; Löwe, J.; Stock, D.; Bochtler, M.; Bartunik, H.D.; Huber, R. Structure of 20S proteasome from yeast at 2.4 Å resolution. Nature 1997, 386, 463–471. [Google Scholar] [CrossRef]
- Wang, X.; Yen, J.; Kaiser, P.; Huang, L. Regulation of the 26S proteasome complex during oxidative stress. Sci. Signal. 2010, 3, ra88. [Google Scholar] [CrossRef] [Green Version]
- Haratake, K.; Sato, A.; Tsuruta, F.; Chiba, T. KIAA0368-deficiency affects disassembly of 26S proteasome under oxidative stress condition. J. Biochem. 2016, 159, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Chemmama, I.E.; Yu, C.; Huszagh, A.; Xu, Y.; Viner, R.; Block, S.A.; Cimermancic, P.; Rychnovsky, S.D.; Ye, Y.; et al. The proteasome-interacting Ecm29 protein disassembles the 26S proteasome in response to oxidative stress. J. Biol. Chem. 2017, 292, 16310–16320. [Google Scholar] [CrossRef] [Green Version]
- Grune, T.; Catalgol, B.; Licht, A.; Ermak, G.; Pickering, A.M.; Ngo, J.K.; Davies, K.J.A. HSP70 mediates dissociation and reassociation of the 26S proteasome during adaptation to oxidative stress. Free Radic. Biol. Med. 2011, 51, 1355–1364. [Google Scholar] [CrossRef] [Green Version]
- Moscovitz, O.; Ben-Nissan, G.; Fainer, I.; Pollack, D.; Mizrachi, L.; Sharon, M. The Parkinson’s-associated protein DJ-1 regulates the 20S proteasome. Nat. Commun. 2015, 6, 6609. [Google Scholar] [CrossRef] [Green Version]
- Moscovitz, O.; Tsvetkov, P.; Hazan, N.; Michaelevski, I.; Keisar, H.; Ben-Nissan, G.; Shaul, Y.; Sharon, M. A Mutually Inhibitory Feedback Loop between the 20S Proteasome and Its Regulator, NQO1. Mol. Cell 2012, 47, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Olshina, M.A.; Arkind, G.; Kumar Deshmukh, F.; Fainer, I.; Taranavsky, M.; Hayat, D.; Ben-Dor, S.; Ben-Nissan, G.; Sharon, M. Regulation of the 20S Proteasome by a Novel Family of Inhibitory Proteins. Antioxid. Redox Signal. 2020, 32, 636–655. [Google Scholar] [CrossRef] [Green Version]
- Abi Habib, J.; De Plaen, E.; Stroobant, V.; Zivkovic, D.; Bousquet, M.P.; Guillaume, B.; Wahni, K.; Messens, J.; Busse, A.; Vigneron, N.; et al. Efficiency of the four proteasome subtypes to degrade ubiquitinated or oxidized proteins. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Myers, N.; Eliav, R.; Adamovich, Y.; Hagai, T.; Adler, J.; Navon, A.; Shaul, Y. NADH Binds and stabilizes the 26S proteasomes independent of ATP. J. Biol. Chem. 2014, 289, 11272–11281. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Powell, S.R.; Wang, X. Enhancement of proteasome function by PA28α overexpression protects against oxidative stress. FASEB J. 2011, 25, 883–893. [Google Scholar] [CrossRef] [Green Version]
- Jung, T.; Höhn, A.; Grune, T. The proteasome and the degradation of oxidized proteins: Part III-Redox regulation of the proteasomal system. Redox Biol. 2014, 2, 388–394. [Google Scholar] [CrossRef] [Green Version]
- Haas, A.L.; Rose, I.A. The Mechanism of Ubiquitin Activating Enzyme. J. Biol. Chem. 1982, 257, 10329–10337. [Google Scholar] [CrossRef]
- Shang, F.; Taylor, A. Ubiquitin-proteasome pathway and cellular responses to oxidative stress. Free Radic. Biol. Med. 2011, 51, 5–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.D.; Hannink, M. Distinct Cysteine Residues in Keap1 Are Required for Keap1-Dependent Ubiquitination of Nrf2 and for Stabilization of Nrf2 by Chemopreventive Agents and Oxidative Stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stankovic-Valentin, N.; Melchior, F. Control of SUMO and Ubiquitin by ROS: Signaling and disease implications. Mol. Asp. Med. 2018, 63, 3–17. [Google Scholar] [CrossRef]
- Yao, D.; Gu, Z.; Nakamura, T.; Shi, Z.-Q.; Ma, Y.; Gaston, B.; Palmer, L.A.; Rockenstein, E.M.; Zhang, Z.; Uehara, T.; et al. Nitrosative Stress Linked to Sporadic Parkinson’s Disease: S-Nitrosylation of Parkin Regulates Its E3 Ubiquitin Ligase Activity. Proc. Natl. Acad. Sci. USA 2004, 101, 10810–10814. [Google Scholar] [CrossRef] [Green Version]
- Ozawa, K.; Komatsubara, A.T.; Nishimura, Y.; Sawada, T.; Kawafune, H.; Tsumoto, H.; Tsuji, Y.; Zhao, J.; Kyotani, Y.; Tanaka, T.; et al. S-nitrosylation regulates mitochondrial quality control via activation of parkin. Sci. Rep. 2013, 3, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, F.; Yao, D.; Shi, Y.; Kabakoff, J.; Wu, W.; Reicher, J.; Ma, Y.; Moosmann, B.; Masliah, E.; Lipton, S.A.; et al. Oxidation of the cysteine-rich regions of parkin perturbs its E3 ligase activity and contributes to protein aggregation. Mol. Neurodegener. 2011, 6, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Marín, I.; Ferrúst, A. Comparative genomics of the RBR family, including the Parkinson’s disease-related gene parkin and the genes of the ariadne subfamily. Mol. Biol. Evol. 2002, 19, 2039–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Gao, J.; Chung, K.K.K.; Huang, H.; Dawson, V.L.; Dawson, T.M. Parkin functions as an E2-dependent ubiquitin-protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc. Natl. Acad. Sci. USA 2000, 97, 13354–13359. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Soda, M.; Takahashi, R. Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J. Biol. Chem. 2000, 275, 35661–35664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimura, H.; Hattori, N.; Kubo, S.I.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000, 25, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Hristova, V.A.; Beasley, S.A.; Rylett, R.J.; Shaw, G.S. Identification of a novel Zn2+-binding domain in the autosomal recessive juvenile Parkinson-related E3 ligase parkin. J. Biol. Chem. 2009, 284, 14978–14986. [Google Scholar] [CrossRef] [Green Version]
- Joselin, A.P.; Hewitt, S.J.; Callaghan, S.M.; Kim, R.H.; Chung, Y.H.; Mak, T.W.; Shen, J.; Slack, R.S.; Park, D.S. ROS-dependent regulation of parkin and DJ-1 localization during oxidative stress in neurons. Hum. Mol. Genet. 2012, 21, 4888–4903. [Google Scholar] [CrossRef] [Green Version]
- Canet-Avilés, R.M.; Wilson, M.A.; Miller, D.W.; Ahmad, R.; McLendon, C.; Bandyopadhyay, S.; Baptista, M.J.; Ringe, D.; Petsko, G.A.; Cookson, M.R. The Parkinson’s disease DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc. Natl. Acad. Sci. USA 2004, 101, 9103–9108. [Google Scholar] [CrossRef] [Green Version]
- Irrcher, I.; Aleyasin, H.; Seifert, E.L.; Hewitt, S.J.; Chhabra, S.; Phillips, M.; Lutz, A.K.; Rousseaux, M.W.C.; Bevilacqua, L.; Jahani-Asl, A.; et al. Loss of the Parkinson’s disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum. Mol. Genet. 2010, 19, 3734–3746. [Google Scholar] [CrossRef] [Green Version]
- Pick, E. The necessity of NEDD8/Rub1 for vitality and its association with mitochondria-derived oxidative stress. Redox Biol. 2020, 37, 101765. [Google Scholar] [CrossRef] [PubMed]
- Bossis, G.; Melchior, F. Regulation of SUMOylation by reversible oxidation of SUMO conjugating enzymes. Mol. Cell 2006, 21, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Stankovic-Valentin, N.; Drzewicka, K.; König, C.; Schiebel, E.; Melchior, F. Redox regulation of SUMO enzymes is required for ATM activity and survival in oxidative stress. EMBO J. 2016, 35, 1312–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, S.; Sun, X.; Xiang, B.; Cang, H.; Kang, X.; Chen, Y.; Li, H.; Shi, G.; Yeh, E.T.H.; Wang, B.; et al. Redox regulation of the stability of the SUMO protease SENP3 via interactions with CHIP and Hsp90. EMBO J. 2010, 29, 3773–3786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Han, Y.; Wang, Y.; Sun, X.; Yan, S.; Yeh, E.T.H.; Chen, Y.; Cang, H.; Li, H.; Shi, G.; et al. SENP3 is responsible for HIF-1 transactivation under mild oxidative stress via p300 de-SUMOylation. EMBO J. 2009, 28, 2748–2762. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Lam, L.S.M.; Lam, L.H.; Chau, S.F.; Ng, T.B.; Au, S.W.N. Molecular basis of the redox regulation of SUMO proteases: A protective mechanism of intermolecular disulfide linkage against irreversible sulfhydryl oxidation. FASEB J. 2008, 22, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Hickey, C.M.; Wilson, N.R.; Hochstrasser, M. Function and regulation of SUMO proteases. Nat. Rev. Mol. Cell Biol. 2012, 13, 755–766. [Google Scholar] [CrossRef] [Green Version]
- Bramasole, L.; Sinha, A.; Gurevich, S.; Radzinski, M.; Klein, Y.; Panat, N.; Gefen, E.; Rinaldi, T.; Jimenez-Morales, D.; Johnson, J.; et al. Proteasome lid bridges mitochondrial stress with Cdc53/Cullin1 NEDDylation status. Redox Biol. 2019, 20, 533–543. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef]
- Moreno, D.F.; Aldea, M. Proteostatic Stress as a Nodal Hallmark of Replicative Aging. Exp. Cell Res. 2020, 394, 112163. [Google Scholar] [CrossRef] [PubMed]
- Leupold, S.; Hubmann, G.; Litsios, A.; Meinema, A.C.; Takhaveev, V.; Papagiannakis, A.; Niebel, B.; Janssens, G.; Siegel, D.; Heinemann, M. Saccharomyces cerevisiae goes through distinct metabolic phases during its replicative lifespan. Elife 2019, 8, e41046. [Google Scholar] [CrossRef]
- He, X.; Memczak, S.; Qu, J.; Belmonte, J.C.I.; Liu, G.H. Single-cell omics in ageing: A young and growing field. Nat. Metab. 2020, 2, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Brandes, N.; Tienson, H.; Lindemann, A.; Vitvitsky, V.; Reichmann, D.; Banerjee, R.; Jakob, U. Time line of redox events in aging postmitotic cells. Elife 2013, 2013, e306. [Google Scholar] [CrossRef]
- Ghosh, D.K.; Roy, A.; Ranjan, A. The ATPase VCP/p97 functions as a disaggregase against toxic Huntingtin-exon1 aggregates. FEBS Lett. 2018, 592, 2680–2692. [Google Scholar] [CrossRef] [Green Version]
- Baek, G.H.; Cheng, H.; Choe, V.; Bao, X.; Shao, J.; Luo, S.; Rao, H. Cdc48: A Swiss Army Knife of Cell Biology. J. Amino Acids 2013, 2013, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Tran, J.R.; Brodsky, J.L. The Cdc48–Vms1 complex maintains 26S proteasome architecture. Biochem. J. 2014, 458, 459–467. [Google Scholar] [CrossRef] [Green Version]
- Saarikangas, J.; Caudron, F.; Prasad, R.; Moreno, D.F.; Bolognesi, A.; Aldea, M.M.; Barral, Y. Compartmentalization of ER-Bound Chaperone Confines Protein Deposit Formation to the Aging Yeast Cell. Curr. Biol. 2017, 27, 773–783. [Google Scholar] [CrossRef] [Green Version]
- Caplan, A.J.; Douglas, M.G. Characterization of YDJ1: A yeast homologue of the bacterial dnaJ protein. J. Cell Biol. 1991, 114, 609–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, S.K.; Lima, C.D. Structure of a Ubiquitin E1-E2 Complex: Insights to E1-E2 Thioester Transfer. Mol. Cell 2013, 49, 884–896. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.S.; Li, M.X.; Liu, J.; Jiao, H.; Xia, J.M.; Shi, X.J.; Zhao, H.; Chu, L.; Liu, J.; Qi, W.; et al. Competitive oxidation and ubiquitylation on the evolutionarily conserved cysteine confer tissue-specific stabilization of Insig-2. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Kolawa, N.; Sweredoski, M.J.; Graham, R.L.J.; Oania, R.; Hess, S.; Deshaies, R.J. Perturbations to the ubiquitin conjugate proteome in yeast δubx mutants identify Ubx2 as a regulator of membrane lipid composition. Mol. Cell. Proteom. 2013, 12, 2791–2803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshihara, E.; Masaki, S.; Matsuo, Y.; Chen, Z.; Tian, H.; Yodoi, J. Thioredoxin/Txnip: Redoxisome, as a redox switch for the pathogenesis of diseases. Front. Immunol. 2013, 4, 514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manohar, S.; Jacob, S.; Wang, J.; Wiechecki, K.A.; Koh, H.W.L.; Simões, V.; Choi, H.; Vogel, C.; Silva, G.M. Polyubiquitin Chains Linked by Lysine Residue 48 (K48) Selectively Target Oxidized Proteins in Vivo. Antioxid. Redox Signal. 2019, 31, 1133–1149. [Google Scholar] [CrossRef]
- Shcherbik, N.; Pestov, D.G. The Impact of Oxidative Stress on Ribosomes: From Injury to Regulation. Cells 2019, 8, 1379. [Google Scholar] [CrossRef] [Green Version]
- Kumsta, C.; Thamsen, M.; Jakob, U. Effects of oxidative stress on behavior, physiology, and the redox thiol proteome of Caenorhabditis elegans. Antioxid. Redox Signal. 2011, 14, 1023–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Li, Y.; Qin, Z.; Guo, S.; Li, Y.; Miao, Y.; Song, C.; Chen, S.; Dai, S. Plant Chloroplast Stress Response: Insights from Thiol Redox Proteomics. Antioxid. Redox Signal. 2020, 33, 35–57. [Google Scholar] [CrossRef] [PubMed]
- Menger, K.E.; James, A.M.; Cochemé, H.M.; Harbour, M.E.; Chouchani, E.T.; Ding, S.; Fearnley, I.M.; Partridge, L.; Murphy, M.P. Fasting, but Not Aging, Dramatically Alters the Redox Status of Cysteine Residues on Proteins in Drosophila melanogaster. Cell Rep. 2015, 11, 1856–1865. [Google Scholar] [CrossRef] [Green Version]
- Petrova, B.; Liu, K.; Tian, C.; Kitaoka, M.; Freinkman, E.; Yang, J.; Orr-Weaver, T.L. Dynamic redox balance directs the oocyte-to-embryo transition via developmentally controlled reactive cysteine changes. Proc. Natl. Acad. Sci. USA 2018, 115, E7978–E7986. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Yi, M.; Liu, Y.; Wang, Q.; Hu, Y.; Deng, H. Glutaredoxin-1 Silencing Induces Cell Senescence via p53/p21/p16 Signaling Axis. J. Proteome Res. 2018, 17, 1091–1100. [Google Scholar] [CrossRef]
- Chiu, J.; Dawes, I.W. Redox control of cell proliferation. Trends Cell Biol. 2012, 22, 592–601. [Google Scholar] [CrossRef]
- Foyer, C.H.; Wilson, M.H.; Wright, M.H. Redox regulation of cell proliferation: Bioinformatics and redox proteomics approaches to identify redox-sensitive cell cycle regulators. Free Radic. Biol. Med. 2018, 122, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Ishibashi, S.; Iglesias-Gonzalez, J.; Chen, Y.; Love, N.R.; Amaya, E. Ca2+-Induced Mitochondrial ROS Regulate the Early Embryonic Cell Cycle. Cell Rep. 2018, 22, 218–231. [Google Scholar] [CrossRef] [Green Version]
- Patterson, J.C.; Joughin, B.A.; van de Kooij, B.; Lim, D.C.; Lauffenburger, D.A.; Yaffe, M.B. ROS and Oxidative Stress Are Elevated in Mitosis during Asynchronous Cell Cycle Progression and Are Exacerbated by Mitotic Arrest. Cell Syst. 2019, 8, 163–167.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Göbl, C.; Morris, V.K.; van Dam, L.; Visscher, M.; Polderman, P.E.; Hartlmüller, C.; de Ruiter, H.; Hora, M.; Liesinger, L.; Birner-Gruenberger, R.; et al. Cysteine oxidation triggers amyloid fibril formation of the tumor suppressor p16INK4A. Redox Biol. 2020, 28, 101316. [Google Scholar] [CrossRef] [PubMed]
- Laun, P.; Pichova, A.; Madeo, F.; Fuchs, J.; Ellinger, A.; Kohlwein, S.; Dawes, I.; Fröhlich, K.-U.; Breitenbach, M. Aged mother cells of Saccharomyces cerevisiae show markers of oxidative stress and apoptosis. Mol. Microbiol. 2004, 39, 1166–1173. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef] [PubMed]
- Marengo, B.; Nitti, M.; Furfaro, A.L.; Colla, R.; De Ciucis, C.; Marinari, U.M.; Pronzato, M.A.; Traverso, N.; Domenicotti, C. Redox homeostasis and cellular antioxidant systems: Crucial players in cancer growth and therapy. Oxid. Med. Cell. Longev. 2016, 2016, 6235641. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | Organism(s) | Type | Reactive Cysteine(s) | Activating Oxidant | Additional Information | References |
|---|---|---|---|---|---|---|
| Hsp33 | Bacteria Trypanosoma Leishmania | ATP-independent | 232, 234, 265, 268 (E. coli) | H2O2 HOCl | [21,122,123,124] | |
| CnoX | Bacteria | ATP-independent | 63 (E. coli) | HOCl | Oxidoreductase activity in various bacterial species, not in E. coli | [48] |
| Get3 TRC40/Asna1 | Yeast Mammals | ATP-independent | 242, 244, 285, 288 | H2O2 | When reduced, mediates the delivery of the thiol-anchoring (TA) proteins to the ER | [49] |
| Ydj1 | Yeast | ATP-dependent | 185, 188 | H2O2 | Part of Hsp40 co-chaperone family | [102,125] |
| Tsa1 Prx1/Prx2 | Yeast, Mammals | ATP-independent | 48, 171 (Tsa1) 47 (Prx1/2) | H2O2 | Active as chaperone only when overoxidized | [106,107] |
| Protein Name | Identified Organism | Reactive Cysteine | Modification Type | Associated Protein | References |
|---|---|---|---|---|---|
| TRAP1 | Human | 501 | S-nitrosylation | Mitochondrial quality control | [143] |
| PDI | Human | 343 | S-nitrosylation | ER quality control | [144] |
| Chs3 | Yeast | Unknown | Palmitoylation | ER protein maturation | [145] |
| Cdc25C | Human | 330, 377 | Disulfide bridge formation | Cell cycle checkpoint control | [146] |
| RhoA | Human | 190 | Prenylation | Protein–protein interaction | [147] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radzinski, M.; Oppenheim, T.; Metanis, N.; Reichmann, D. The Cys Sense: Thiol Redox Switches Mediate Life Cycles of Cellular Proteins. Biomolecules 2021, 11, 469. https://doi.org/10.3390/biom11030469
Radzinski M, Oppenheim T, Metanis N, Reichmann D. The Cys Sense: Thiol Redox Switches Mediate Life Cycles of Cellular Proteins. Biomolecules. 2021; 11(3):469. https://doi.org/10.3390/biom11030469
Chicago/Turabian StyleRadzinski, Meytal, Tal Oppenheim, Norman Metanis, and Dana Reichmann. 2021. "The Cys Sense: Thiol Redox Switches Mediate Life Cycles of Cellular Proteins" Biomolecules 11, no. 3: 469. https://doi.org/10.3390/biom11030469
APA StyleRadzinski, M., Oppenheim, T., Metanis, N., & Reichmann, D. (2021). The Cys Sense: Thiol Redox Switches Mediate Life Cycles of Cellular Proteins. Biomolecules, 11(3), 469. https://doi.org/10.3390/biom11030469