
Actin-Resistant DNase1L2 as a Potential Therapeutics for CF Lung Disease

 ,
, 
 , , ,
, , ,  and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
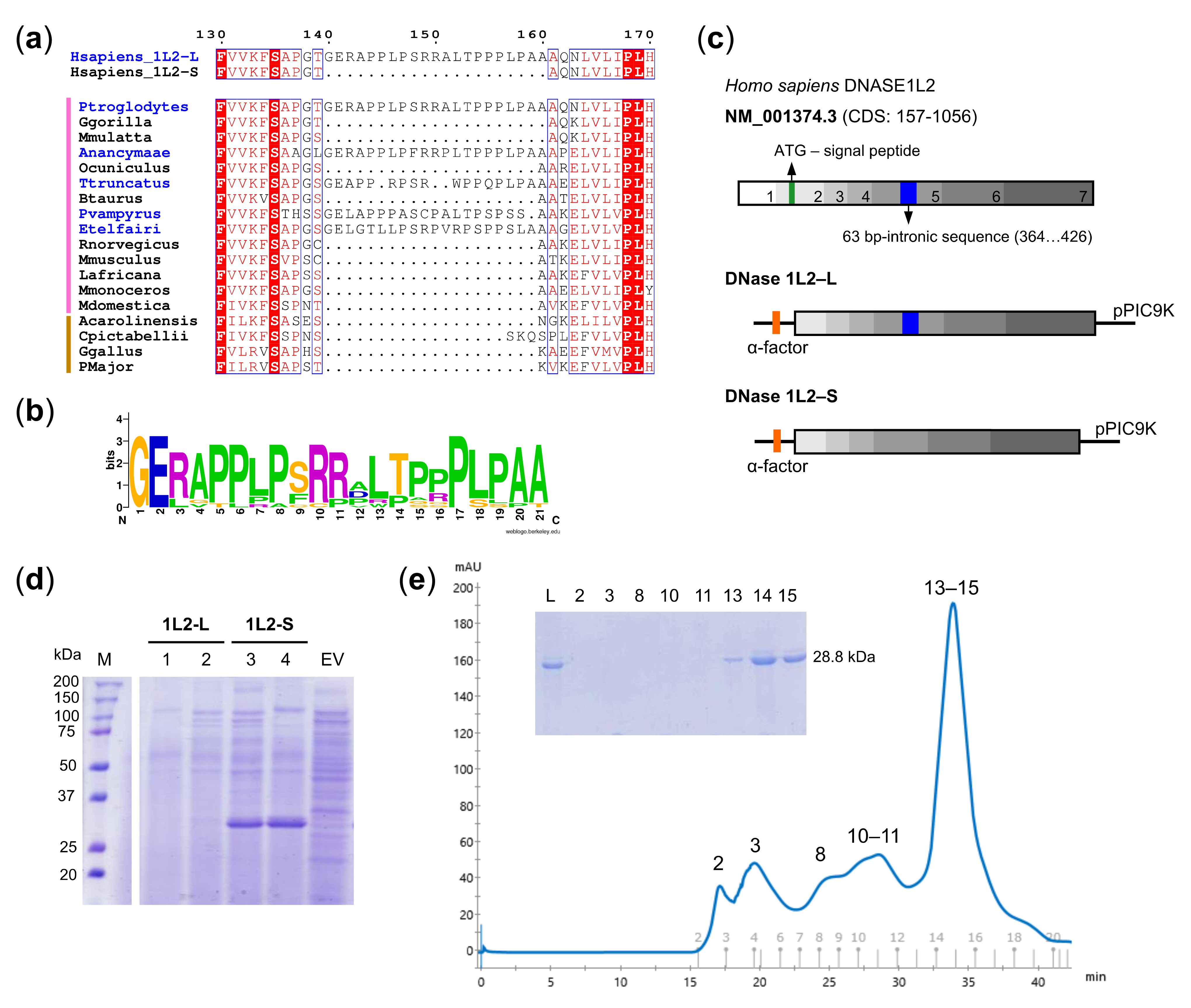
2.1. Bioinformatics
2.2. Recombinant Expression in Escherichia Coli
2.3. Integration in Pichia pastoris
2.4. Recombinant Expression in P. pastoris
2.5. Protein Purification
2.6. DNA Plasmid Preparation
2.7. DNase Activity Assays
2.8. Viscosity Measurements
2.9. Cysteine Reduction and Alkylation
2.10. Protein PEGylation
3. Results and Discussion
3.1. Expression and Purification of Recombinant DNase1L2 from Pichia Pastoris
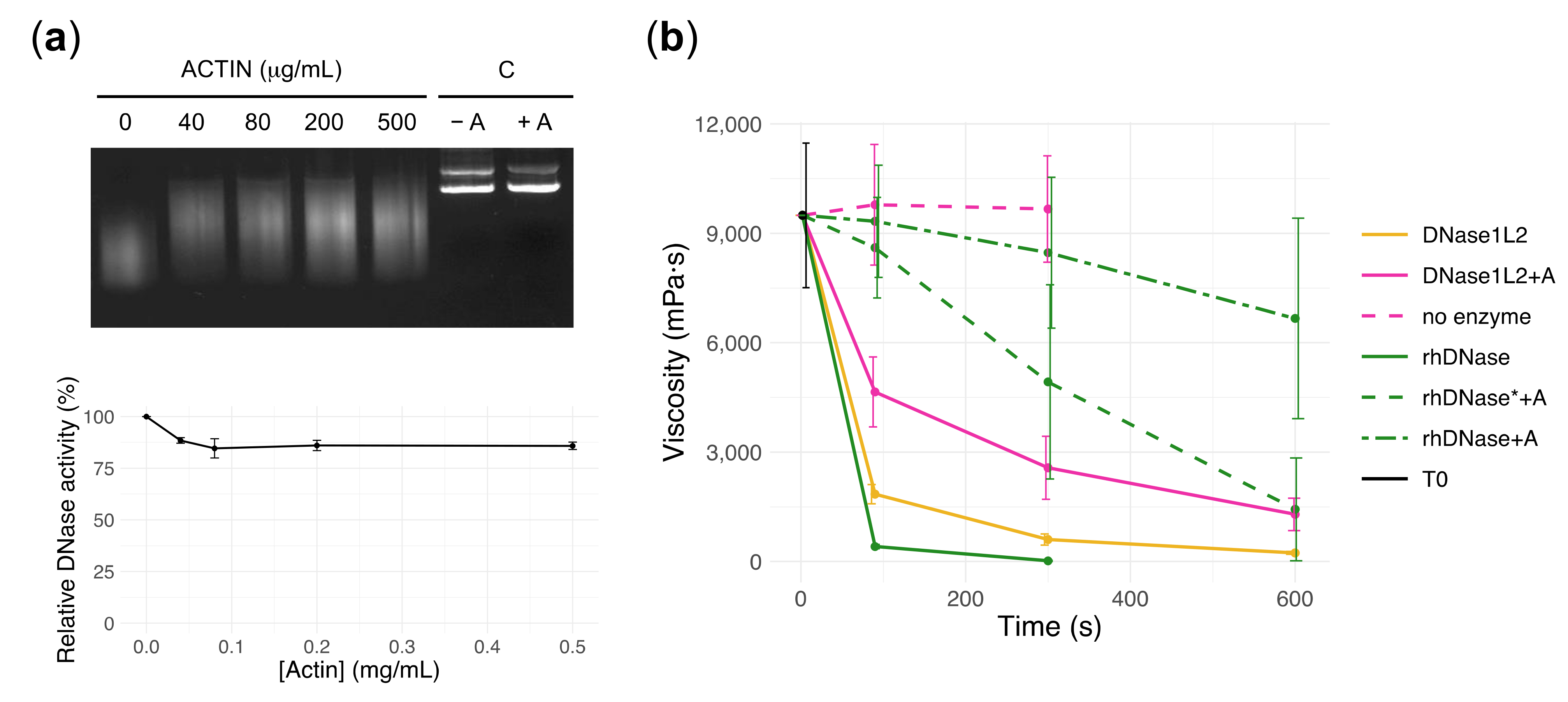
3.2. Endonuclease Activity of DNase1L2 on Purified Plasmid DNA
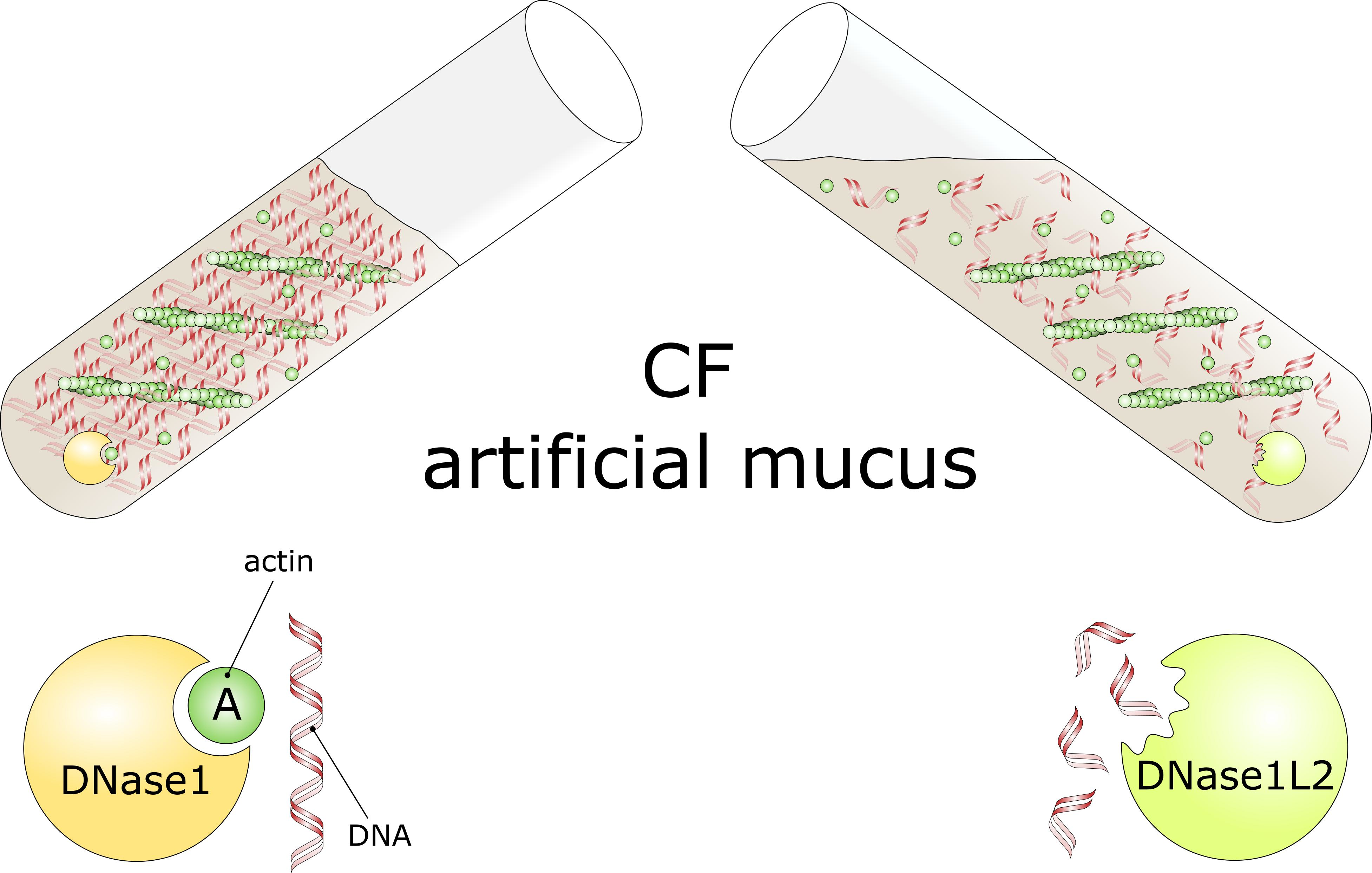
3.3. DNase1L2 Reduces Viscosity of CF Artificial Mucus with Marked Resistance to Actin Inhibition
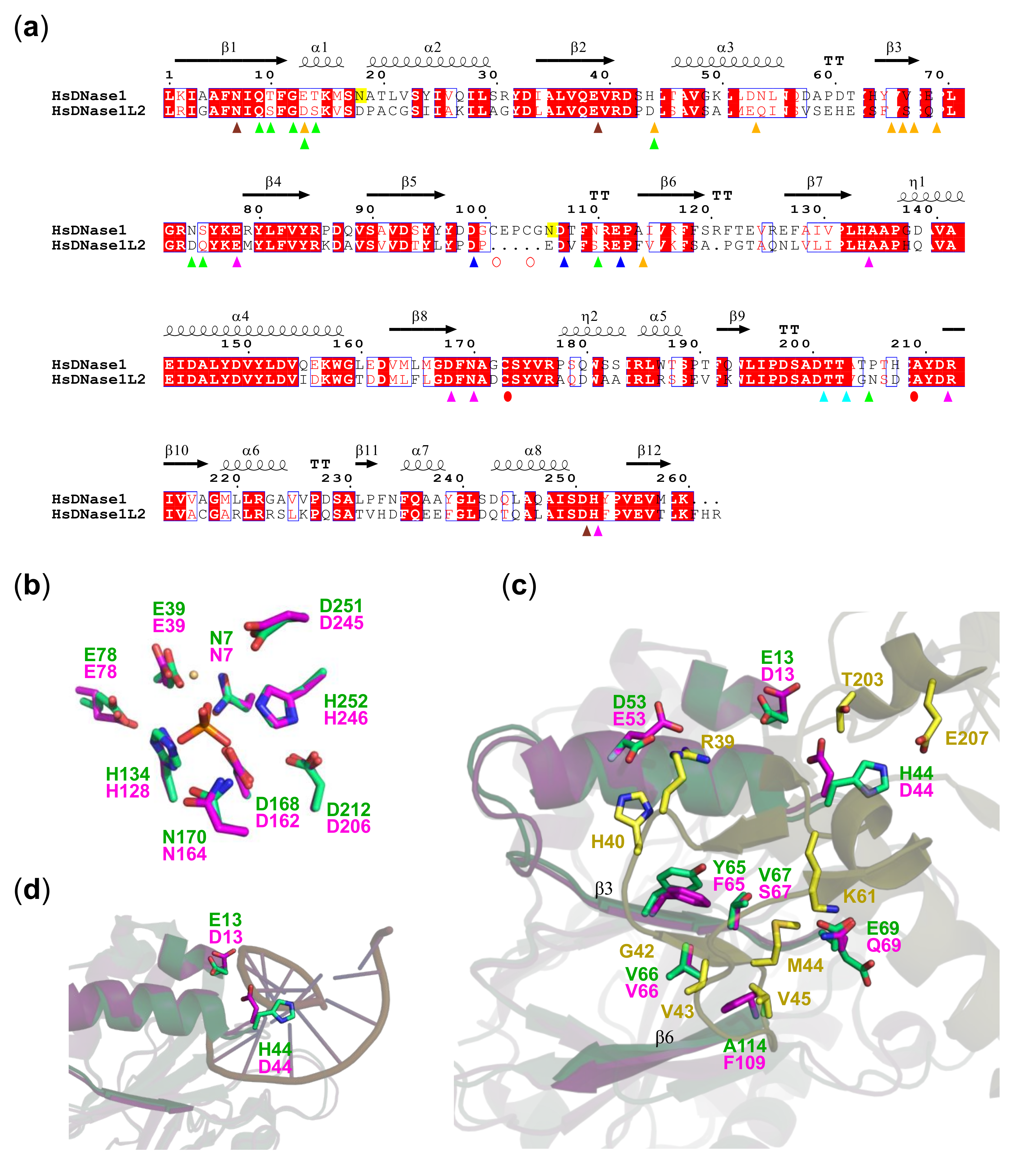
3.4. DNase1L2 Structural Model Shows Conservation of rhDNase Active Site Residues but Significant Differences in the Actin-Binding Interface
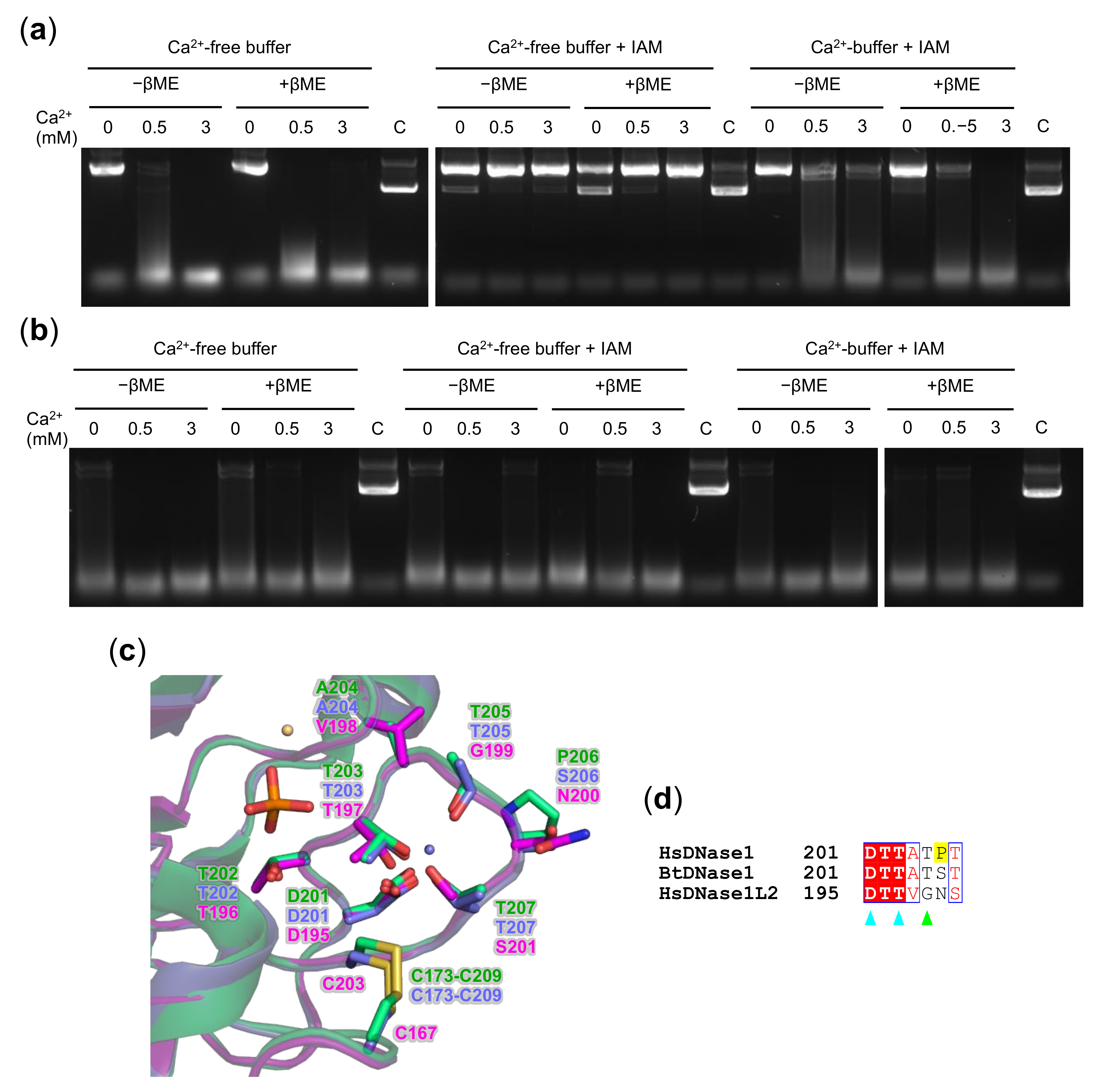
3.5. Oxidation State of DNase1L2 Conserved Cysteines
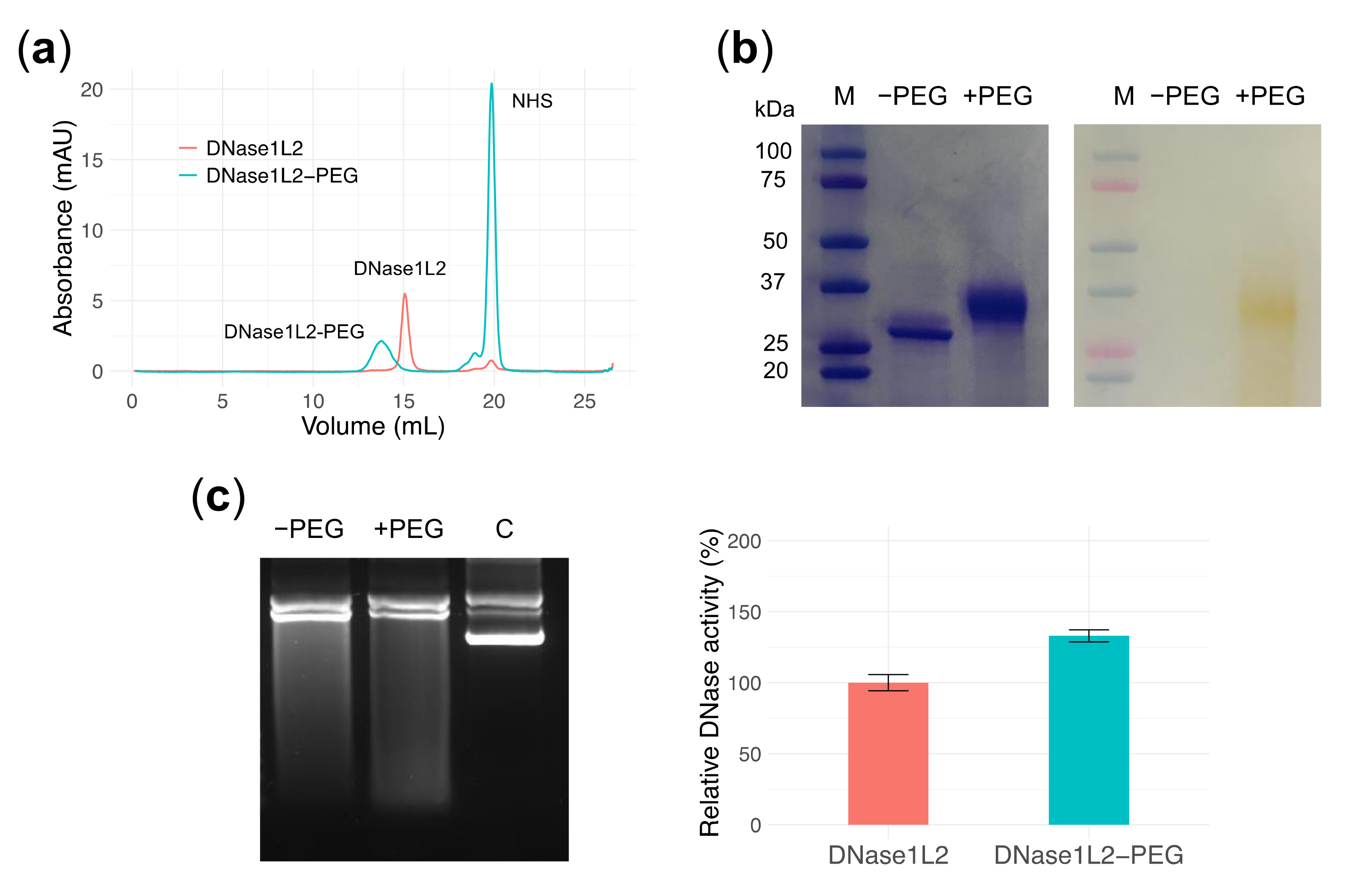
3.6. PEGylation Does Not Perturb DNase1L2 Endonuclease Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Mall, M.A.; Hartl, D. CFTR: Cystic fibrosis and beyond. Eur. Respir. J. 2014, 44, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.B.; Drumm, M.; Konstan, M.W. Cystic fibrosis. Am. J. Respir. Crit. Care Med. 1996, 154, 1229–1256. [Google Scholar] [CrossRef]
- Davis, P.B. Cystic fibrosis since 1938. Am. J. Respir. Crit. Care Med. 2006, 173, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Lethem, M.I.; James, S.L.; Marriott, C.; Burke, J.F. The origin of DNA associated with mucus glycoproteins in cystic fibrosis sputum. Eur. Respir. J. 1990, 3, 19–23. [Google Scholar]
- Vasconcellos, C.; Allen, P.; Wohl, M.; Drazen, J.; Janmey, P.; Stossel, T. Reduction in viscosity of cystic fibrosis sputum in vitro by gelsolin. Science 1994, 263, 969–971. [Google Scholar] [CrossRef] [PubMed]
- Gibson, R.L.; Burns, J.L.; Ramsey, B.W. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 918–951. [Google Scholar] [CrossRef] [PubMed]
- Castellani, C.; Duff, A.J.; Bell, S.C.; Heijerman, H.G.; Munck, A.; Ratjen, F.; Sermet-Gaudelus, I.; Southern, K.W.; Barben, J.; Flume, P.A.; et al. ECFS best practice guidelines: The 2018 revision. J. Cyst. Fibros. 2018, 17, 153–178. [Google Scholar] [CrossRef] [PubMed]
- Shak, S.; Capon, D.J.; Hellmiss, R.; Marsters, S.A.; Baker, C.L. Recombinant human DNase I reduces the viscosity of cystic fibrosis sputum. Proc. Natl. Acad. Sci. USA 1990, 87, 9188–9192. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, H.J.; Borowitz, D.S.; Christiansen, D.H.; Morris, E.M.; Nash, M.L.; Ramsey, B.W.; Rosenstein, B.J.; Smith, A.L.; Wohl, M.E. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N. Engl. J. Med. 1994, 331, 637–642. [Google Scholar] [CrossRef]
- Suri, R. The use of human deoxyribonuclease (rhDNase) in the management of cystic fibrosis. BioDrugs 2005, 19, 135–144. [Google Scholar] [CrossRef]
- Parrish, J.E.; Ciccodicola, A.; Wehhert, M.; Cox, G.F.; Chen, E.; Nelson, D.L. A muscle-specific DNase I-like gene in human Xq28. Hum. Mol. Genet. 1995, 4, 1557–1564. [Google Scholar] [CrossRef] [PubMed]
- Pergolizzi, R.; Appierto, V.; Bosetti, A.; DeBellis, G.L.; Rovida, E.; Biunno, I. Cloning of a gene encoding a DNase I-like endonuclease in the human Xq28 region. Gene 1996, 168, 267–270. [Google Scholar] [CrossRef]
- Rodriguez, A.M.; Rodin, D.; Nomura, H.; Morton, C.C.; Weremowicz, S.; Schneider, M.C. Identification, localization, and expression of two novel human genes similar to deoxyribonuclease I. Genomics 1997, 42, 507–513. [Google Scholar] [CrossRef]
- Laskowski, M. 12 Deoxyribonuclease I. In Hydrolysis; Elsevier: Amsterdam, The Netherlands, 1971; pp. 289–311. [Google Scholar]
- Moore, S. 15 Pancreatic DNase. In Biology of Aminoacyl-tRNA Synthetases; Elsevier: Amsterdam, The Netherlands, 1981; pp. 281–296. [Google Scholar]
- Melgar, E.; Goldthwait, D.A. Deoxyribonucleic acid nucleases. II. The effects of metals on the mechanism of action of deoxyribonuclease I. J. Biol. Chem. 1968, 243, 4409–4416. [Google Scholar] [CrossRef]
- Junowicz, E.; Spencer, J.H. Studies on bovine pancreatic deoxyribonuclease A. Biochim. Biophys. Acta Nucleic Acids Protein Synth. 1973, 312, 85–102. [Google Scholar] [CrossRef]
- Shiokawa, D.; Tanuma, S. Characterization of human DNase I family endonucleases and activation of DNase gamma during apoptosis. Biochemistry 2001, 40, 143–152. [Google Scholar] [CrossRef]
- Napirei, M.; Ricken, A.; Eulitz, D.; Knoop, H.; Mannherz, H.G. Expression pattern of the deoxyribonuclease 1 gene: Lessons from the Dnase1 knockout mouse. Biochem. J. 2004, 380, 929–937. [Google Scholar] [CrossRef]
- Keyel, P.A. Dnases in health and disease. Dev. Biol. 2017, 429, 1–11. [Google Scholar] [CrossRef]
- Shiokawa, D.; Matsushita, T.; Kobayashi, T.; Matsumoto, Y.; Tanuma, S.-I. Characterization of the human DNAS1L2 gene and the molecular mechanism for its transcriptional activation induced by inflammatory cytokines. Genomics 2004, 84, 95–105. [Google Scholar] [CrossRef]
- Fischer, H.; Eckhart, L.; Mildner, M.; Jaeger, K.; Buchberger, M.; Ghannadan, M.; Tschachler, E. DNase1L2 degrades nuclear DNA during corneocyte formation. J. Invest. Dermatol. 2007, 127, 24–30. [Google Scholar] [CrossRef]
- Fischer, H.; Szabo, S.; Scherz, J.; Jaeger, K.; Rossiter, H.; Buchberger, M.; Ghannadan, M.; Hermann, M.; Theussl, H.-C.; Tobin, D.J.; et al. Essential role of the keratinocyte-specific endonuclease DNase1L2 in the removal of nuclear DNA from hair and nails. J. Invest. Dermatol. 2011, 131, 1208–1215. [Google Scholar] [CrossRef]
- Eckhart, L.; Fischer, H.; Barken, K.B.; Tolker-Nielsen, T.; Tschachler, E. DNase1L2 suppresses biofilm formation by Pseudomonas aeruginosa and Staphylococcus aureus. Br. J. Dermatol. 2007, 156, 1342–1345. [Google Scholar] [CrossRef]
- Lazarides, E.; Lindberg, U. Actin is the naturally occurring inhibitor of deoxyribonuclease I. Proc. Natl. Acad. Sci. USA 1974, 71, 4742–4746. [Google Scholar] [CrossRef]
- Christopher, F.; Chase, D.; Stein, K.; Milne, R. rhDNase therapy for the treatment of cystic fibrosis patients with mild to moderate lung disease. J. Clin. Pharm Ther. 1999, 24, 415–426. [Google Scholar] [CrossRef][Green Version]
- Cobos, N.; Danés, I.; Gartner, S.; González, M.; Liñán, S.; Arnau, J.M. DNase use in the daily care of cystic fibrosis: Who benefits from it and to what extent? Results of a cohort study of 199 patients in 13 centres. DNase National Study Group. Eur. J. Pediatr. 2000, 159, 176–181. [Google Scholar] [CrossRef]
- Ulmer, J.S.; Herzka, A.; Toy, K.J.; Baker, D.L.; Dodge, A.H.; Sinicropi, D.; Shak, S.; Lazarus, R.A. Engineering actin-resistant human DNase I for treatment of cystic fibrosis. Proc. Natl. Acad. Sci. USA 1996, 93, 8225–8229. [Google Scholar] [CrossRef]
- Zahm, J.M.; Debordeaux, C.; Maurer, C.; Hubert, D.; Dusser, D.; Bonnet, N.; Lazarus, R.A.; Puchelle, E. Improved activity of an actin-resistant DNase I variant on the cystic fibrosis airway secretions. Am. J. Respir. Crit. Care Med. 2001, 163, 1153–1157. [Google Scholar] [CrossRef]
- Veronese, F.M.; Pasut, G. PEGylation, successful approach to drug delivery. Drug Discov. Today 2005, 10, 1451–1458. [Google Scholar] [CrossRef]
- Guichard, M.-J.; Kinoo, D.; Aubriot, A.-S.; Bauwens, N.; Gougué, J.; Vermeulen, F.; Lebecque, P.; Leal, T.; Vanbever, R. Impact of PEGylation on the mucolytic activity of recombinant human deoxyribonuclease I in cystic fibrosis sputum. Clin. Sci. 2018, 132, 1439–1452. [Google Scholar] [CrossRef] [PubMed]
- Mahri, S.; Rondon, A.; Wilms, T.; Bosquillon, C.; Vanbever, R. Biodistribution and elimination pathways of PEGylated recombinant human deoxyribonuclease I after pulmonary delivery in mice. J. Control. Release 2021, 329, 1054–1065. [Google Scholar] [CrossRef]
- Koussoroplis, S.J.; Paulissen, G.; Tyteca, D.; Goldansaz, H.; Todoroff, J.; Barilly, C.; Uyttenhove, C.; Van Snick, J.; Cataldo, D.; Vanbever, R. PEGylation of antibody fragments greatly increases their local residence time following delivery to the respiratory tract. J. Control. Release 2014, 187, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef]
- Gupta, R.; Jung, E.; Brunak, S. Prediction of N-glycosylation Sites in Human Proteins. 2004. Available online: http://www.cbs.dtu.dk/services/NetNGlyc/ (accessed on 31 January 2021).
- Bordoli, L.; Kiefer, F.; Arnold, K.; Benkert, P.; Battey, J.; Schwede, T. Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 2009, 4, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the expasy server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Birnboim, H.C.; Doly, J. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 1979, 7, 1513–1523. [Google Scholar] [CrossRef]
- Birnboim, H. A rapid alkaline extraction method for the isolation of plasmid DNA. In Recombinant DNA Part B; Elsevier: Amsterdam, The Netherlands, 1983; Volume 100, pp. 243–255. [Google Scholar]
- Craparo, E.F.; Porsio, B.; Sardo, C.; Giammona, G.; Cavallaro, G. Pegylated Polyaspartamide-Polylactide-Based Nanoparticles Penetrating Cystic Fibrosis Artificial Mucus. Biomacromolecules 2016, 17, 767–777. [Google Scholar] [CrossRef]
- Ueki, M.; Takeshita, H.; Utsunomiya, N.; Chino, T.; Oyama, N.; Hasegawa, M.; Kimura-Kataoka, K.; Fujihara, J.; Iida, R.; Yasuda, T. Survey of single-nucleotide polymorphisms in the gene encoding human deoxyribonuclease I-like 2 producing loss of function potentially implicated in the pathogenesis of parakeratosis. PLoS ONE 2017, 12, e0175083. [Google Scholar] [CrossRef] [PubMed]
- Karbalaei, M.; Rezaee, S.A.; Farsiani, H. Pichia pastoris: A highly successful expression system for optimal synthesis of heterologous proteins. J. Cell Physiol. 2020, 235, 5867–5881. [Google Scholar] [CrossRef]
- Bretthauer, R.K.; Castellino, F.J. Glycosylation of Pichia pastoris-derived proteins. Biotechnol. Appl. Biochem. 1999, 30, 193–200. [Google Scholar] [PubMed]
- Oefner, C.; Suck, D. Crystallographic refinement and structure of DNase I at 2 A resolution. J. Mol. Biol. 1986, 192, 605–632. [Google Scholar] [CrossRef]
- Wiberg, J.S. On the mechanism of metal activation of deoxyribonuclease I. Arch. Biochem. Biophys. 1958, 73, 337–358. [Google Scholar] [CrossRef]
- Desreux, V.; Hacha, R.; Fredericq, E. Activation of deoxyribonucleases by divalent cations. J. Gen. Physiol. 1962, 45, 93–102. [Google Scholar] [CrossRef]
- Sanders, N.N.; Franckx, H.; De Boeck, K.; Haustraete, J.; De Smedt, S.C.; Demeester, J. Role of magnesium in the failure of rhDNase therapy in patients with cystic fibrosis. Thorax 2006, 61, 962–968. [Google Scholar] [CrossRef]
- Sanders, N.; Rudolph, C.; Braeckmans, K.; De Smedt, S.C.; Demeester, J. Extracellular barriers in respiratory gene therapy. Adv. Drug Deliv. Rev. 2009, 61, 115–127. [Google Scholar] [CrossRef]
- Parsiegla, G.; Noguere, C.; Santell, L.; Lazarus, R.A.; Bourne, Y. The structure of human DNase I bound to magnesium and phosphate ions points to a catalytic mechanism common to members of the DNase I-like superfamily. Biochemistry 2012, 51, 10250–10258. [Google Scholar] [CrossRef]
- Coakley, R.D.; Grubb, B.R.; Paradiso, A.M.; Gatzy, J.T.; Johnson, L.G.; Kreda, S.M.; O’Neal, W.K.; Boucher, R.C. Abnormal surface liquid pH regulation by cultured cystic fibrosis bronchial epithelium. Proc. Natl. Acad. Sci. USA 2003, 100, 16083–16088. [Google Scholar] [CrossRef]
- Song, Y.; Salinas, D.; Nielson, D.W.; Verkman, A.S. Hyperacidity of secreted fluid from submucosal glands in early cystic fibrosis. Am. J. Physiol. Cell Physiol. 2006, 290, C741–C749. [Google Scholar] [CrossRef]
- Pezzulo, A.A.; Tang, X.X.; Hoegger, M.J.; Abou Alaiwa, M.H.; Ramachandran, S.; Moninger, T.O.; Karp, P.H.; Wohlford-Lenane, C.L.; Haagsman, H.P.; van Eijk, M.; et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 2012, 487, 109–113. [Google Scholar] [CrossRef]
- Xie, Y.; Lu, L.; Tang, X.X.; Moninger, T.O.; Huang, T.J.; Stoltz, D.A.; Welsh, M.J. Acidic submucosal gland ph and elevated protein concentration produce abnormal cystic fibrosis mucus. Dev. Cell 2020, 54, 488–500. [Google Scholar] [CrossRef]
- Campbell, V.W.; Jackson, D.A. The effect of divalent cations on the mode of action of DNase I. The initial reaction products produced from covalently closed circular DNA. J. Biol. Chem. 1980, 255, 3726–3735. [Google Scholar] [CrossRef]
- Price, P.A. The essential role of Ca2+ in the activity of bovine pancreatic deoxyribonuclease. J. Biol. Chem. 1975, 250, 1981–1986. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Lu, S.-C.; Liao, T.-H. The distinctive functions of the two structural calcium atoms in bovine pancreatic deoxyribonuclease. Protein Sci. 2002, 11, 659–668. [Google Scholar] [CrossRef]
- Watts, R.G.; Howard, T.H. Mechanisms for actin reorganization in chemotactic factor-activated polymorphonuclear leukocytes. Blood 1993, 81, 2750–2757. [Google Scholar] [CrossRef]
- Kabsch, W.; Vandekerckhove, J. Structure and function of actin. Annu. Rev. Biophys. Biomol. Struct. 1992, 21, 49–76. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Tsifansky, M.D.; Wu, C.-J.; Yang, H.I.; Schmidt, G.; Yeo, Y. Inhalable antibiotic delivery using a dry powder co-delivering recombinant deoxyribonuclease and ciprofloxacin for treatment of cystic fibrosis. Pharm. Res. 2010, 27, 151–160. [Google Scholar] [CrossRef]
- Thomas, S.R.; Ray, A.; Hodson, M.E.; Pitt, T.L. Increased sputum amino acid concentrations and auxotrophy of Pseudomonas aeruginosa in severe cystic fibrosis lung disease. Thorax 2000, 55, 795–797. [Google Scholar] [CrossRef]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Gallo Cassarino, T.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Mannherz, H.G.; Suck, D.; Pai, E.F.; Holmes, K.C. Atomic structure of the actin: DNase I complex. Nature 1990, 347, 37–44. [Google Scholar] [CrossRef]
- Pan, C.Q.; Dodge, T.H.; Baker, D.L.; Prince, W.S.; Sinicropi, D.V.; Lazarus, R.A. Improved potency of hyperactive and actin-resistant human DNase I variants for treatment of cystic fibrosis and systemic lupus erythematosus. J. Biol. Chem. 1998, 273, 18374–18381. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.Q.; Lazarus, R.A. Engineering hyperactive variants of human deoxyribonuclease I by altering its functional mechanism. Biochemistry 1997, 36, 6624–6632. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.Q.; Lazarus, R.A. Hyperactivity of human DNase I variants. Dependence on the number of positively charged residues and concentration, length, and environment of DNA. J. Biol. Chem. 1998, 273, 11701–11708. [Google Scholar] [CrossRef] [PubMed]
- Weston, S.A.; Lahm, A.; Suck, D. X-ray structure of the DNase I-d(GGTATACC)2 complex at 2.3 A resolution. J. Mol. Biol. 1992, 226, 1237–1256. [Google Scholar] [CrossRef]
- Price, P.A.; Stein, W.H.; Moore, S. Effect of divalent cations on the reduction and re-formation of the disulfide bonds of deoxyribonuclease. J. Biol. Chem. 1969, 244, 929–932. [Google Scholar] [CrossRef]
- Chen, W.-J.; Lee, I.-S.; Chen, C.-Y.; Liao, T.-H. Biological functions of the disulfides in bovine pancreatic deoxyribonuclease. Protein Sci. 2004, 13, 875–883. [Google Scholar] [CrossRef]
- Pan, C.Q.; Lazarus, R.A. Ca2+-dependent activity of human DNase I and its hyperactive variants. Protein Sci. 1999, 8, 1780–1788. [Google Scholar] [CrossRef]
- Harris, J.M.; Chess, R.B. Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2003, 2, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Grigoletto, A.; Tedeschini, T.; Canato, E.; Pasut, G. The evolution of polymer conjugation and drug targeting for the delivery of proteins and bioactive molecules. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020, e1689. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Lai, S.K.; Suk, J.S.; Pace, A.; Cone, R.; Hanes, J. Addressing the PEG mucoadhesivity paradox to engineer nanoparticles that “slip” through the human mucus barrier. Angew. Chem. Int. Ed. Engl. 2008, 47, 9726–9729. [Google Scholar] [CrossRef]
- Kim, A.J.; Boylan, N.J.; Suk, J.S.; Hwangbo, M.; Yu, T.; Schuster, B.S.; Cebotaru, L.; Lesniak, W.G.; Oh, J.S.; Adstamongkonkul, P.; et al. Use of single-site-functionalized PEG dendrons to prepare gene vectors that penetrate human mucus barriers. Angew. Chem. Int. Ed. Engl. 2013, 52, 3985–3988. [Google Scholar] [CrossRef] [PubMed]
- Chakraborti, S.; Sarkar, J.; Pramanik, P.K.; Chakraborti, T. Role of proteases in lung disease: A brief overview. In Proteases in Human Diseases; Chakraborti, S., Chakraborti, T., Dhalla, N.S., Eds.; Springer: Singapore, 2017; pp. 333–374. [Google Scholar]
- Zhang, C.; Desai, R.; Perez-Luna, V.; Karuri, N. PEGylation of lysine residues improves the proteolytic stability of fibronectin while retaining biological activity. Biotechnol. J. 2014, 9, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Freches, D.; Patil, H.P.; Machado Franco, M.; Uyttenhove, C.; Heywood, S.; Vanbever, R. PEGylation prolongs the pulmonary retention of an anti-IL-17A Fab’ antibody fragment after pulmonary delivery in three different species. Int. J. Pharm. 2017, 521, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Niven, R.W.; Whitcomb, K.L.; Shaner, L.; Ip, A.Y.; Kinstler, O.B. The pulmonary absorption of aerosolized and intratracheally instilled rhG-CSF and monoPEGylated rhG-CSF. Pharm. Res. 1995, 12, 1343–1349. [Google Scholar] [CrossRef] [PubMed]
- Davoodian, K.; Ritchings, B.W.; Ramphal, R.; Bubb, M.R. Gelsolin activates DNase I in vitro and cystic fibrosis sputum. Biochemistry 1997, 36, 9637–9641. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delfino, D.; Mori, G.; Rivetti, C.; Grigoletto, A.; Bizzotto, G.; Cavozzi, C.; Malatesta, M.; Cavazzini, D.; Pasut, G.; Percudani, R. Actin-Resistant DNase1L2 as a Potential Therapeutics for CF Lung Disease. Biomolecules 2021, 11, 410. https://doi.org/10.3390/biom11030410
Delfino D, Mori G, Rivetti C, Grigoletto A, Bizzotto G, Cavozzi C, Malatesta M, Cavazzini D, Pasut G, Percudani R. Actin-Resistant DNase1L2 as a Potential Therapeutics for CF Lung Disease. Biomolecules. 2021; 11(3):410. https://doi.org/10.3390/biom11030410
Chicago/Turabian StyleDelfino, Danila, Giulia Mori, Claudio Rivetti, Antonella Grigoletto, Gloria Bizzotto, Cristian Cavozzi, Marco Malatesta, Davide Cavazzini, Gianfranco Pasut, and Riccardo Percudani. 2021. "Actin-Resistant DNase1L2 as a Potential Therapeutics for CF Lung Disease" Biomolecules 11, no. 3: 410. https://doi.org/10.3390/biom11030410
APA StyleDelfino, D., Mori, G., Rivetti, C., Grigoletto, A., Bizzotto, G., Cavozzi, C., Malatesta, M., Cavazzini, D., Pasut, G., & Percudani, R. (2021). Actin-Resistant DNase1L2 as a Potential Therapeutics for CF Lung Disease. Biomolecules, 11(3), 410. https://doi.org/10.3390/biom11030410