
Eicosapentaenoic Acid Inhibits KRAS Mutant Pancreatic Cancer Cell Growth by Suppressing Hepassocin Expression and STAT3 Phosphorylation

 ,
,  ,
, 
Abstract
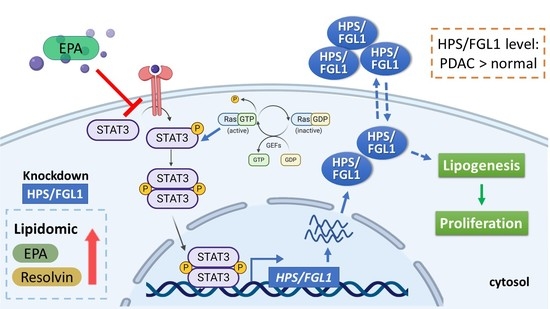
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Ras Activation
2.4. Lentiviral Production and Infection
2.5. Western Blotting
2.6. Cell Viability Assay
2.7. Cell Cycle Analysis
2.8. Lipidomic Analysis
2.9. Statistical Analysis
3. Results
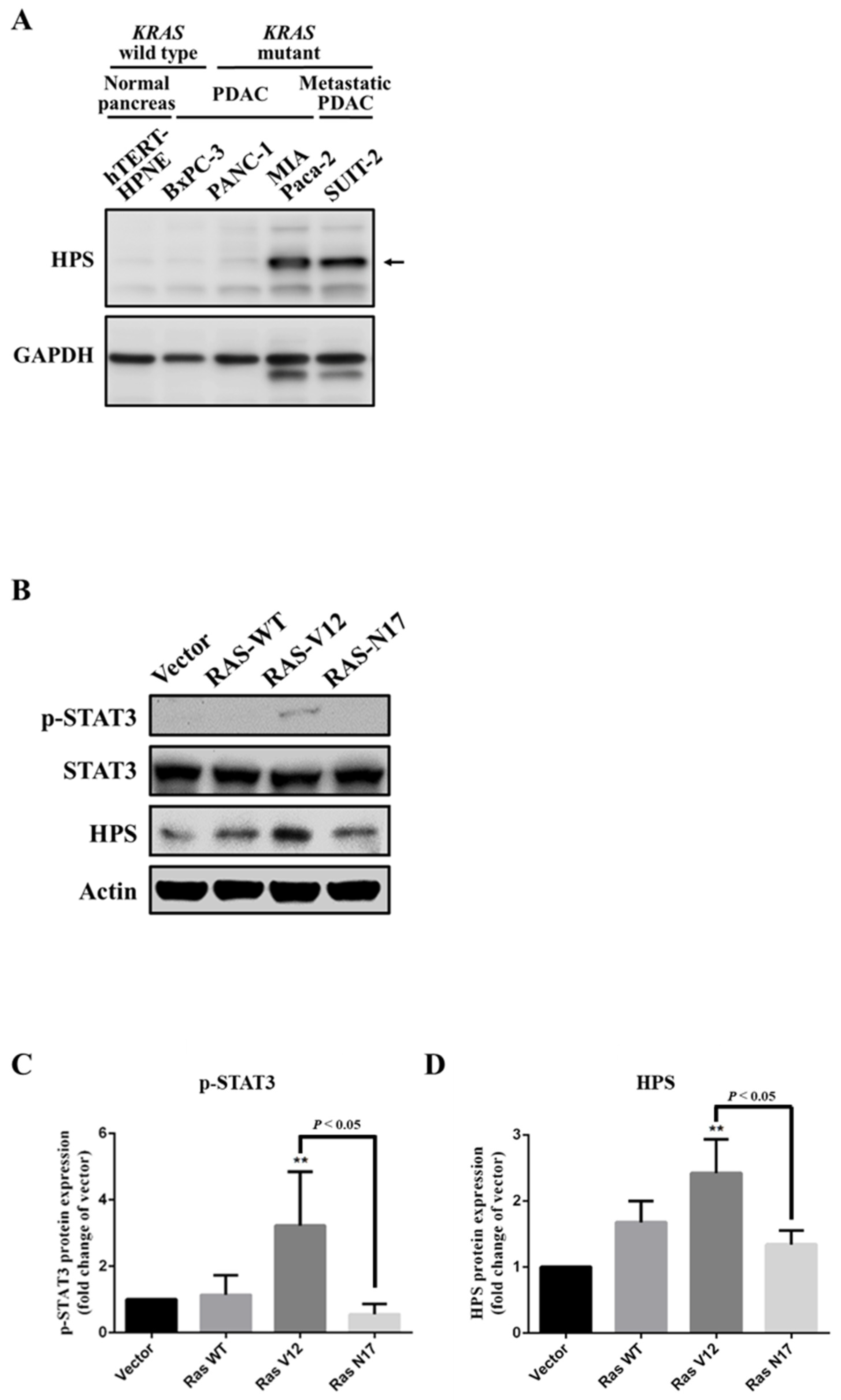
3.1. Expression of HPS in KRAS-Mutant Pancreatic Cancer Cells
3.2. Knockdown of HPS in SUIT-2 Cells Decreased Cell Growth and Induced Cell Cycle Arrest
3.3. Effects of HPS on Lipid Contents and Composition in SUIT-2 Cells
3.4. EPA Decreases Cell Survival and HPS Expression of SUIT-2 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA: Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-W.; Health Promotion Administration (Eds.) 2019 Health Promotion Administration Annual Report; Health Promotion Administration: Taipei City, Taiwan, 2019. [Google Scholar]
- Nollmann, F.I.; Ruess, D.A. Targeting Mutant KRAS in Pancreatic Cancer: Futile or Promising? Biomedicines 2020, 8, 281. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, S.; Ellenrieder, V.; Fernandez-Zapico, M.E. Oncogenic transcription factors: Cornerstones of inflammation-linked pancreatic carcinogenesis. Gut 2013, 62, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Gukovsky, I.; Li, N.; Todoric, J.; Gukovskaya, A.; Karin, M. Inflammation, autophagy, and obesity: Common features in the pathogenesis of pancreatitis and pancreatic cancer. Gastroenterology 2013, 144, 1199–1209. [Google Scholar] [CrossRef]
- Pupo, E.; Avanzato, D.; Middonti, E.; Bussolino, F.; Lanzetti, L. KRAS-Driven Metabolic Rewiring Reveals Novel Actionable Targets in Cancer. Front. Oncol. 2019, 9, 848–857. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Yang, G.; Yang, J.; Ren, B.; Wang, H.; Chen, G.; Zhao, F.; You, L.; Wang, W.; Zhao, Y. Metabolism of pancreatic cancer: Paving the way to better anticancer strategies. Mol. Cancer 2020, 19, 50–69. [Google Scholar] [CrossRef]
- Rozeveld, C.N.; Johnson, K.M.; Zhang, L.; Razidlo, G.L. KRAS controls pancreatic cancer cell lipid metabolism and invasive potential through the lipase HSL. Cancer Res. 2020, 80, 4932–4945. [Google Scholar] [CrossRef]
- Yao, X.; Tian, Z. Saturated, Monounsaturated and Polyunsaturated Fatty Acids Intake and Risk of Pancreatic Cancer: Evidence from Observational Studies. PLoS ONE 2015, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, V.C.; Hassing, M.R.; Lewandowski, P.A. Marine polyunsaturated fatty acids and cancer therapy. Br. J. Cancer 2013, 108, 486–492. [Google Scholar] [CrossRef]
- Zhang, W.; Long, Y.; Zhang, J.; Wang, C. Modulatory effects of EPA and DHA on proliferation and apoptosis of pancreatic cancer cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2007, 27, 547–550. [Google Scholar] [CrossRef]
- Wang, T.M.; Hsieh, S.C.; Chen, J.W.; Chiang, A.N. Docosahexaenoic acid and eicosapentaenoic acid reduce C-reactive protein expression and STAT3 activation in IL-6-treated HepG2 cells. Mol. Cell. Biochem. 2013, 377, 97–106. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Contino, G.; Deshpande, V.; Tzatsos, A.; Conrad, C.; Benes, C.H.; Levy, D.E.; Settleman, J.; Engelman, J.A.; Bardeesy, N. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res. 2011, 71, 5020–5029. [Google Scholar] [CrossRef]
- Hara, H.; Uchida, S.; Yoshimura, H.; Aoki, M.; Toyoda, Y.; Sakai, Y.; Morimoto, S.; Fukamachi, H.; Shiokawa, K.; Hanada, K. Isolation and characterization of a novel liver-specific gene, hepassocin, upregulated during liver regeneration. Biochim. Biophys. Acta 2000, 1492, 31–44. [Google Scholar] [CrossRef]
- Yan, J.; Ying, H.; Gu, F.; He, J.; Li, Y.L.; Liu, H.M.; Xu, Y.H. Cloning and characterization of a mouse liver-specific gene mfrep-1, up-regulated in liver regeneration. Cell Res. 2002, 12, 353–361. [Google Scholar] [CrossRef]
- Li, C.Y.; Cao, C.Z.; Xu, W.X.; Cao, M.M.; Yang, F.; Dong, L.; Yu, M.; Zhan, Y.Q.; Gao, Y.B.; Li, W.; et al. Recombinant human hepassocin stimulates proliferation of hepatocytes in vivo and improves survival in rats with fulminant hepatic failure. Gut 2010, 59, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.M.; Xu, W.X.; Li, C.Y.; Cao, C.Z.; Wang, Z.D.; Yao, J.W.; Yu, M.; Zhan, Y.Q.; Wang, X.H.; Tang, L.J.; et al. Hepassocin regulates cell proliferation of the human hepatic cells L02 and hepatocarcinoma cells through different mechanisms. J. Cell. Biochem. 2011, 112, 2882–2890. [Google Scholar] [CrossRef]
- Yu, H.T.; Yu, M.; Li, C.Y.; Zhan, Y.Q.; Xu, W.X.; Li, Y.H.; Li, W.; Wang, Z.D.; Ge, C.H.; Yang, X.M. Specific expression and regulation of hepassocin in the liver and down-regulation of the correlation of HNF1alpha with decreased levels of hepassocin in human hepatocellular carcinoma. J. Biol. Chem. 2009, 284, 13335–13347. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.T.; Lu, F.H.; Ou, H.Y.; Su, Y.C.; Hung, H.C.; Wu, J.S.; Yang, Y.C.; Wu, C.L.; Chang, C.J. The role of Hepassocin in the development of non-alcoholic fatty liver disease. J. Hepatol. 2013, 59, 1065–1072. [Google Scholar] [CrossRef]
- Zhang, Y.; Qiao, H.X.; Zhou, Y.T.; Hong, L.; Chen, J.H. Fibrinogenlikeprotein 1 promotes the invasion and metastasis of gastric cancer and is associated with poor prognosis. Mol. Med. Rep. 2018, 18, 1465–1472. [Google Scholar] [CrossRef]
- Freitas, R.D.S.; Campos, M.M. Protective Effects of Omega-3 Fatty Acids in Cancer-Related Complications. Nutrients 2019, 11, 945. [Google Scholar] [CrossRef] [PubMed]
- Laviano, A.; Rianda, S.; Molfino, A.; Rossi Fanelli, F. Omega-3 fatty acids in cancer. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 156–161. [Google Scholar] [CrossRef]
- Calder, P.C.; Yaqoob, P. Understanding omega-3 polyunsaturated fatty acids. Postgrad Med. 2009, 121, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Shan, K.; Chen, H.; Chen, Y.Q. n-3 Polyunsaturated Fatty Acids and their Role in Cancer Chemoprevention. Curr. Pharm. Rep. 2015, 1, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.A.; Mourtzakis, M.; Mazurak, V.C. n-3 polyunsaturated fatty acids: The potential role for supplementation in cancer. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Black, H.S.; Rhodes, L.E. Potential Benefits of Omega-3 Fatty Acids in Non-Melanoma Skin Cancer. J. Clin. Med. 2016, 5, 23. [Google Scholar] [CrossRef]
- Fuentes, N.R.; Mlih, M.; Barhoumi, R.; Fan, Y.Y.; Hardin, P.; Steele, T.J.; Behmer, S.; Prior, I.A.; Karpac, J.; Chapkin, R.S. Long-Chain n-3 Fatty Acids Attenuate Oncogenic KRas-Driven Proliferation by Altering Plasma Membrane Nanoscale Proteolipid Composition. Cancer Res. 2018, 78, 3899–3912. [Google Scholar] [CrossRef]
- Ding, Y.; Mullapudi, B.; Torres, C.; Mascarinas, E.; Mancinelli, G.; Diaz, A.M.; McKinney, R.; Barron, M.; Schultz, M.; Heiferman, M.; et al. Omega-3 Fatty Acids Prevent Early Pancreatic Carcinogenesis via Repression of the AKT Pathway. Nutrients 2018, 10, 1289. [Google Scholar] [CrossRef]
- Park, M.; Lim, J.W.; Kim, H. Docoxahexaenoic Acid Induces Apoptosis of Pancreatic Cancer Cells by Suppressing Activation of STAT3 and NF-kappaB. Nutrients 2018, 10, 1621. [Google Scholar] [CrossRef]
- Al-Jawadi, A.; Rasha, F.; Ramalingam, L.; Alhaj, S.; Moussa, H.; Gollahon, L.; Dharmawardhane, S.; Moustaid-Moussa, N. Protective effects of eicosapentaenoic acid in adipocyte-breast cancer cell cross talk. J. Nutr. Biochem. 2020, 75, 108244–108252. [Google Scholar] [CrossRef]
- Inoue-Yamauchi, A.; Itagaki, H.; Oda, H. Eicosapentaenoic acid attenuates obesity-related hepatocellular carcinogenesis. Carcinogenesis 2018, 39, 28–35. [Google Scholar] [CrossRef]
- Nagathihalli, N.S.; Castellanos, J.A.; Lamichhane, P.; Messaggio, F.; Shi, C.; Dai, X.; Rai, P.; Chen, X.; VanSaun, M.N.; Merchant, N.B. Inverse Correlation of STAT3 and MEK Signaling Mediates Resistance to RAS Pathway Inhibition in Pancreatic Cancer. Cancer Res. 2018, 78, 6235–6246. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Huang, J. The expanded role of fatty acid metabolism in cancer: New aspects and targets. Precis. Clin. Med. 2019, 2, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Byon, C.H.; Hardy, R.W.; Ren, C.C.; Ponnazhagan, S.; Welch, D.R.; McDonald, J.M.; Chen, Y.B. Free fatty acids enhance breast cancer cell migration through plasminogen activator inhibitor-1 and SMAD4. Lab. Investig. 2009, 89, 1221–1228. [Google Scholar] [CrossRef]
- Gouw, A.M.; Eberlin, L.S.; Margulis, K.; Sullivan, D.K.; Toal, G.G.; Tong, L.; Zare, R.N.; Felsher, D.W. Oncogene KRAS activates fatty acid synthase, resulting in specific ERK and lipid signatures associated with lung adenocarcinoma. Proc. Natl. Acad. Sci. USA 2017, 114, 4300–4305. [Google Scholar] [CrossRef]
- Flavin, R.; Peluso, S.; Nguyen, P.L.; Loda, M. Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol. 2010, 6, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, M.; Braun, S.M.G.; Zurkirchen, L.; von Schoultz, C.; Zamboni, N.; Arauzo-Bravo, M.J.; Kovacs, W.J.; Karalay, O.; Suter, U.; Machado, R.A.C.; et al. Metabolic control of adult neural stem cell activity by Fasn-dependent lipogenesis. Nature 2013, 493, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Loftus, T.M.; Jaworsky, D.E.; Frehywot, G.L.; Townsend, C.A.; Ronnett, G.V.; Lane, M.D.; Kuhajda, F.P. Reduced food intake and body weight in mice treated with fatty acid synthase inhibitors. Science 2000, 288, 2379–2381. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Xiao, L.; Sugiura, H.; Huang, X.; Ali, A.; Kuro-o, M.; Deberardinis, R.J.; Boothman, D.A. Metabolic reprogramming during TGFbeta1-induced epithelial-to-mesenchymal transition. Oncogene 2015, 34, 3908–3916. [Google Scholar] [CrossRef]
- Glatzel, D.K.; Koeberle, A.; Pein, H.; Loser, K.; Stark, A.; Keksel, N.; Werz, O.; Muller, R.; Bischoff, I.; Furst, R. Acetyl-CoA carboxylase 1 regulates endothelial cell migration by shifting the phospholipid composition. J. Lipid Res. 2018, 59, 298–311. [Google Scholar] [CrossRef]
- Ye, B.; Yin, L.; Wang, Q.; Xu, C. ACC1 is overexpressed in liver cancers and contributes to the proliferation of human hepatoma Hep G2 cells and the rat liver cell line BRL 3A. Mol. Med. Rep. 2019, 19, 3431–3440. [Google Scholar] [CrossRef]
- Jones, J.E.; Esler, W.P.; Patel, R.; Lanba, A.; Vera, N.B.; Pfefferkorn, J.A.; Vernochet, C. Inhibition of Acetyl-CoA Carboxylase 1 (ACC1) and 2 (ACC2) Reduces Proliferation and De Novo Lipogenesis of EGFRvIII Human Glioblastoma Cells. PLoS ONE 2017, 12, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Tadros, S.; Shukla, S.K.; King, R.J.; Gunda, V.; Vernucci, E.; Abrego, J.; Chaika, N.V.; Yu, F.; Lazenby, A.J.; Berim, L.; et al. De Novo Lipid Synthesis Facilitates Gemcitabine Resistance through Endoplasmic Reticulum Stress in Pancreatic Cancer. Cancer Res. 2017, 77, 5503–5517. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Ruiz, C.; Bhalla, K.; Haley, J.A.; Li, Q.K.; Acquaah-Mensah, G.; Montal, E.; Sudini, K.R.; Skoulidis, F.; Wistuba, I.I.; et al. De novo lipogenesis represents a therapeutic target in mutant Kras non-small cell lung cancer. FASEB J. 2018, 32, 7018–7027. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.K.; Shankar, S.; Srivastava, R.K. STAT3 as an emerging molecular target in pancreatic cancer. Gastrointest. Cancer Targets Ther. 2014, 4, 115–122. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Times | Cell Lines | n-3 PUFA | n-6 PUFA | |||
|---|---|---|---|---|---|---|
| ALA | EPA | DHA | LA | AA | ||
| 48 h | SUIT-2 | 88.5 | 51.8 | 140.4 | 99.8 | 32.8 |
| hTERP-HPNE | 571.0 | 1354.0 | 307.2 | 230.3 | 292.0 | |
| 72 h | SUIT-2 | 37.2 | 46.5 | 51.7 | 53.7 | 7.0 |
| hTERP-HPNE | 109.4 | 160.8 | 221.9 | 134.4 | 122.3 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiu, C.-F.; Hsu, M.-I.; Yeh, H.-Y.; Park, J.M.; Shen, Y.-S.; Tung, T.-H.; Huang, J.-J.; Wu, H.-T.; Huang, S.-Y. Eicosapentaenoic Acid Inhibits KRAS Mutant Pancreatic Cancer Cell Growth by Suppressing Hepassocin Expression and STAT3 Phosphorylation. Biomolecules 2021, 11, 370. https://doi.org/10.3390/biom11030370
Chiu C-F, Hsu M-I, Yeh H-Y, Park JM, Shen Y-S, Tung T-H, Huang J-J, Wu H-T, Huang S-Y. Eicosapentaenoic Acid Inhibits KRAS Mutant Pancreatic Cancer Cell Growth by Suppressing Hepassocin Expression and STAT3 Phosphorylation. Biomolecules. 2021; 11(3):370. https://doi.org/10.3390/biom11030370
Chicago/Turabian StyleChiu, Ching-Feng, Ming-I Hsu, Hsiu-Yen Yeh, Ji Min Park, Yu-Shiuan Shen, Te-Hsuan Tung, Jun-Jie Huang, Hung-Tsung Wu, and Shih-Yi Huang. 2021. "Eicosapentaenoic Acid Inhibits KRAS Mutant Pancreatic Cancer Cell Growth by Suppressing Hepassocin Expression and STAT3 Phosphorylation" Biomolecules 11, no. 3: 370. https://doi.org/10.3390/biom11030370
APA StyleChiu, C.-F., Hsu, M.-I., Yeh, H.-Y., Park, J. M., Shen, Y.-S., Tung, T.-H., Huang, J.-J., Wu, H.-T., & Huang, S.-Y. (2021). Eicosapentaenoic Acid Inhibits KRAS Mutant Pancreatic Cancer Cell Growth by Suppressing Hepassocin Expression and STAT3 Phosphorylation. Biomolecules, 11(3), 370. https://doi.org/10.3390/biom11030370