
Syndecans and Pancreatic Ductal Adenocarcinoma





Abstract
1. Introduction
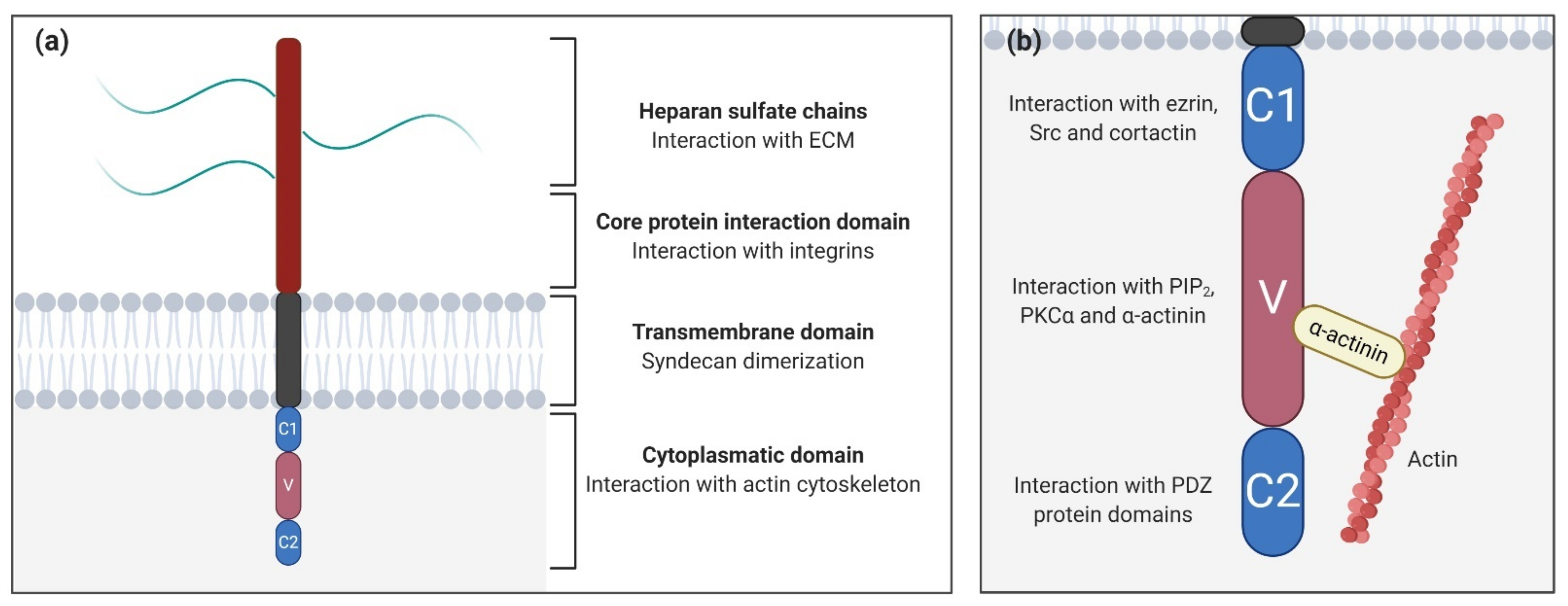
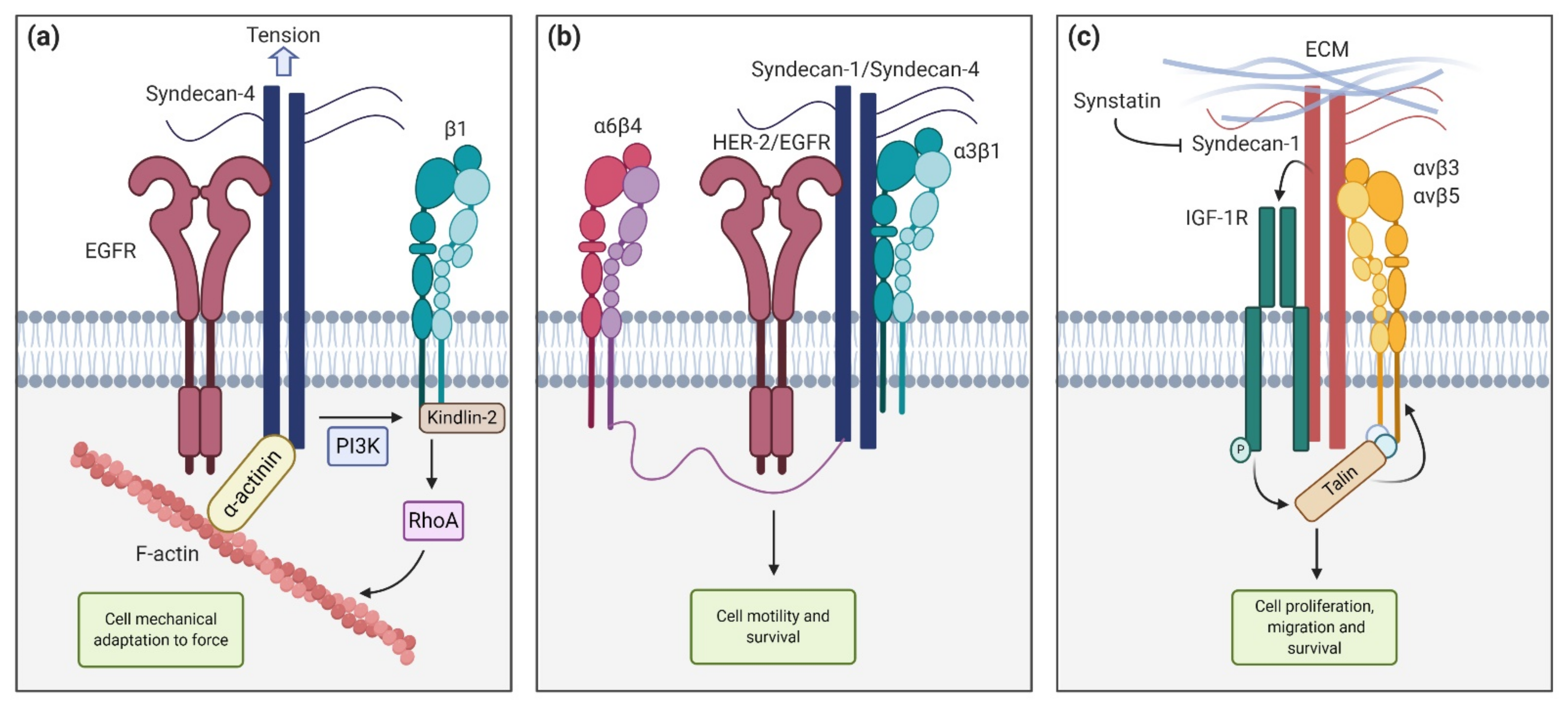
2. Syndecan Structure and Interactions
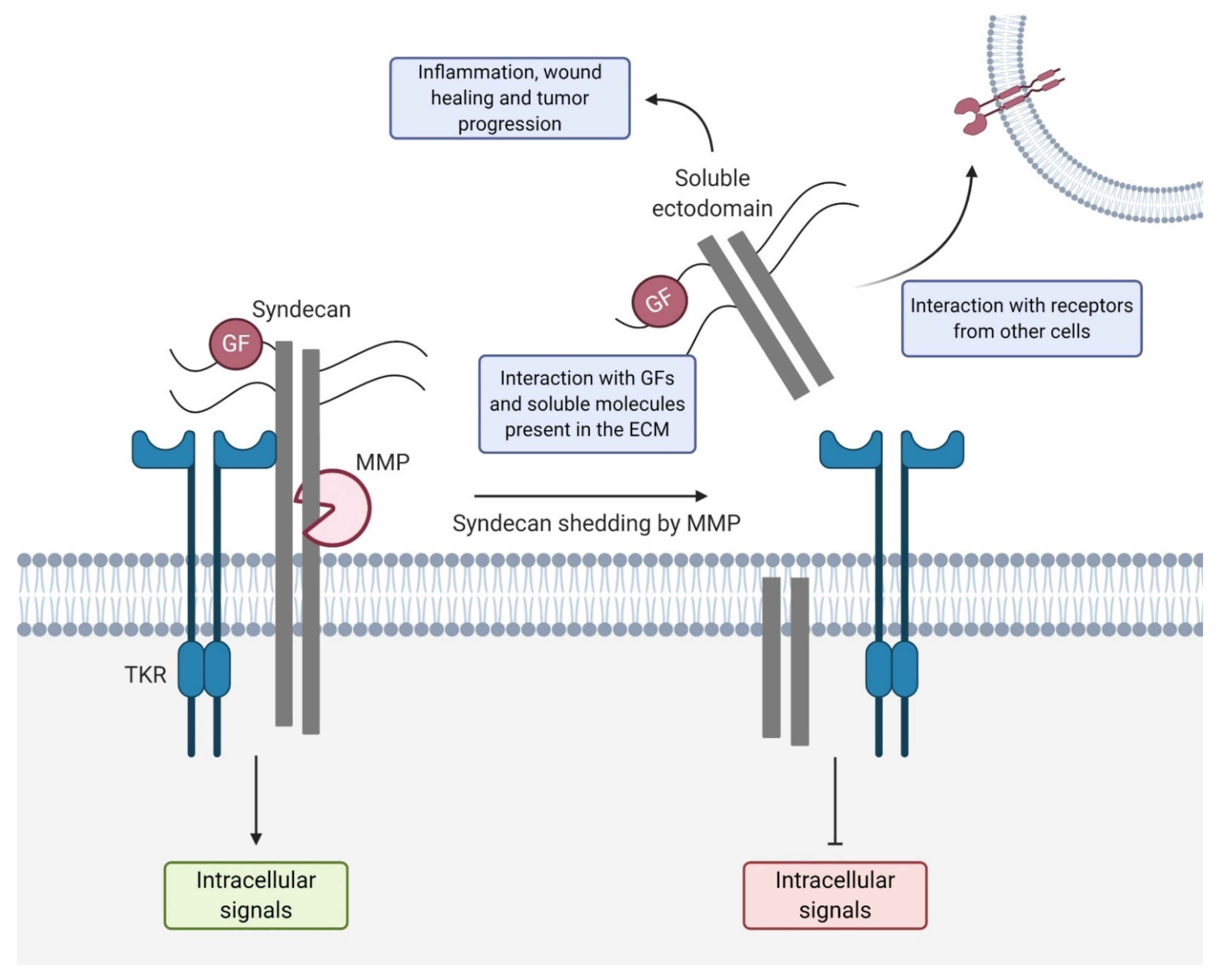
3. Syndecan Shedding
3.1. Syndecan Shedding during Wound Healing
3.2. Syndecan Shedding during Tumor Development
3.3. Syndecan Shedding during Bacterial Pathogenesis
4. Syndecans in Cancer
4.1. Syndecans in PDAC
4.1.1. Syndecan-1
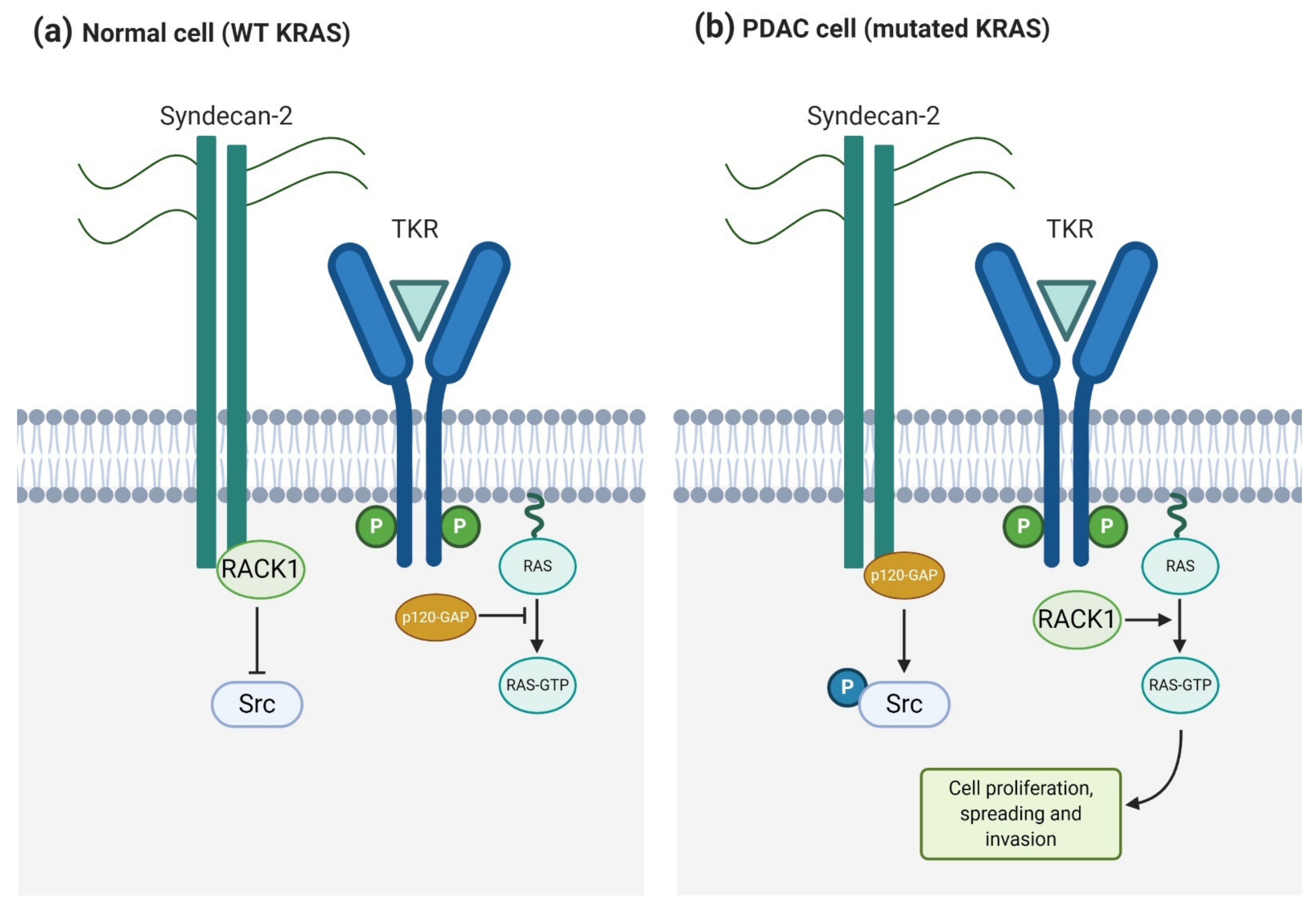
4.1.2. Syndecan-2
4.1.3. Syndecan-3 and -4
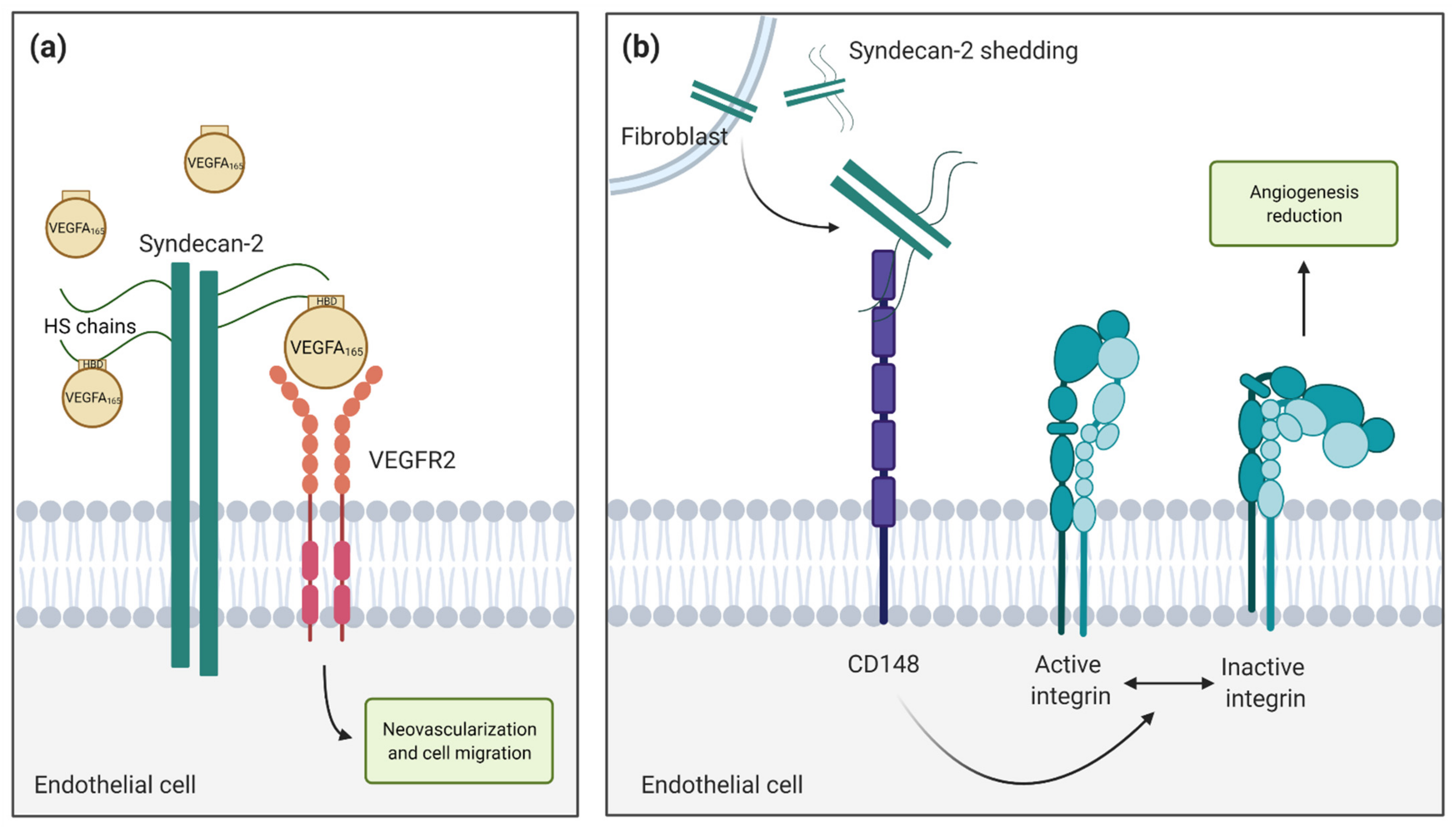
5. Syndecan-2 in Angiogenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mice KO | Phenotype | Conclusion |
|---|---|---|
| SDC1−/− | Reduced Wnt1-induced hyperplasia and inhibition of the Wnt pathway | Syndecan-1 promotes tumorigenesis via the Wnt pathway in breast cancer [18] |
| SDC2−/− | Retinal vascular problems Deficient wound healing | Syndecan-2 is a key angiogenic element [139] |
| SDC4−/− | Delayed skin wound healing and angiogenesis after injury | Syndecan-4 plays important roles in wound healing and angiogenesis [89] |
6. Future Perspectives
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Morales-Oyarvide, V.; Babic, A.; Clish, C.B.; Kraft, P.; Bao, Y.; Qian, Z.R.; Rubinson, D.A.; Ng, K.M.; Giovannucci, E.L.; et al. Cigarette Smoking and Pancreatic Cancer Survival. J. Clin. Oncol. 2017, 35, 1822–1828. [Google Scholar] [CrossRef] [PubMed]
- Goral, V. Pancreatic Cancer: Pathogenesis and Diagnosis. Asian Pac. J. Cancer Prev. 2015, 16, 5619–5624. [Google Scholar] [CrossRef]
- Grant, T.J.; Hua, K.; Singh, A. Molecular Pathogenesis of Pancreatic Cancer. Prog. Mol. Biol. Transl. Sci. 2016, 144, 241–275. [Google Scholar] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor-Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Bray, D.; Lewis, J.; Raff, M.; Roberts, K.; Watson, J.D. Molecular Biology of the Cell, 3rd ed.; W.W Norton & Co.: New York, NY, USA, 1994. [Google Scholar]
- Hynes, R.O. The Extracellular Matrix: Not Just Pretty Fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef]
- Klein, G.; Vellenga, E.; Fraaije, M.W.; Kamps, W.A.; de Bont, E.S.J.M. The Possible Role of Matrix Metalloproteinase (MMP)-2 and MMP-9 in Cancer, e.g., Acute Leukemia. Crit. Rev. Oncol. Hematol. 2004, 50, 87–100. [Google Scholar] [CrossRef]
- Laklai, H.; Miroshnikova, Y.A.; Pickup, M.W.; Collisson, E.A.; Kim, E.; Barrett, A.S.; Hill, R.C.; Lakins, N.J.; Schlaepfer, D.; Mouw, J.K.; et al. Genotype Tunes Pancreatic Ductal Adenocarcinoma Tissue Tension to Induce Matricellular-Fibrosis and Tumor Progression. Nat. Med. 2016, 22, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Weniger, M.; Honselmann, K.C.; Liss, A.S. The Extracellular Matrix and Pancreatic Cancer: A Complex Relationship. Cancers 2018, 10, 316. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, A.; Malvi, P.; Wajapeyee, N. Heparan Sulfate and Heparan Sulfate Proteoglycans in Cancer Initiation and Progression. Front. Endocrinol. 2018, 9, 483. [Google Scholar] [CrossRef]
- Christianson, H.C.; Belting, M. Heparan Sulfate Proteoglycan as a Cell-Surface Endocytosis Receptor. Matrix Biol. 2014, 35, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.R.; Schuksz, M.; Esko, J.D. Heparan Sulphate Proteoglycans Fine-Tune Mammalian Physiology. Nature 2007, 446, 1030–1037. [Google Scholar] [CrossRef]
- Knelson, E.H.; Nee, J.C.; Blobe, G.C. Heparan Sulfate Signalling in Cancer. Trends Biochem. Sci. 2014, 39, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Xian, X.; Gopal, S.; Couchman, J.R. Syndecans as Receptors and Organizers of the Extracellular Matrix. Cell Tissue Res. 2010, 339, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Saunders, S.; Jalkanen, M.; O’Farrell, S.; Bernfield, M. Molecular Cloning of Syndecan, an Integral Membrane Proteoglycan. J. Cell Biol. 1989, 108, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Beauvais, D.L.M.; Rapraeger, A.C. Syndecan-1-Mediated Cell Spreading Requires Signaling by αVβ3 Integrins in Human Breast Carcinoma Cells. Exp. Cell Res. 2003, 286, 219–232. [Google Scholar] [CrossRef]
- Alexander, C.M.; Reichsman, F.; Hinkes, M.T.; Lincecum, J.; Becker, K.A.; Cumberledge, S. Syndecan-1 Is Required for Wnt-1-Induced Mammary Tumorigenesis in Mice. Nat. Genet. 2000, 25, 329–332. [Google Scholar] [CrossRef]
- Malek-Hosseini, Z.; Jelodar, S.; Talei, A.; Ghaderi, A.; Doroudchi, M. Elevated Syndecan-1 Levels in the Sera of Patients with Breast Cancer Correlate with Tumor Size. Breast Cancer 2017, 24, 742–747. [Google Scholar] [CrossRef]
- Szarvas, T.; Reis, H.; vom Dorp, F.; Tschirdewahn, S.; Niedworok, C.; Nyirady, P. Soluble Syndecan-1 (SDC1) Serum Level as an Independent Pre-Operative Predictor of Cancer-Specific Survival in Prostate Cancer. Prostate 2016, 76, 977–985. [Google Scholar] [CrossRef]
- Wei, H.T.; Guo, E.N.; Dong, B.G.; Chen, L.S. Prognostic and Clinical Significance of Syndecan-1 in Colorectal Cancer: A Meta-Analysis. BMC Gastroenterol. 2015, 15, 152. [Google Scholar] [CrossRef] [PubMed]
- Conejo, J.R.; Kleeff, J.; Koliopanos, A.; Matsuda, K.; Zhu, Z.W.; Goecke, H. Syndecan-1 Expression Is up-Regulated in Pancreatic but Not in Other Gastrointestinal Cancers. Int. J. Cancer 2000, 88, 12–20. [Google Scholar] [CrossRef]
- Liu, J.; Yan, L.; Kapoor, A.; Hou, P.; Chen, Z.; Feng, N. Syndecan1 Is a Critical Mediator of Macropinocytosis in Pancreatic Cancer. Nature 2019, 568, 410–414. [Google Scholar]
- Marynen, P.; Zhang, J.; Cassiman, J.J.; Van den Berghe, H.; David, G. Partial Primary Structure of the 48- and 90-Kilodalton Core Proteins of Cell Surface-Associated Heparan Sulfate Proteoglycans of Lung Fibroblasts. Prediction of an Integral Membrane Domain and Evidence for Multiple Distinct Core Proteins at the Cell Surfa. J. Biol. Chem. 1989, 264, 7017–7024. [Google Scholar] [CrossRef]
- Chen, E.; Hermanson, S.; Ekker, S.C. Syndecan-2 Is Essential for Angiogenic Sprouting During Zebrafish Development. Blood 2004, 103, 1710–1719. [Google Scholar] [CrossRef]
- Noguer, O.; Villena, J.; Lorita, J.; Vilaró, S.; Reina, M. Syndecan-2 Downregulation Impairs Angiogenesis in Human Microvascular Endothelial Cells. Exp. Cell Res. 2009, 315, 795–808. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, T.; Abiatari, I.; Raulefs, S.; Sauliunaite, D.; Erkan, M.; Kong, B.; Fries, H.; Michalski, C.W.; Kleeff, J. Syndecan-2 Promotes Perineural Invasion and Cooperates With K-Ras to Induce an Invasive Pancreatic Cancer Cell Phenotype. Mol. Cancer 2012, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Vicente, C.M.; Ricci, R.; Nader, H.B.; Toma, L. Syndecan-2 Is Upregulated in Colorectal Cancer Cells Through Interactions with Extracellular Matrix Produced by Stromal Fibroblasts. BMC Cell Biol. 2013, 14, 25. [Google Scholar] [CrossRef]
- Munesue, S.; Kusano, Y.; Oguri, K.; Itano, N.; Yoshitomi, Y.; Nakanishi, H.; Yamashina, I.; Okayama, M. The Role of Syndecan-2 in Regulation of Actin-Cytoskeletal Organization of Lewis Lung Carcinoma-Derived Metastatic Clones. Biochem. J. 2002, 363, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Carey, D.J.; Evans, D.M.; Stahl, R.C.; Asundi, V.K.; Conner, K.J.; Garbes, P. Molecular Cloning and Characterization of N-Syndecan, a Novel Transmembrane Heparan Sulfate Proteoglycan. J. Cell Biol. 1992, 117, 191–201. [Google Scholar] [CrossRef]
- Gould, S.E.; Upholt, W.B.; Kosher, R.A. Syndecan 3: A Member of the Syndecan Family of Membrane-Intercalated Proteoglycans That Is Expressed in High Amounts at the Onset of Chicken Limb Cartilage Differentiation. Proc. Natl. Acad. Sci. USA 1992, 89, 3271–3275. [Google Scholar] [CrossRef]
- Reizes, O.; Lincecum, J.; Wang, Z.; Goldberger, O.; Huang, L.; Kaksonen, M. Transgenic Expression of Syndecan-1 Uncovers a Physiological Control of Feeding Behavior by Syndecan-3. Cell 2001, 106, 105–116. [Google Scholar] [CrossRef]
- Patterson, A.M.; Cartwright, A.; David, G.; Fitzgeraldm, O.; Bresnihan, B.; Ashton, B.A.; Middleton, J. Differential Expression of Syndecans and Glypicans in Chronically Inflamed Synovium. Ann. Rheum. Dis. 2008, 67, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Arokiasamy, S.; Balderstone, M.J.M.; De Rossi, G.; Whiteford, J.R. Syndecan-3 in Inflammation and Angiogenesis. Front. Immunol. 2020, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- De Witte, L.; Bobardt, M.; Chatterji, U.; Degeest, G.; David, G.; Geijtenbeek, T.B.H. Syndecan-3 Is a Dendritic Cell-Specific Attachment Receptor For HIV-1. Proc. Natl. Acad. Sci. USA 2007, 104, 19464–19469. [Google Scholar] [CrossRef] [PubMed]
- David, G.; van der Schueren, B.; Marynen, P.; Cassiman, J.J.; van den Berghe, H. Molecular Cloning of Amphiglycan, a Novel Integral Membrane Heparan Sulfate Proteoglycan Expressed by Epithelial and FI-Broblastic Cells. J. Cell Biol. 1992, 118, 961–969. [Google Scholar] [CrossRef]
- Kojima, T.; Shworak, N.W.; Rosenberg, R.D. Molecular Cloning and Expression of Two Distinct cDNA-Encoding Heparan Sulfate Proteoglycan Core Proteins from a Rat en-Dothelial Cell Line. J. Biol. Chem. 1992, 267, 4870–4877. [Google Scholar] [CrossRef]
- Chronopoulos, A.; Thorpe, S.D.; Cortes, E.; Lachowski, D.; Rice, A.J.; Mykuliak, V.V. Syndecan-4 Tunes Cell Mechanics by Activating the Kindlin-Integrin-RhoA Pathway. Nat. Mater. 2020, 19, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Couchman, J.R. Syndecans: Proteoglycan Regulators of Cell-Surface Microdomains? Nat. Rev. Mol. Cell Biol. 2003, 4, 926–937. [Google Scholar] [CrossRef]
- Keum, E.; Kim, Y.; Kim, J.; Kwon, S.; Lim, Y.; Han, I. Syndecan-4 Regulates Localization, Activity and Stability of Protein Kinase C-α. Biochem. J. 2004, 378, 1007–1014. [Google Scholar] [CrossRef]
- Greene, D.K.; Tumova, S.; Couchman, J.R.; Woods, A. Syndecan-4 Associates With α-Actinin. J. Biol. Chem. 2003, 278, 7617–7623. [Google Scholar] [CrossRef]
- Afratis, N.; Gialeli, C.; Nikitovic, D.; Tsegenidis, T.; Karousou, E.; Theocharis, A.D.; Pavao, M.S.; Tzanakakis, G.N.; Karamanos, N.K. Glycosaminoglycans: Key Players in Cancer Cell Biology and Treatment. FEBS J. 2012, 279, 1177–1197. [Google Scholar] [CrossRef]
- Yoneda, A.; Couchman, J.R. Regulation of Cytoskeletal Organization by Syndecan Transmembrane Proteoglycans. Matrix Biol. 2003, 22, 25–33. [Google Scholar] [CrossRef]
- Whiteford, J.R.; Behrends, V.; Kirby, H.; Kusche-Gullberg, M.; Muramatsu, T.; Couchman, J.R. Syndecans Promote Integrin-Mediated Adhesion of Mesenchymal Cells in Two Distinct Pathways. Exp. Cell. Res. 2007, 313, 3902–3913. [Google Scholar] [CrossRef]
- Roper, J.A.; Williamson, R.C.; Bass, M.D. Syndecan and Integrin Interactomes: Large Complexes in Small Spaces. Curr Opin. Struct Biol. 2012, 22, 583–590. [Google Scholar] [CrossRef]
- Beauvais, D.L.M.; Burbach, B.J.; Rapraeger, A.C. The Syndecan-1 Ectodomain Regulates αVβ3 Integrin Activily in Human Mammary Carcinoma Cells. J. Cell Biol. 2004, 167, 171–181. [Google Scholar] [CrossRef] [PubMed]
- McQuade, K.J.; Beauvais, D.L.M.; Burbach, B.J.; Rapraeger, A.C. Syndecan-1 Regulates αvβ5 Integrin Activity in B82L Fibroblasts. J. Cell Sci. 2006, 119, 2445–2456. [Google Scholar] [CrossRef]
- Whiteford, J.R.; Xian, X.; Chaussade, C.; Vanhaesebroeck, B.; Nourshargh, S.; Couchman, J.R. Syndecan-2 Is a Novel Ligand for the Protein Tyrosine Phosphatase Receptor CD148. Mol. Biol. Cell 2011, 22, 3609–3624. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Lee, E.; Kwon, S.; Park, H.; Yi, J.Y.; Kim, S.; Han, I.E.; Yun, Y.; Oh, E.S. Transmembrane Domain-Induced Oligomerization Is Crucial for the Functions of Syndecan-2 and Syndecan-4. J. Biol. Chem. 2005, 280, 42573–42579. [Google Scholar] [CrossRef] [PubMed]
- Multhaupt, H.A.; Yoneda, A.; Whiteford, J.R.; Oh, E.S.; Lee, W.; Couchman, J.R. Syndecan Signaling: When, Where and Why? J. Physiol. Pharmacol. 2009, 60, 31–38. [Google Scholar] [PubMed]
- Kinnunen, A.; Kinnunen, T.; Kaksonen, M.; Nolo, R.; Panula, P.; Rauvala, H. N-syndecan and HB-GAM (Heparin-Binding Growth-Associated Molecule) associate with early axonal tracts in the rat brain. Eur. J. Neurosci. 1998, 10, 635–648. [Google Scholar] [CrossRef]
- Granés, F.; Berndt, C.; Roy, C.; Mangeat, P.; Reina, M.; Vilaró, S. Identification of a Novel Ezrin-Binding Site in Syndecan-2 Cytoplasmic Domain. FEBS Lett. 2003, 547, 212–216. [Google Scholar] [CrossRef]
- Kinnunen, T.; Kaksonen, M.; Saarinen, J.; Kalkkinen, N.; Peng, H.B.; Rauvala, H. Cortactin-Src Kinase Signaling Pathway Is Involved in N-Syndecan- Dependent Neurite Outgrowth. J. Biol. Chem. 1998, 273, 10702–10708. [Google Scholar] [CrossRef]
- Zhang, H.G.; Grizzle, W.E. Exosomes: A Novel Pathway of Local and Distant Intercellular Communication That Facilitates the Growth and Metastasis of Neoplastic Lesions. Am. J. Pathol. 2014, 184, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, A.; Murakami, M.; Gao, Y.; Simons, M. Phosphatidylinositol-4,5-Bisphosphate Mediates the Interaction of Syndecan-4 With Protein Kinase C. Biochemistry 1999, 38, 15871–15877. [Google Scholar] [CrossRef]
- Whiteford, J.R.; Ko, S.; Lee, W.; Couchman, J.R. Structural and Cell Adhesion Properties of Zebrafish Syndecan-4 Are Shared with Higher Vertebrates. J. Biol. Chem. 2008, 283, 29322–29330. [Google Scholar] [CrossRef]
- Lim, S.T.; Longley, R.L.; Couchman, J.R.; Woods, A. Direct Binding of Syndecan-4 Cytoplasmic Domain to the Catalytic Domain of Protein Kinase Cα (PKCα) Increases Focal Adhesion Localization of PKCα. J. Biol Chem. 2003, 278, 13795–13802. [Google Scholar] [CrossRef] [PubMed]
- Dovas, A.; Choi, Y.; Yoneda, A.; Multhaupt, H.A.B.; Kwon, S.H.; Kang, D. Serine 34 Phosphorylation of Rho Guanine Dissociation Inhibitor (RhoGDIα) Links Signaling from Conventional Protein Kinase C to RhoGTPase in Cell Adhesion. J. Biol. Chem. 2010, 285, 23296–23308. [Google Scholar] [CrossRef]
- Koo, B.K.; Jung, Y.S.; Shin, J.; Han, I.; Mortier, E.; Zimmermann, P. Structural Basis of Syndecan-4 Phosphorylation as a Molecular Switch to Regulate Signaling. J. Mol. Biol. 2006, 355, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Kim, S.; Lee, J.; Ko, S.G.; Lee, W.; Han, I.O.; Woods, A.; Oh, E.S. The Oligomeric Status of Syndecan-4 Regulates Syndecan-4 Interaction With α-Actinin. Eur. J. Cell Biol. 2008, 87, 807–815. [Google Scholar] [CrossRef]
- Couchman, J.R. Transmembrane Signaling Proteoglycans. Annu. Rev. Cell Dev. Biol. 2010, 26, 89–114. [Google Scholar] [CrossRef]
- Gitay-Goren, H.; Soker, S.; Vlodavsky, I.; Neufeld, G. The Binding of Vascular Endothelial Growth Factor to Its Receptors Is Dependent on Cell Surface-Associated Heparin-Like Molecules. J. Biol. Chem. 1992, 267, 6093–6098. [Google Scholar] [CrossRef]
- Allen, B.L.; Filla, M.S.; Rapraeger, A.C. Role of Heparan Sulfate as a Tissue-Specific Regulator of FGF-4 and FGF Receptor Recognition. J. Cell. Biol. 2001, 155, 845–857. [Google Scholar] [CrossRef]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan Sulfate Proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–33. [Google Scholar] [CrossRef]
- Wang, H.; Jin, H.; Rapraeger, A.C. Syndecan-1 and Syndecan-4 Capture Epidermal Growth Factor Receptor Family Members and the α3β1 Integrin via Binding Sites in Their Ecto-Domains: Novel Synstatins Prevent Kinase Capture and Inhibitα6β4-Integrindependent Epithelial Cell Motility. J. Biol. Chem. 2015, 290, 26103–26113. [Google Scholar] [CrossRef]
- Beauvais, D.M.; Ell, B.J.; McWhorter, A.R.; Rapraeger, A.C. Syndecan-1 Regulates αvβ3 and αvβ5 Integrin Activation During Angiogenesis and Is Blocked by Synstatin, a Novel Peptide Inhibitor. J. Exp. Med. 2009, 206, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Beauvais, D.L.M.; Rapraeger, A.C. Syndecan-1 Couples the Insulin-Like Growth Factor-1 Receptor to Inside-Out Integrin Activation. J. Cell Sci. 2010, 123, 3796–3807. [Google Scholar] [CrossRef]
- Rapraeger, A.C. Synstatin: A Selective Inhibitor of the Syndecan-1-Coupled IGF1R-αvβ3 Integrin Complex in Tumorigenesis and Angiogenesis. FEBS J. 2013, 280, 2207–2215. [Google Scholar] [CrossRef]
- Avalos, A.M.; Valdivia, A.D.; Muñoz, N.; Herrera-Molina, R.; Tapia, J.C.; Lavandero, S. Neuronal Thy-1 Induces Astrocyte Adhesion by Engaging Syndecan-4 in a Cooperative Interaction With αVβ3 Integrin That Activates PKCα and RhoA. J. Cell Sci. 2009, 122, 3462–3471. [Google Scholar] [CrossRef]
- Mostafavi-Pour, Z.; Askari, J.A.; Parkinson, S.J.; Parker, P.J.; Ng, T.T.; Humphries, M.J. Integrin-Specific Signaling Pathways Controlling Focal Adhesion Formation and Cell Migration. J. Cell Biol. 2003, 161, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Fiore, V.F.; Ju, L.; Chen, Y.; Zhu, C.; Barker, T.H. Dynamic Catch of a Thy-1-α5 β1 + Syndecan-4 Trimolecular Complex. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Mao, D.; Zhang, Y.; Lu, H.; Zhang, H. Molecular Basis Underlying Inhibition of Metastasis of Gastric Cancer by Anti-VEGFa Treatment. Tumor Biol. 2014, 35, 8217–8223. [Google Scholar] [CrossRef]
- Rao, V.H.; Kansal, V.; Stoupa, S.; Agrawal, D.K. MMP-1 and MMP-9 Regulate Epidermal Growth Factor-Dependent Collagen Loss in Human Carotid Plaque Smooth Muscle Cells. Physiol. Rep. 2014, 2, e00224. [Google Scholar] [CrossRef]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73. [Google Scholar]
- Nagase, H.; Visse, R.; Murphy, G. Structure and Function of Matrix Metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef]
- Visse, R.; Nagase, H. Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases: Structure, Function, and Biochemistry. Circ. Res. 2003, 92, 827–839. [Google Scholar] [CrossRef]
- Gopal, S. Syndecans in Inflammation at a Glance. Front. Immunol. 2020, 11, 1–8. [Google Scholar] [CrossRef]
- Subramanian, S.V.; Fitzgerald, M.L.; Bernfield, M. Regulated Shedding of Syndecan-1 and-4 Ectodomains by Thrombin and Growth Factor Receptor Activation. J. Biol. Chem. 1997, 272, 14713–14720. [Google Scholar] [CrossRef]
- Brule, S.; Charnaux, N.; Sutton, A.; Ledoux, D.; Chaigneau, T.; Saffar, L.; Gattegno, L. The Shedding of Syndecan-4 and Syndecan-1 From Hela Cells and Human Primary Macrophages Is Accelerated by SDF-1/CXCL12 and Mediated by the Matrix Metalloproteinase-9. Glycobiology 2006, 16, 488–501. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hayashida, A.; Bennett, A.E.; Hollingshead, S.K.; Pyong, W.P. Streptococcus Pneumoniae Sheds Syndecan-1 Ectodomains Through ZmpC, a Metalloproteinase Virulence Factor. J. Biol. Chem. 2007, 282, 159–167. [Google Scholar] [CrossRef]
- Jalkanen, M.; Rapraeger, A.; Saunders, S.; Bernfield, M. Cell Surface Proteoglycan of Mouse Mammary Epithelial Cells Is Shed by Cleavage of Its Matrix-Binding Ectodomain From Its Membrane-Associated Domain. J. Cell. Biol. 1987, 105, 3087–3096. [Google Scholar] [CrossRef]
- Wang, J.B.; Guan, J.; Shen, J.; Zhou, L.; Zhang, Y.J.; Si, Y.F.; Yang, L.; Jian, X.-h.; Sheng, Y. Insulin Increases Shedding of Syndecan-1 in the Serum of Patients with Type 2 Diabetes Mellitus. Diabetes Res. Clin. Pr. 2009, 86, 83–88. [Google Scholar] [CrossRef]
- Yang, Y.; MacLeod, V.; Miao, H.Q.; Theus, A.; Zhan, F.; Shaughnessy, J.D.; Sawyer, J.; Li, J.P.; Zcharia, E.; Vlodavsky, I.; et al. Heparanase Enhances Syndecan-1 Shedding: A Novel Mechanism for Stimulation of Tumor Growth and Metastasis. J. Biol. Chem. 2007, 282, 13326–13333. [Google Scholar] [CrossRef] [PubMed]
- Manon-Jensen, T.; Itoh, Y.; Couchman, J.R. Proteoglycans in Health and Disease: The Multiple Roles of Syndecan Shedding. FEBS J. 2010, 277, 3876–3889. [Google Scholar] [CrossRef]
- Götte, M.; Joussen, A.M.; Klein, C.; Andre, P.; Wagner, D.D.; Hinkes, M.T. Role of Syndecan-1 in Leukocyte-Endothelial Interactions in the Ocular Vasculature. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1135–1141. [Google Scholar]
- Götte, M. Syndecans in Inflammation. FASEB J. 2003, 17, 575–591. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Song, H.K.; Hwang, E.S.; Lee, A.R.; Han, D.S.; Kim, S.E. Up-Regulation of Syndecan-2 in Proximal Colon Correlates with Acute Inflammation. FASEB J. 2019, 33, 11381–11395. [Google Scholar] [CrossRef] [PubMed]
- Vuong, T.T.; Reine, T.M.; Sudworth, A.; Jenssen, T.G.; Kolset, S.O. Syndecan-4 Is a Major Syndecan in Primary Human Endothelial Cells In Vitro, Modulated by Inflammatory Stimuli and Involved in Wound Healing. J. Histochem. Cytochem. 2015, 63, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Echtermeyer, F.; Streit, M.; Wilcox-Adelman, S.; Saoncella, S.; Denhez, F.; Detmar, M. Delayed Wound Repair and Impaired Angiogenesis in Mice Lacking Syndecan-4. J Clin Investig. 2001, 107, 9–14. [Google Scholar] [CrossRef]
- Chen, P.; Abacherli, L.E.; Nadler, S.T.; Wang, Y.; Li, Q.; Parks, W.C. MMP7 Shedding of Syndecan-1 Facilitates Re-Epithelialization by Affecting α2β1 Integrin Activation. PLoS ONE 2009, 4, e6565. [Google Scholar] [CrossRef] [PubMed]
- Purushothaman, A.; Uyama, T.; Kobayashi, F.; Yamada, S.; Sugahara, K.; Rapraeger, A.C. Heparanase-Enhanced Shedding of Syndecan-1 by Myeloma Cells Promotes Endothelial Invasion and Angiogenesis. Blood 2010, 115, 2449–2457. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, J.; Bollmann, M. Soluble Syndecans: Biomarkers for Diseases and Therapeutic Options. Br. J. Pharmacol. 2019, 176, 67–81. [Google Scholar] [CrossRef]
- Park, P.W.; Foster, T.J.; Nishi, E.; Duncan, S.J.; Klagsbrun, M.; Chen, Y. Activation of Syndecan-1 Ectodomain Shedding by Staphylococcus aureus α-Toxin and β-Toxin. J. Biol. Chem. 2004, 279, 251–258. [Google Scholar] [CrossRef]
- Haynes, A.; Ruda, F.; Oliver, J.; Hamood, A.N.; Griswold, J.A.; Park, P.W. Syndecan 1 Shedding Contributes to Pseudomonas Aeruginosa Sepsis. Infect Immun. 2005, 73, 7914–7921. [Google Scholar] [CrossRef]
- Sanderson, R.D.; Yang, Y. Syndecan-1: A Dynamic Regulator of the Myeloma Microenvironment. Clin. Exp. Metastasis 2008, 25, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Alexander, C.M.; Friedl, A. Induction of Syndecan-1 Expression in Stromal Fibroblasts Promotes Proliferation of Human Breast Cancer Cells. Cancer Res. 2004, 64, 612–621. [Google Scholar] [CrossRef]
- Jagannath, S.; Heffner, L.T.; Ailawadhi, S.; Munshi, N.C.; Zimmerman, T.M.; Rosenblatt, J. Indatuximab Ravtansine (BT062) Monotherapy in Patients with Relapsed and/or Refractory Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2019, 19, 372–380. [Google Scholar] [CrossRef]
- Lim, H.C.; Couchman, J.R. Syndecan-2 Regulation of Morphology in Breast Carcinoma Cells Is Dependent on RhoGTPases. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 2482–2490. [Google Scholar] [CrossRef]
- Farnedi, A.; Rossi, S.; Bertani, N.; Gulli, M.; Silini, E.M.; Mucignat, M.T.; Poli, T.; Sesenna, E.; Lanfranco, D.; Montebugnoli, L.; et al. Proteoglycan-Based Diversification of Disease Outcome in Head and Neck Cancer Patients Identifies NG2/CSPG4 and Syndecan-2 as Unique Relapse and Overall Survival Predicting Factors. BMC Cancer 2015, 15, 352. [Google Scholar] [CrossRef]
- Diamantopoulou, Z.; Kitsou, P.; Menashi, S.; Courty, J.; Katsoris, P. Loss of Receptor Protein Tyrosine Phosphatase β/ζ (RPTPβ/ζ) Promotes Prostate Cancer Metastasis. J. Biol. Chem. 2012, 287, 40339–40349. [Google Scholar] [CrossRef] [PubMed]
- Roskams, T.; De Vos, R.; David, G.; Van Damme, B.; Desmet, V. Heparan Sulphate Proteoglycan Expression in Human Primary Liver Tumours. J. Pathol. 1998, 185, 290–297. [Google Scholar] [CrossRef]
- Tsonis, A.I.; Afratis, N.; Gialeli, C.; Ellina, M.I.; Piperigkou, Z.; Skandalis, S.S. Evaluation of the Coordinated Actions of Estrogen Receptors with Epidermal Growth Factor Receptor and Insulin-Like Growth Factor Receptor in the Expression of Cell Surface Heparan Sulfate Proteoglycans and Cell Motility in Breast Cancer Cells. FEBS J. 2013, 280, 2248–2259. [Google Scholar] [CrossRef] [PubMed]
- Mundhenke, C.; Meyer, K.; Drew, S.; Friedl, A. Heparan Sulfate Proteoglycans as Regulators of Fibroblast Growth Factor-2 Receptor Binding in Breast Carcinomas. Am. J. Pathol. 2002, 160, 185–194. [Google Scholar] [CrossRef]
- Storz, P.; Crawford, H.C. Carcinogenesis of Pancreatic Ductal Adenocarcinoma. Gastroenterology 2020, 158, 2072–2081. [Google Scholar] [CrossRef]
- Juuti, A.; Nordling, S.; Lundin, J.; Louhimo, J.; Haglund, C. Syndecan-1 Expression—A Novel Prognostic Marker in Pancreatic Cancer. Oncology 2005, 68, 97–106. [Google Scholar] [CrossRef]
- Koliopanos, A.; Friess, H.; Kleeff, J.; Shi, X.; Liao, Q.; Pecker, I. Heparanase Expression in Primary and Metastatic Pancreatic Cancer. Cancer Res. 2001, 61, 4655–4659. [Google Scholar] [PubMed]
- Rohloff, J.; Zinke, J.; Schoppmeyer, K.; Tannapfel, A.; Witzigmann, H.; Mössner, J. Heparanase Expression Is a Prognostic Indicator for Postoperative Survival in Pancreatic Adenocarcinoma. Br. J. Cancer 2002, 86, 1270–1275. [Google Scholar] [CrossRef]
- Hoffmann, A.C.; Mori, R.; Vallbohmer, D.; Brabender, J.; Drebber, U.; Baldus, S.E. High Expression of Heparanase Is Significantly Associated with Dedifferentiation and Lymph Node Metastasis in Patients With Pancreatic Ductal Adenocarcinomas and Correlated to PDGFA and via HIF1a to HB-EGF and bFGF. J. Gastrointest. Surg. 2008, 12, 1674–1681. [Google Scholar] [CrossRef] [PubMed]
- Meirovitz, A.; Hermano, E.; Lerner, I.; Zcharia, E.; Pisano, C.; Peretz, T.; Elkin, M. Role of Heparanase in Radiation-Enhanced Invasiveness of Pancreatic Carcinoma. Cancer Res. 2011, 71, 2772–2780. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, H.; Chen, C.; Li, J.; He, J.; Fu, X. The HPA/SDC1 Axis Promotes Invasion and Metastasis of Pancreatic Cancer Cells by Activating EMT via FGF2 Upregulation. Oncol. Lett. 2019, 19, 211–220. [Google Scholar] [CrossRef]
- Ding, K.; Lopez-Burks, M.; Sánchez-Duran, J.A.; Korc, M.; Lander, A.D. Growth Factor-Induced Shedding of Syndecan-1 Confers Glypican-1 Dependence on Mitogenic Responses of Cancer Cells. J. Cell Biol. 2005, 171, 729–738. [Google Scholar] [CrossRef]
- Ramani, V.C.; Sanderson, R.D. Chemotherapy Stimulates Syndecan-1 Shedding: A Potentially Negative Effect of Treatment That May Promote Tumor Relapse. Matrix Biol. 2014, 35, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, H.; Anttonen, A.; Eriksson, M.; Mäkitaro, R.; Alfthan, H.; Kinnula, V.; Sirpa, L. Soluble Syndecan-1 and Serum Basic Fibroblast Growth Factor Are New Prognostic Factors in Lung Cancer. Cancer Res. 2002, 62, 5210–5217. [Google Scholar] [PubMed]
- Yamamoto, H.; Itoh, F.; Iku, S.; Adachi, Y.; Fukushima, H.; Sasaki, S.; Mukaiya, M.; Hirata, K.; Imai, K. Expression of Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases in Human Pancreatic Adenocarcinomas: Clinicopathologic and Prognostic Significance of Matrilysin Expression. J. Clin. Oncol. 2001, 19, 1118–1127. [Google Scholar] [CrossRef]
- Crawford, H.C.; Scoggins, C.R.; Washington, M.K.; Matrisian, L.M.; Leach, S.D. Matrix Metalloproteinase-7 Is Expressed by Pancreatic Cancer Precursors and Regulates Acinar-to-Ductal Metaplasia in Exocrine Pancreas. J. Clin. Investig. 2002, 109, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Blaine, S.A.; Qiao, D.; Friedl, A. Shedding of Syndecan-1 by Stromal Fibroblasts Stimulates Human Breast Cancer Cell Proliferation via FGF2 Activation. J. Biol. Chem. 2007, 282, 14906–14915. [Google Scholar] [CrossRef]
- Nikolova, V.; Koo, C.-Y.; Ibrahim, S.A.; Wang, Z.; Spillmann, D.; Dreier, R. Differential Roles for Membrane-Bound and Soluble Syndecan-1 (CD138) in Breast Cancer Progression. Carcinogenesis 2009, 30, 397–407. [Google Scholar] [CrossRef]
- Wu, Y.; Huang, H.; Fervers, B.; Lu, L. Syndecan-1 and KRAS Gene Expression Signature Associates with Patient Survival in Pancreatic Cancer. Pancreas 2020, 49, 1187–1194. [Google Scholar] [CrossRef]
- Huang, J.W.; Chen, C.L.; Chuang, N.N. P120-GAP Associated with Syndecan-2 to Function as an Active Switch Signal for Src Upon Transformation with Oncogenic Ras. Biochem. Biophys. Res. Commun. 2005, 329, 855–862. [Google Scholar] [CrossRef]
- Wheeler, D.L.; Iida, M.; Dunn, E.F. The Role of Src in Solid Tumors. Oncologist 2009, 14, 667. [Google Scholar] [CrossRef]
- Yao, J.; Zhang, L.L.; Huang, X.M.; Li, W.Y.; Gao, S.G. Pleiotrophin and N-Syndecan Promote Perineural Invasion and Tumor Progression in an Orthotopic Mouse Model of Pancreatic Cancer. World J. Gastroenterol. 2017, 23, 3907–3914. [Google Scholar] [CrossRef]
- Yao, J.; Li, W.Y.; Li, S.G.; Feng, X.S.; Gao, S.G. Midkine Promotes Perineural Invasion in Human Pancreatic Cancer. World J. Gastroenterol. 2014, 20, 3018–3024. [Google Scholar] [CrossRef] [PubMed]
- Brioudes, E.; Alibashe-Ahmed, M.; Lavallard, V.; Berney, T.; Bosco, D. Syndecan-4 Is Regulated by IL-1β in β-Cells and Human Islets. Mol. Cell Endocrinol. 2020, 510, 110815. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.Y.C.; Whitelock, J.; Poole-Warren, L. Syndecan-4 Is Associated with Beta-Cells in the Pancreas and the min6 Beta-Cell Line. Histochem. Cell Biol. 2012, 138, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic Reaction in Pancreatic Cancer: Role of Pancreatic Stellate Cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef]
- Fogh, B.S.; Multhaupt, H.A.B.; Couchman, J.R. Protein Kinase C, Focal Adhesions and the Regulation of Cell Migration. J. Histochem. Cytochem. 2014, 62, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Gopal, S.; Bober, A.; Whiteford, J.R.; Multhaupt, H.A.B.; Yoneda, A.; Couchman, J.R. Heparan Sulfate Chain Valency Controls Syndecan-4 Function in Cell Adhesion. J Biol. Chem. 2010, 285, 14247–14258. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, D.A.; Campbell, I.S.; Critchley, D.R. Talins and Kindlins: Partners in Integrin-Mediated Adhesion. Nat. Rev. Mol. 2013, 14, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Takehara, Y.; Kawase, T.; Terashima, K.; Ohkawa, Y.; Hirose, Y.; Koda, A.; Hyodo, N.; Ushio, T.; Hirai, Y.; et al. Feasibility of Magnetic Resonance Elastography for the Pancreas at 3T. J. Magn. Reson. Imaging 2016, 43, 384–390. [Google Scholar] [CrossRef]
- Rubiano, A.; Delitto, D.; Han, S.; Gerber, M.; Galitz, C.; Trevino, J.; Thomas, R.M.; Hughes, S.J.; Simmons, C.S. Viscoelastic Properties of Human Pancreatic Tumors and in Vitro Constructs to Mimic Mechanical Properties. Acta Biomater. 2017, 67, 331–340. [Google Scholar] [CrossRef]
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.P.; Hernandez, A.D.R. Matrix Stiffness Induces Epithelial—Mesenchymal Transition and Promotes Chemoresistance in Pancreatic Cancer Cells. Oncogenesis 2017, 6, e342–e352. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J.; Shing, Y. Angiogenesis. J. Biol. Chem. 1992, 267, 10931–10934. [Google Scholar] [CrossRef]
- Hanahan, D.; Folkman, J. Patterns and Emerging Mechanisms of the Angiogenic Switch during Tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef]
- Yancopoulos, G.D.; Davis, S.; Gale, N.W.; Rudge, J.S.; Wiegand, S.J.; Holash, J. Vascular-Specific Growth Factors and Blood Vessel Formation. Nature 2000, 407, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Essner, J.J.; Chen, E.; Ekker, S.C. Syndecan-2. Int. J. Biochem. Cell Biol. 2006, 38, 152–156. [Google Scholar]
- Clasper, S.; Vekemans, S.; Fiore, M.; Plebanski, M.; Wordsworth, P.; David, G. Inducible Expression of the Cell Surface Heparan Sulfate Proteoglycan Syndecan-2 (Fibroglycan) on Human Activated Macro-Phages Can Regulate Fibroblast Growth Factor Action. J. Biol. Chem. 1999, 274, 24113–24123. [Google Scholar] [CrossRef] [PubMed]
- Noguer, O.; Reina, M. Is Syndecan-2 a Key Angiogenic Element? Sci. World J. 2009, 9, 729–732. [Google Scholar] [CrossRef]
- Grünewald, F.S.; Prota, A.E.; Giese, A.; Ballmer-Hofer, K. Structure-Function Analysis of VEGF Receptor Activation and the Role of Coreceptors in Angiogenic Signaling. Biochim. Biophys. Acta Proteins Proteom. 2010, 1804, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Corti, F.; Wang, Y.; Rhodes, J.M.; Atri, D.; Archer-Hartmann, S.; Zhang, J. N-Terminal Syndecan-2 Domain Selectively Enhances 6-O Heparan Sulfate Chains Sulfation and Promotes VEGFA165-Dependent Neovascularization. Nat. Commun. 2019, 10, 1–14. [Google Scholar]
- Oh, E.S.; Couchman, J.R. Syndecans-2 and-4; Close Cousins, but Not Identical Twins. Mol. Cells 2004, 17, 181–187. [Google Scholar]
- Ono, K.; Hattori, H.; Takeshita, S.; Kurita, A.; Ishihara, M. Structural Features in Heparin That Interact with VEGF165 and Modulate Its Biological Activity. Glycobiology 1999, 9, 705–711. [Google Scholar] [CrossRef]
- Robinson, C.J.; Mulloy, B.; Gallagher, J.T.; Stringer, S.E. VEGF165-Binding Sites Within Heparan Sulfate Encompass Two Highly Sulfated Domains and Can Be Liberated by K5 Lyase. J. Biol. Chem. 2006, 281, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- De Rossi, G.; Evans, A.R.; Kay, E.; Woodfin, A.; McKay, T.R.; Nourshargh, S. Shed Syndecan-2 Inhibits Angiogenesis. J. Cell Sci. 2014, 127, 4788–4799. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Malesci, A.; Tommasini, M.A.; Bonato, C.; Bocchia, P.; Bersani, M.; Zerbi, A. Determination of CA 19-9 Antigen in Serum and Pancreatic Juice for Differential Diagnosis of Pancreatic Adenocarcinoma from Chronic Pancreatitis. Gastroenterology 1987, 92, 60–67. [Google Scholar] [CrossRef]
- Torphy, R.J.; Fujiwara, Y.; Schulick, R.D. Pancreatic Cancer Treatment: Better, but a Long Way to Go. Surg. Today 2020, 50, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Bamlet, W.R.; Oberg, A.L.; Chaffee, K.G.; Donahue, G.; Cao, J.; Chari, S.; Garcia, B.A.; Petersen, G.M.; Zaret, K.S. Detection of Early Pancreatic Ductal Adenocarcinoma With thrombospondin-2 and CA19-9 Blood Markers. Sci. Transl. Med. 2017, 9, eaah5583. [Google Scholar] [CrossRef] [PubMed]
- Regel, I.; Mayerle, J.; Mahajan, U.M. Current Strategies and Future Perspectives for Precision Medicine in Pancreatic Cancer. Cancers 2020, 12, 1024. [Google Scholar] [CrossRef]
- Iovanna, J.; Mallmann, M.C.; Gonçalves, A.; Turrini, O.; Dagorn, J.C. Current Knowledge on Pancreatic Cancer. Front. Oncol. 2012, 2. [Google Scholar] [CrossRef]
- Mutgan, A.C.; Besikcioglu, H.E.; Wang, S.; Friess, H.; Ceyhan, G.O.; Demir, I.E. Insulin/IGF-Driven Cancer Cell-Stroma Crosstalk as a Novel Therapeutic Target in Pancreatic Cancer. Mol. Cancer 2018, 17, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Tao, X.; Li, C.B.; Wang, C.M. MicroRNA-494 Acts as a Tumor Suppressor in Pancreatic Cancer, Inhibiting Epithelial-Mesenchymal Transition, Migration and Invasion by Binding to SDC1. Int. J. Oncol. 2018, 53, 1204–1214. [Google Scholar] [CrossRef]
- Li, L.; Li, Z.; Kong, X.; Xie, D.; Jia, Z.; Jiang, W. Down-Regulation of MicroRNA-494 via Loss of SMAD4 Increases FOXM1 and β-Catenin Signaling in Pancreatic Ductal Adenocarcinoma Cells. Gastroenterology 2014, 147, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, X.; Zhu, S.; Zhang, J.G.; Yang, M.; Qin, Q. Ectopic Expression of MIR-494 Inhibited the Proliferation, Invasion and Chemoresistance of Pancreatic Cancer by Regulating SIRT1 and c-Myc. Gene Ther. 2015, 22, 729–738. [Google Scholar] [CrossRef]
- Kimbrough, C.W.; Hudson, S.; Khanal, A.; Egger, M.E.; McNally, L.R. Orthotopic Pancreatic Tumors Detected by Optoacoustic Tomography Using Syndecan-1. J. Surg. Res. 2015, 193, 246–254. [Google Scholar] [CrossRef][Green Version]
- Hrabar, D.; Aralica, G.; Gomerčić, M.; Ljubičić, N.; Krušlin, B.; Tomas, D. Epithelial and Stromal Expression of Syndecan-2 in Pancreatic Carcinoma. Anticancer Res. 2010, 30, 2749–2753. [Google Scholar]
- Griffith, L.G.; Swartz, M.A. Capturing Complex 3d Tissue Physiology In Vitro. Nat. Rev. Mol. Cell Biol. 2006, 7, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Gagliano, N.; Sforza, C.; Sommariva, M.; Menon, A.; Conte, V.; Sartori, P. 3D-Spheroids: What Can They Tell Us About Pancreatic Ductal Adenocarcinoma Cell Phenotype? Exp. Cell. Res. 2017, 357, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Betriu, N.; Semino, C.E. Development of a 3D Co-Culture System as a Cancer Model Using a Self-Assembling Peptide Scaffold. Gels 2018, 4, 65. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Muiños, T.; Recha-Sancho, L.; López-Chicón, P.; Castells-Sala, C.; Mata, A.; Semino, C.E. Bimolecular Based Heparin and Self-Assembling Hydrogel for Tissue Engineering Applications. Acta Biomater. 2015, 16, 35–48. [Google Scholar] [CrossRef] [PubMed]





| Syndecan | Main Location | Function |
|---|---|---|
| Syndecan-1 | Epithelial and plasma cells [16] | Cooperates with several integrins (αvβ3, αvβ5, α2β1, α3β1, and α6β4) through the core protein. Plays roles in matrix remodeling, cell adhesion and spreading, migration, and cytoskeleton rearrangements [17]. Present in breast [18,19], prostate [20], colorectal [21], and pancreatic [22,23] cancers |
| Syndecan-2 | Mesenchymal cells [24] | Important regulator of angiogenesis [25,26]. Present in some cancers such as PDAC [27] and colon cancer [28]. Regulates actin cytoskeleton organization, especially in lung cancer [29] |
| Syndecan-3 | Brain, nervous system, and cartilage [30,31] | Important for brain development as well as in feeding behaviors (is upregulated in the hypothalamus in response to food deprivation] [32]. Plays roles in rheumatoid arthritis disease [33], angiogenesis [34], and HIV-1 infection [35] |
| Syndecan-4 | Most tissues [36,37] | Plays roles in cell mechanics [38] and induces focal adhesion formation [39] and cytoskeleton organization [40,41] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Betriu, N.; Bertran-Mas, J.; Andreeva, A.; Semino, C.E. Syndecans and Pancreatic Ductal Adenocarcinoma. Biomolecules 2021, 11, 349. https://doi.org/10.3390/biom11030349
Betriu N, Bertran-Mas J, Andreeva A, Semino CE. Syndecans and Pancreatic Ductal Adenocarcinoma. Biomolecules. 2021; 11(3):349. https://doi.org/10.3390/biom11030349
Chicago/Turabian StyleBetriu, Nausika, Juan Bertran-Mas, Anna Andreeva, and Carlos E. Semino. 2021. "Syndecans and Pancreatic Ductal Adenocarcinoma" Biomolecules 11, no. 3: 349. https://doi.org/10.3390/biom11030349
APA StyleBetriu, N., Bertran-Mas, J., Andreeva, A., & Semino, C. E. (2021). Syndecans and Pancreatic Ductal Adenocarcinoma. Biomolecules, 11(3), 349. https://doi.org/10.3390/biom11030349