1. Introduction
Camelid single-domain antibodies (sdAbs) [
1], also referred to as nanobodies
® (a trademark of Ablynx), are employed in fundamental research and clinical diagnostics, and as promising candidates for therapeutic applications. Due to their small size of ~15 kDa, easy genetic accessibility and low production cost, they are ideal tools for many fields of life sciences [
1,
2,
3,
4,
5]. Furthermore, sdAbs can be easily selected from phage-, bacterial-, or yeast-display libraries, allowing for a specific selection of binders against desired target proteins [
6,
7,
8]. Although the selection of sdAbs from immune libraries generated from peripheral blood mononuclear cells (PBMCs) of immunized alpacas, llamas or camels is a straightforward process, provoking the required immune responses in these animals can be challenging. This is the case, in particular, for toxic or highly conserved proteins. In addition, immunizing animals is a time-consuming process with intrinsically unpredictable outcomes regarding the nature, quality and specificity of the sdAbs obtained. To overcome these limitations, some fully synthetic libraries have been developed, typically based on commonly found immunoglobulin frameworks present in camelid sdAbs [
9,
10]. While most sdAbs have been selected and optimized to display high affinity towards their antigens (for examples, see [
11,
12,
13,
14,
15]), the generation of mid- and low-affinity sdAbs is of growing interest [
16]. For instance, reversible binding to a specific target protein can be advantageous not only for in vivo and therapeutic use [
16] but also for biochemical applications, enabling binding and elution under physiological conditions and thereby preserving the structure and function of selected target proteins [
16,
17].
Recently, we introduced the ALFA system, comprising the rationally designed ALFA-tag and a set of highly versatile sdAbs binding ALFA-tagged proteins with extraordinary specificity regardless of the position of the tag on the protein. The high-affinity sdAb (NbALFA) shows tight binding to ALFA-tagged proteins (K
d~25 pM) with a very slow off-rate and is ideal for applications requiring stable interactions [
15]. It may, however, be suboptimal for a range of biochemical applications, as it fails to release targets under physiological conditions. In order to solve this problem, we introduced a second sdAb (NbALFA
PE for “peptide-elutable”) showing a strongly enhanced off-rate (K
d~11 nM). An agarose-based resin featuring immobilized NbALFA
PE (ALFA Selector
PE) allows for an efficient competitive elution of ALFA-tagged proteins under physiological conditions. In summary, the ALFA system is ideally suited for a broad spectrum of applications, including high-resolution microscopy (e.g., fluorescence microscopy, STED microscopy, DNA-PAINT, etc.) [
18,
19,
20], in vivo detection and manipulation of living cells [
21,
22], intracellular proteins [
22,
23], and also advanced biochemical experimentation [
15,
24]. Therefore, it offers a superior and versatile alternative to most common epitope tag systems [
25].
While competitive elution from ALFA Selector
PE works exceptionally well in batch or stopped-flow elution protocols performed at room temperature, the elution efficiency is impaired at lower temperatures. This may limit the application of the ALFA system, especially for the purification of temperature-sensitive targets or delicate protein complexes, or in the case that it is essential to perfectly preserve structure and function. Therefore, we set out to complement the ALFA system by adding a third sdAb that allows for the efficient capture and elution of proteins at 4 °C and under physiological buffer conditions while maintaining the exquisite specificity and favorable biochemical properties of the existing NbALFA variants [
15]. To achieve this, we combined a structure-guided protein engineering approach with a novel off-rate-driven phage display selection. We demonstrate that the applied rational design principles in combination with an application-specific selection protocol can lead to affinity reagents fulfilling the experimental requirements in a directed fashion. We finally present NbALFA
CE (for “cold-elutable”), a new member of the ALFA system, which proves to be an ideal tool for the purification and elution of ALFA-tagged target proteins at cold temperatures in physiological buffer.
2. Materials and Methods
2.1. Animal Handling—Immunizations
All the work involving animal experiments was conducted in compliance with ethical regulations for animal research and testing. All experiments conducted did not require ethical approval, but were communicated to and accepted by the local authorities (LAVES, Niedersachsen, Germany). Two alpacas were immunized six times at 14-day intervals with a total of 0.5 mg of ALFA peptide conjugated to keyhole limpet hemocyanin (KLH). The first immunization was performed using complete Freund’s adjuvant; for all the following immunizations, incomplete Freund’s adjuvant was used. Five days after the last immunization, 100 mL of peripheral blood was taken from each animal and immediately supplemented with 5000 IU/mL of heparin to prevent clotting.
2.2. Preparation of Phagemid Libraries
Two independent phagemid libraries were constructed for the selection of ALFA-specific binders that can be peptide-eluted at 4 °C. The first library was prepared from total ribonucleic acid (RNA) isolated from peripheral blood mononuclear cells (PBMCs) obtained from fresh alpaca blood. The PBMCs were isolated using Ficoll-Paque PLUS (GE Healthcare, Uppsala, Sweden). Subsequently, total RNA was isolated using a NucleoSpin RNA plus kit (Macherey Nagel, Düren, Germany). The obtained RNA was used for a reverse transcription reaction using Superscript IV Reverse Transcriptase (Thermo Fisher Scientific, Waltham, MA, USA). SdAb-encoding sequences were amplified by a two-step nested polymerase chain reaction (PCR) using the primers CaLl 01 and CaLl 02 [
26] and primers F1 and R1 (
Table S1) in a second PCR reaction. The final PCR product was cloned into a pHen2-derived phagemid vector and transformed into TG1 cells, yielding an sdAb library with a complexity of ~2 × 10
8 individual clones.
For the preparation of the second sdAb library, a structure-guided approach was chosen. Based on the previously published structure of NbALFA bound to an ALFA peptide ([
15], PDB: 6I2G), various mutations were introduced to lower the binding affinity of NbALFA and NbALFA
PE towards their substrate while ideally retaining their binding specificity. For that, the respective coding sequences were cloned into a pHen2-derived phagemid vector. Then, in both templates, cysteine residue 24 (numbering according to PDB: 6I2G) was mutated to serine. In all four plasmids, all CDR3 residues forming direct contacts with the ALFA peptide were randomized by saturation mutagenesis using degenerate NNK codons, resulting in a library with a theoretical complexity of 6.4 × 10
5 individual sdAbs. All mutagenesis steps were carried out using primer-directed PCR mutagenesis. The combined pool of mutant phagemids was transformed into TG1 cells, yielding ~1.2 × 10
9 individual clones.
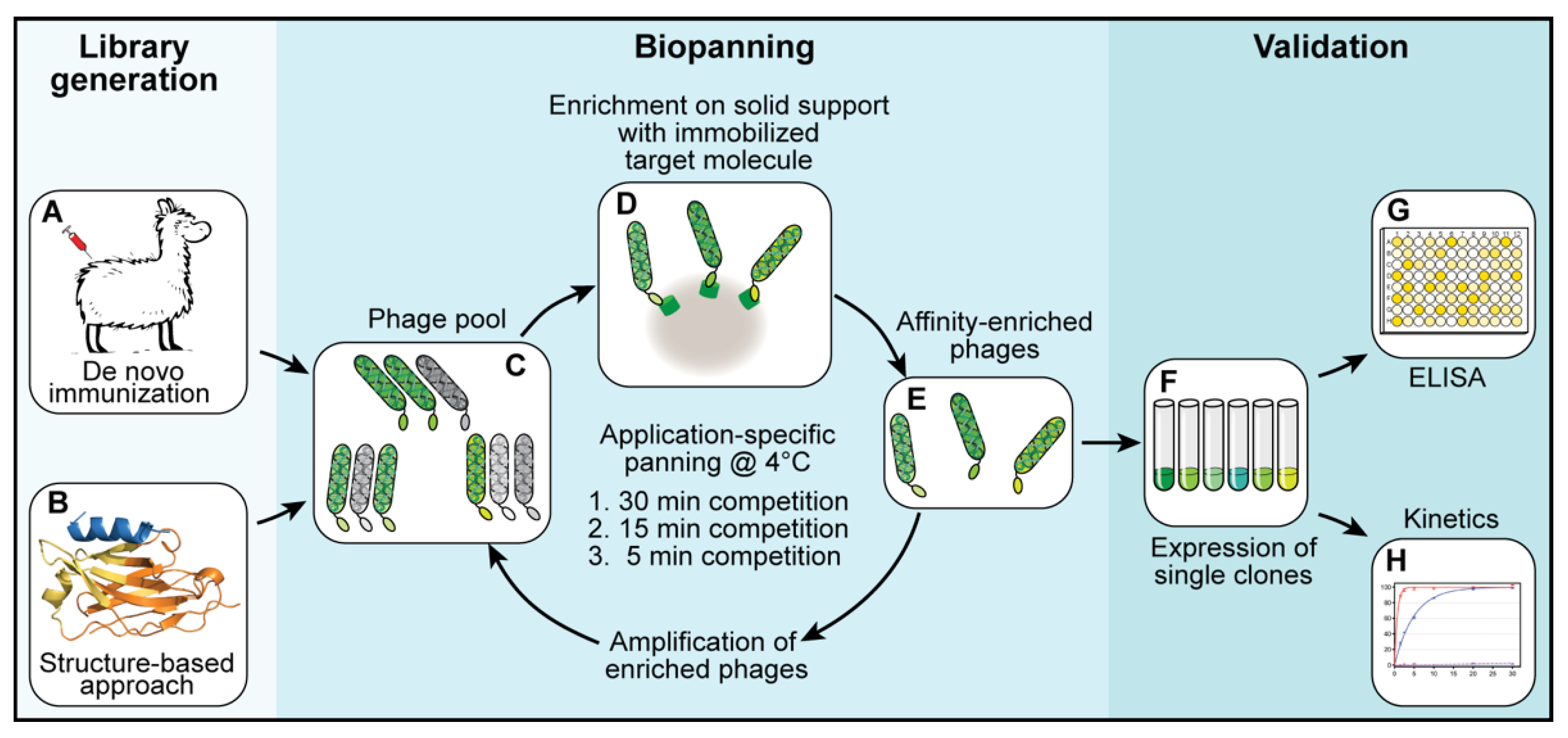
2.3. Selection of Binders—Biopanning
For each library, three subsequent biopanning steps were performed (
Figure 1C–E). In each biopanning step, the experimental conditions were adjusted and the stringency, increased. For the first biopanning step, 100 µL of streptavidin-coated MyOne DynaBeads (Thermo Fisher Scientific, Waltham, MA, USA) were loaded with a total of 200 pmol biotinylated target protein (shGFP2-ALFA). The mixture was incubated for 1 h at room temperature, washed three times with 1 mL of phosphate-buffered saline (PBS) and added to a phage suspension containing 2.0 × 10
12 phages pre-blocked with 20 µg/mL bovine serum albumin (BSA). The suspension was incubated for 1 h at room temperature, transferred to a 4 °C environment and washed once at 4 °C for 30 min with 10 mL of PBS. The remaining binders were eluted at 4 °C with 0.5 mL of PBS containing a 1000-fold excess of ALFA peptide (200 pmol target protein, 200 nmol ALFA peptide). Eluted phages were used to infect
E. coli TG1 cells for 1 h at 37 °C. The infected TG1 cells were grown overnight at 37 °C. The next day, the culture was diluted and grown to an optical density (OD) of 0.6 before infection with a MK13KO7 helper phage for 1 h at 37 °C. The phages were selected by adding kanamycin (50 µg/mL) and grown overnight at 30 °C. The next day, the phages were purified from the cleared culture supernatants by repeated precipitation with 5% PEG-8000 and 1.5 M NaCl (final concentrations in suspension: 1% PEG-8000 and 300 mM NaCl) and resuspended in a final volume of 4 mL of resuspension buffer. The second biopanning step was performed in a similar way while reducing the amount of target protein (ALFA-biotin) immobilized on the beads to 20 pmol and the elution time to 15 min at 4 °C. For the last biopanning step, 20 pmol of biotin-ALFA was used as a bait. The elution time was reduced to 5 min at 4 °C.
After three rounds of selection, the obtained sdAb-containing libraries were cloned into a pQE-derived expression vector. A total of 96 single clones per library were expressed. Crude lysates were prepared and tested by enzyme-linked immunosorbent assay (ELISA) for their binding properties and their ability to be peptide-eluted at 4 °C. Positive clones were sequenced, aligned and grouped into families. Representative clones were further analyzed biochemically.
2.4. Protein Expression and Purification
Recombinant proteins were expressed in
E. coli from pQE-derived expression vectors (
Table S2). The recombinant proteins ALFA-shGFP2, shGFP2-ALFA, NbALFA, NbALFA
PE and NbALFA
CE were expressed as N-terminal His
14-bdSUMO fusion proteins. In general,
E. coli transformed with the respective plasmids were cultured in terrific broth (TB) medium until an OD of 4.0 was reached. Protein expression was induced by the addition of 0.3 mM isopropyl thiogalactopyranoside (IPTG) at 23 °C, overnight. Before the cells were harvested by centrifugation, the cultures were supplemented with 5 mM ethyleneimine tetraacetate (EDTA) and 1 mM phenylmethylsulfonyl fluoride (PMSF). Subsequently, the cells were lysed in LS buffer (50 mM Tris/HCl, pH 7.5; 300 mM NaCl; 5 mM EDTA) supplemented with 15 mM imidazole/HCl, pH 7.5, and 10 mM dithiothreitol (DTT). The crude lysates were cleared of remaining cellular debris and bound to Ni
2+-chelate beads for 1 h at 4 °C. The protein-loaded beads were washed extensively, and the proteins were cleaved on column with 100 nM bdSENP1 [
27] for 1 h at 4 °C. All proteins were subsequently subjected to size exclusion chromatography. Coupling to an SH-reactive, agarose-based resin was performed using an ectopic cysteine fused to the C-terminus of the respective NbALFA variants via a hydrophilic linker. Product numbers of the resulting Selector resins are summarized in
Table S3.
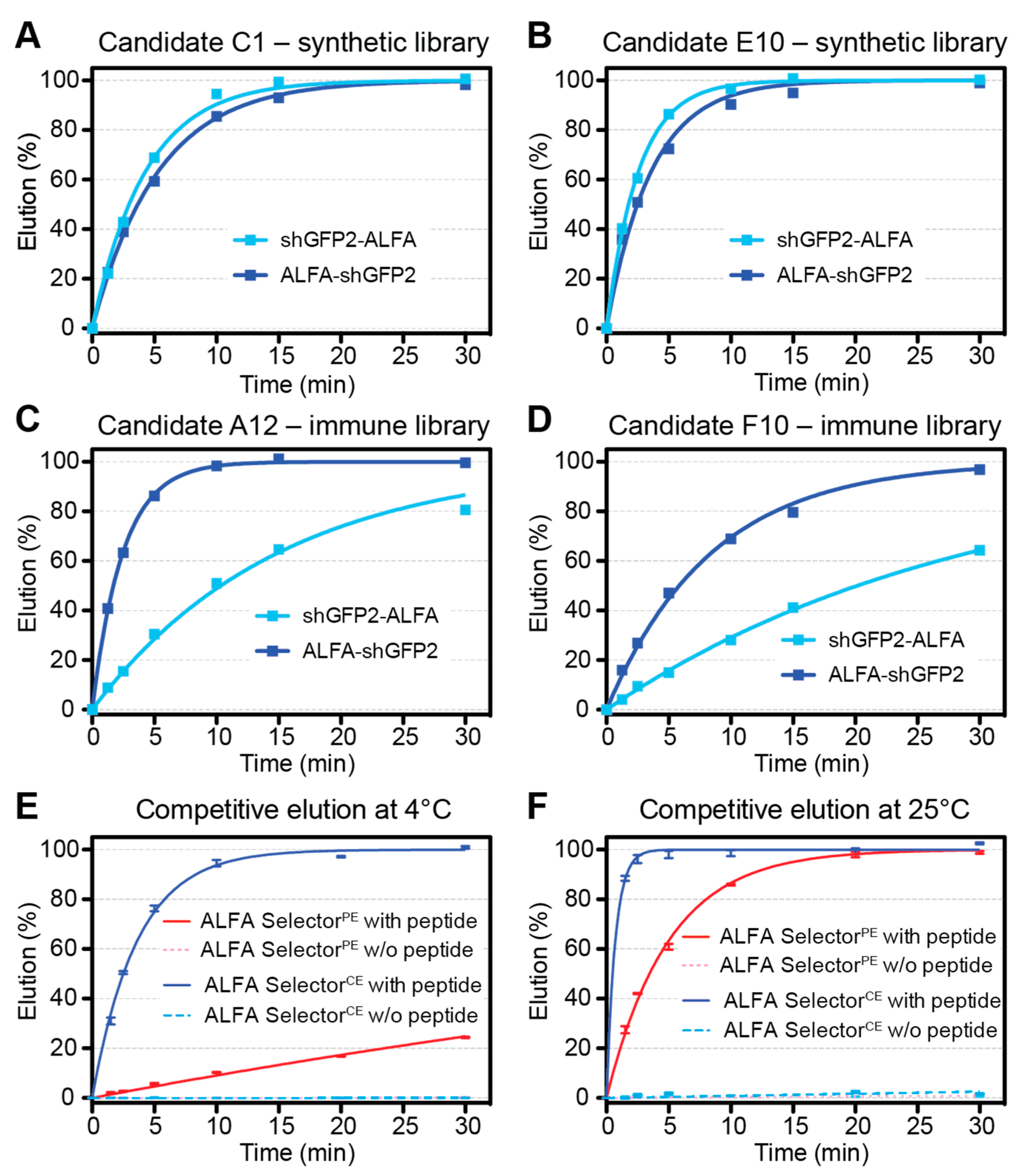
2.5. Off-Rate Assays
For an estimation of the off-rates for ALFA Selector
PE and analogous resins coupled to NbALFA
CE candidates, 20 µL of substrate-saturated resin was washed four times with PBS and resuspended in 200 µL of PBS containing 200 µM ALFA peptide. At given time points, the progression of elution was quantified by measuring the fluorescence of the GFP-tagged protein released into the supernatant (QBit 3.0; Thermo-Fischer Scientific, Waltham, MA, USA). After the kinetic measurements, all reactions were adjusted to 200 µM peptide concentrations and incubated for 30 min at 30 °C. The fluorescence values obtained after such post-elution were set to 100%. For the experiments shown in
Figure 2E,F, each data point represents the average of four independent experiments performed in parallel. The statistical analyses and curve fittings were performed using GraphPad Prism 5.0.
2.6. Titration of ALFA Peptide
For each experiment, 250 µL of ALFA SelectorCE was saturated with shGFP2-ALFA, extensively washed with PBS containing 0.02% Triton X-100 (TX-100) and packed in a 1 mL syringe equipped with a porous bottom filter frit. For stopped-flow elution at 22 or 4 °C, 70 µL aliquots of PBS + 0.02% TX-100 containing various concentrations of ALFA peptide were added either every 1.5 min (effective flow rate, 0.18 column volumes (CV)/min) or every 5 min (effective flow rate, 0.056 CV/min) and allowed to enter the column by gravity flow while collecting the eluate. In each fraction, the released protein was quantified fluorometrically (QBit 3.0; Thermo-Fischer Scientific, Waltham, MA, USA).
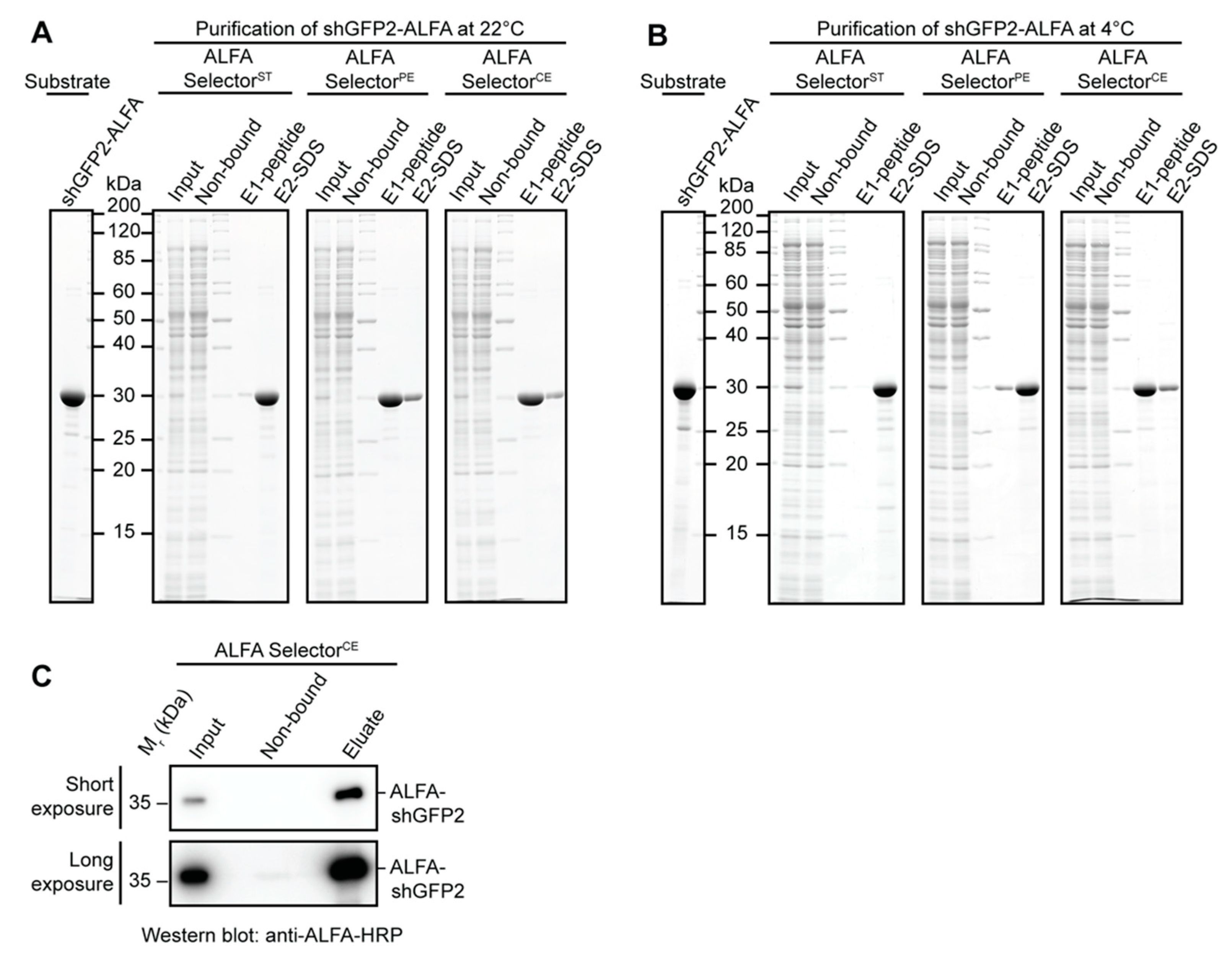
2.7. Affinity Purification from E. coli and HeLa Lysates
Affinity-purification experiments with E. coli or HeLa lysates were performed with a defined amount of ALFA-tagged target protein. Therefore, cleared E. coli lysate or HeLa S100 lysate was blended with 3 µM purified ALFA-tagged target protein (ALFA-shGFP2, shGFP2-ALFA). For each experiment, 25 µL of the indicated ALFA SelectorST/PE/CE (NanoTag Biotechnologies, Göttingen, Germany, Cat No. N1511, N1510, N1512) was incubated with 1 mL of lysate containing the indicated ALFA-tagged target protein for 1 h at room temperature or at 4 °C. After binding, the resins were washed three times in batch with 1 mL of PBS, transferred to a MiniSpin column and washed twice with 0.6 mL of PBS. The resin was then resuspended in 50 µL of PBS containing 200 µM ALFA peptide and incubated for 15 min at room temperature or at 4 °C. All Selector resins were additionally incubated in SDS sample buffer to remove the remaining proteins, heating the samples to 95 °C for 5 min. As specificity controls, the indicated ALFA Selectors were incubated with E. coli or HeLa lysate lacking any ALFA-tagged target protein. The samples were resolved by SDS-PAGE and analyzed by Coomassie staining.
2.8. Affinity Purification of Low-Abundant Proteins from HeLa Lysates
A volume of 50 mL of HeLa S100 extract was blended with 100 nM shGFP2-ALFA and applied to 1 mL of ALFA Selector
CE resin (NanoTag Biotechnologies, Göttingen, Germany, Cat No. N1512) at a flow rate of 0.8–1.0 mL/min. The resin was washed with 10 CV of PBS and eluted by a stepwise addition of PBS containing 1 mM ALFA peptide (250 µL every 3 min) at room temperature. Eluate fractions containing the target protein were pooled. All samples were resolved by SDS-PAGE and further analyzed by Western Blotting. The ALFA-tagged protein was detected using an ALFA-specific HRP-coupled sdAb (NanoTag Biotechnologies, Göttingen, Germany, Cat No. N1501-HRP;
Table S3). Western blotting was essentially performed as described before by Götzke et al. in 2019 [
15].
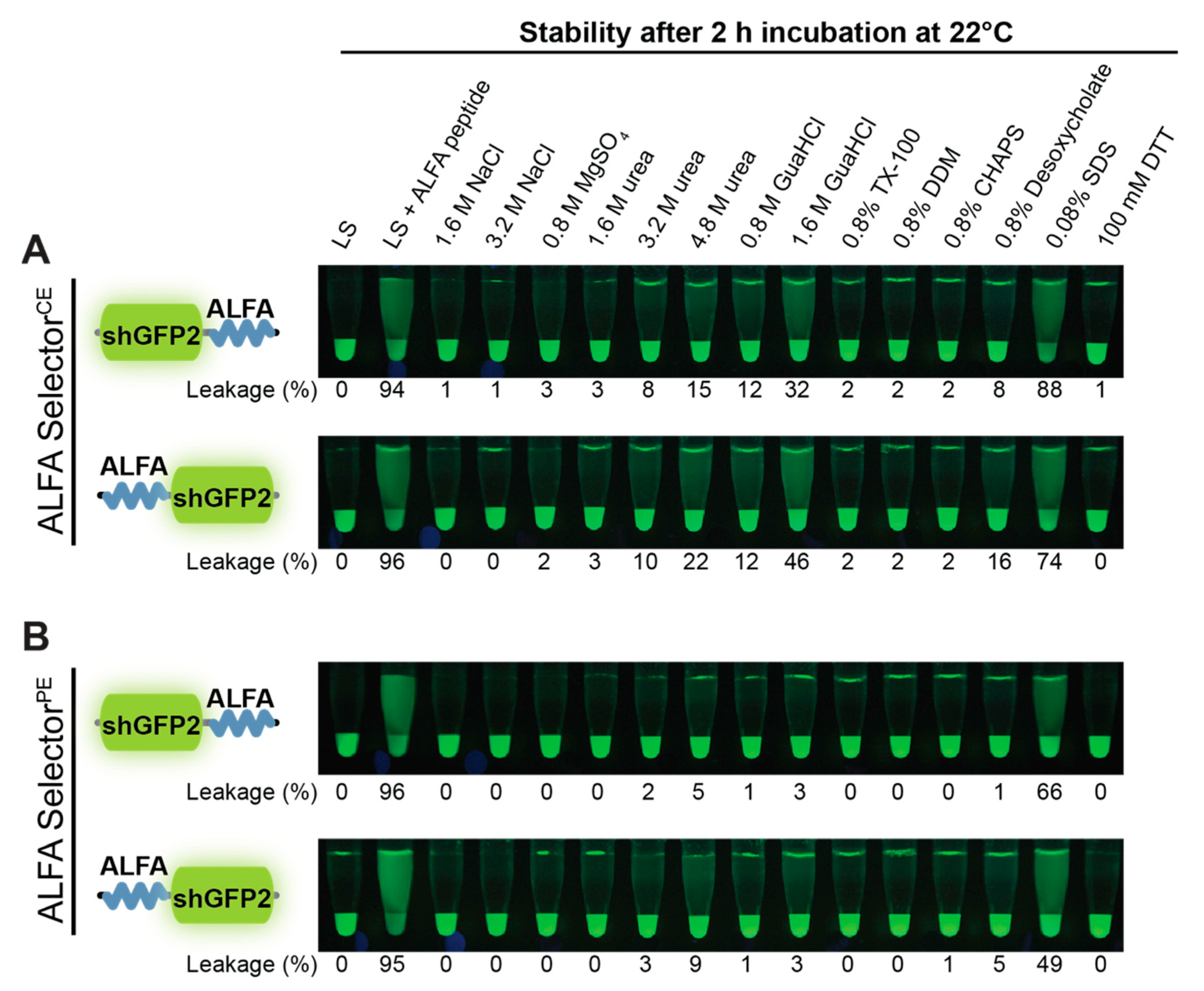
2.9. Resistance to Stringent Washing and pH
A volume of 20 µL of ALFA SelectorPE or ALFA SelectorCE was saturated with either ALFA-shGFP2 or shGFP2-ALFA. The beads were washed four times with Tris-buffered saline (TBS: 25 mM Tris/HCl, pH 7.5; 150 mM NaCl; 2 mM EDTA) and subsequently incubated in 200 µL of the indicated solutions or buffers for 2 h at room temperature with shaking. For post-elution, 250 µM ALFA peptide was added, and the reactions were further incubated for 30 min at 25 °C. The eluates were quantified using a fluorometer (QBit 3.0; Thermo-Fischer Scientific, Waltham, MA, USA) before and after post-elution with ALFA peptide. Pictures were taken upon UV illumination using a Nikon D700 camera equipped with a 105 mm macro lens (Nikon, Minato, Japan).
2.10. Regeneration of ALFA SelectorCE
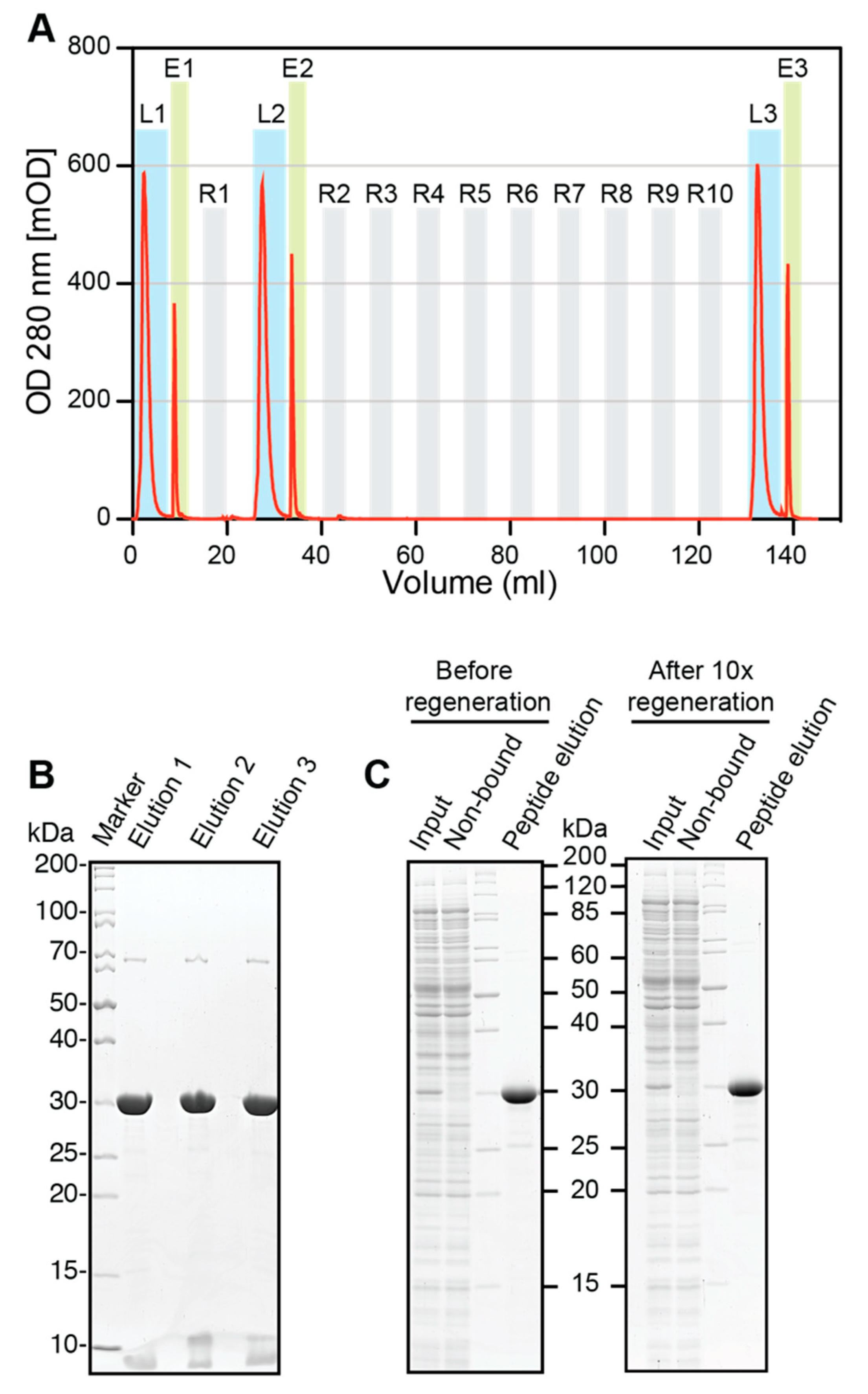
Regeneration experiments were performed using an Äkta FPLC (Amersham Biosciences, Little Chalfont, UK). A 0.5 mL volume of ALFA SelectorCE was packed in a glass column and equilibrated with PBS before saturating it with ALFA-shGFP2. The loaded columns were washed with 10 CV of PBS, and the proteins were eluted in flow (0.1 mL/min) with PBS containing 1 mM ALFA peptide (elution buffer). Eluate fractions containing the target protein were pooled. The protein content was quantified by measuring the absorbance at 280 nm and additionally analyzed by SDS-PAGE. The column was regenerated with 10 CV of glycine pH2.2 (100 mM glycine, pH 2.2; 150 mM NaCl) and re-equilibrated with 10 CV of PBS. After the first regeneration, binding of ALFA-shGFP2 and elution were repeated as described above. The resin was subjected to nine additional regeneration cycles with glycine, before the final target protein loading and elution was performed. All samples were resolved by SDS-PAGE and visualized by Coomassie staining. The experiment was repeated with fresh resin using 100 mM NaOH instead of glycine for regeneration. The elution profiles were plotted using GraphPad Prism 5.0.
4. Discussion
We recently described the ALFA system comprising the ALFA-tag, a novel epitope tag and a set of ALFA-specific sdAbs, NbALFA and NbALFA
PE [
15]. NbALFA shows an extremely stable and robust association with ALFA-tagged proteins and is ideally suited for in vivo protein manipulations, highly sensitive affinity capturing and advanced detection and imaging techniques. By contrast, NbALFA
PE was optimized for competitive elution at ambient temperature, and can be used for the purification of proteins or demanding living cell isolation under physiological conditions [
15]. To broaden the versatility of the ALFA system even further, we, here, aimed at developing NbALFA
CE, a new sdAb combining high selectivity and stable binding of ALFA-tagged proteins with an efficient competitive elution at 4 °C under physiological buffer conditions.
To find sdAbs fulfilling these virtually diverging goals, we started two independent phage-display screens based on different libraries. The first library was created by conventional immunization of alpacas, while the second library was designed by introducing mutations into NbALFA and NbALFAPE by a rational, structure-guided approach. After applying stringent selection criteria, both libraries yielded NbALFACE candidates that allowed stable binding of ALFA-tagged proteins while permitting an efficient competitive elution at 4 °C. In addition to these requested features we could directly screen for, optimal NbALFACE candidates should ideally show comparable binding strength and elution kinetics for target proteins harboring N- or C-terminal ALFA-tags.
Surprisingly, however, we noticed striking differences depending on the origin of the clones analyzed. Both NbALFACE candidates derived from the immunized library showed a strong preference for proteins harboring a C-terminal ALFA-tag. By contrast, clones originating from the semisynthetic library showed remarkably similar elution kinetics irrespective of the localization of the ALFA-tag within the target protein—just as described for NbALFA and NbALFAPE, indicating that the NbALFACE candidates originating from the semisynthetic library preserved this feature.
The consistent recognition of target proteins tagged at either terminus is, therefore, not necessarily self-evident. By generating a semisynthetic library based on sdAbs that already possess this requested feature, we aimed to increase the chances of also finding these properties in the descendant clones. We believe that sdAb clones with similar properties may also be present in the immunized library. However, it is hard to envision a selection procedure that would result in sdAbs featuring these properties in a stringent and reproducible manner. Therefore, once a well-characterized sdAb displaying some of the desired features and biophysical properties is available, using this sdAb as the precursor of a new library seems to be an effective alternative to proceeding with a conventional de novo discovery process. In analogy, this general approach can potentially be applied to finetune certain binding properties (e.g., binding strength) while making sure the sdAb keeps other desired properties, such as its biochemical properties, its charge distribution or its specificity. Such an approach is most promising if a direct screening for a rare feature is not feasible or would require unreasonable effort.
The final NbALFA
CE displays four modifications with respect to NbALFA. In comparison to the NbALFA
PE, featuring intermediate affinity, the off-rate of NbALFA
CE is substantially enhanced, which is a prerequisite for achieving an efficient competitive elution at 4 °C (
Figure 2E,F). The resin can, nevertheless, efficiently capture ALFA-tagged proteins even from dilute samples using a simple gravity-flow purification system (
Figure 3C). As expected, the higher off-rate also influences the biochemical stability of the complexes formed with ALFA-tagged proteins. NbALFA
CE is, therefore, slightly more sensitive than NbALFA
PE towards denaturing reagents such as urea or guanidinium hydrochloride and certain detergents (
Figure 4). However, under physiological buffer conditions or even at drastically elevated salt concentrations, any spontaneous leakage of target protein from an NbALFA
CE-coupled resin (ALFA Selector
CE) is negligible (
Figure 2E,F and
Figure 4).
Although the N- and C-terminal ALFA-tagged proteins behaved in a strikingly similar way in the kinetic off-rate assays performed with ALFA Selector
CE, we observed subtle differences regarding their biochemical properties and pull-down behavior, especially under nonphysiological conditions (
Figure 4).The observed differences in biochemical behavior are marginal for our model substrate shGFP2. However, as for any tag, it may be advisable to test the optimal position of the tag on other proteins or protein complexes, as steric effects or target-specific surface properties might influence its accessibility.
When varying the concentration of the ALFA peptide during elution from ALFA Selector
CE, we observed the sharpest elution profiles in stopped-flow and flow mode if 1 mM peptide was used. At this concentration, the excess of peptide over resin-bound sdAb is sufficiently high to ensure a nearly off-rate-limited elution. Therefore, higher peptide concentrations will not increase the elution speed further. When using larger elution volumes, as generally performed in “batch” mode, peptide concentrations of only 200 µM are fully sufficient (
Figure 2), as a similar excess of peptide over the resin-bound sdAb is achieved. While competitive peptide elution at 4 °C from ALFA Selector
CE requires 15–20 min for completion, elution at room temperature can even be performed using gravity flow (
Figure 3C,
Figure S1C and
Figure 5).
At all tested temperatures, rapid and complete elution from ALFA Selector
CE can also be achieved by applying acidic or basic conditions (
Figure 5 and
Figure S4), which was exploited to perform cleaning-in-place (CIP) regeneration. After 10 cycles of regeneration, the binding capacity and specificity of ALFA Selector
CE remained largely unaffected (
Figure 5B,C and
Figure S4B,C). ALFA Selector
CE can therefore be reused several times without a loss of performance.
In summary, by introducing the novel, cold-elutable NbALFACE, the same ALFA-tagged protein can be bound by three alternative NbALFA variants featuring different affinities, thereby increasing the versatility of the ALFA system even further. ALFA SelectorCE combines a highly specific and biochemically stable binding of ALFA-tagged proteins with the ability to readily elute at low temperatures under physiological buffer conditions. Thereby, the newly characterized ALFA SelectorCE is an ideal tool for the clean, single-step purification of temperature-labile targets and applications requiring perfect structural and functional conservation, such as for cryo-electron microscopy or the purification of sensitive enzymes, respectively.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}