
Demystifying Chronic Kidney Disease of Unknown Etiology (CKDu): Computational Interaction Analysis of Pesticides and Metabolites with Vital Renal Enzymes


Abstract
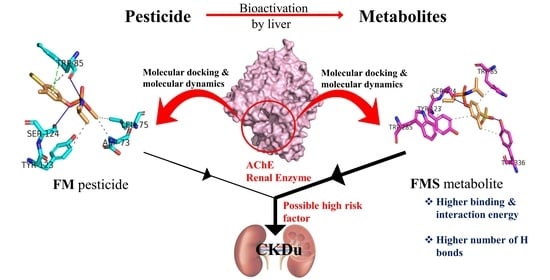
1. Introduction
2. Materials and Methods
2.1. Ligand Preparation
2.2. Protein Structure Preparation
2.3. Molecular Docking Details
2.4. Molecular Dynamics Simulation Details
2.5. Validation of the Theoretical Approach
2.6. Analysis
3. Results
3.1. PDB Structure Refinement and Model Validation
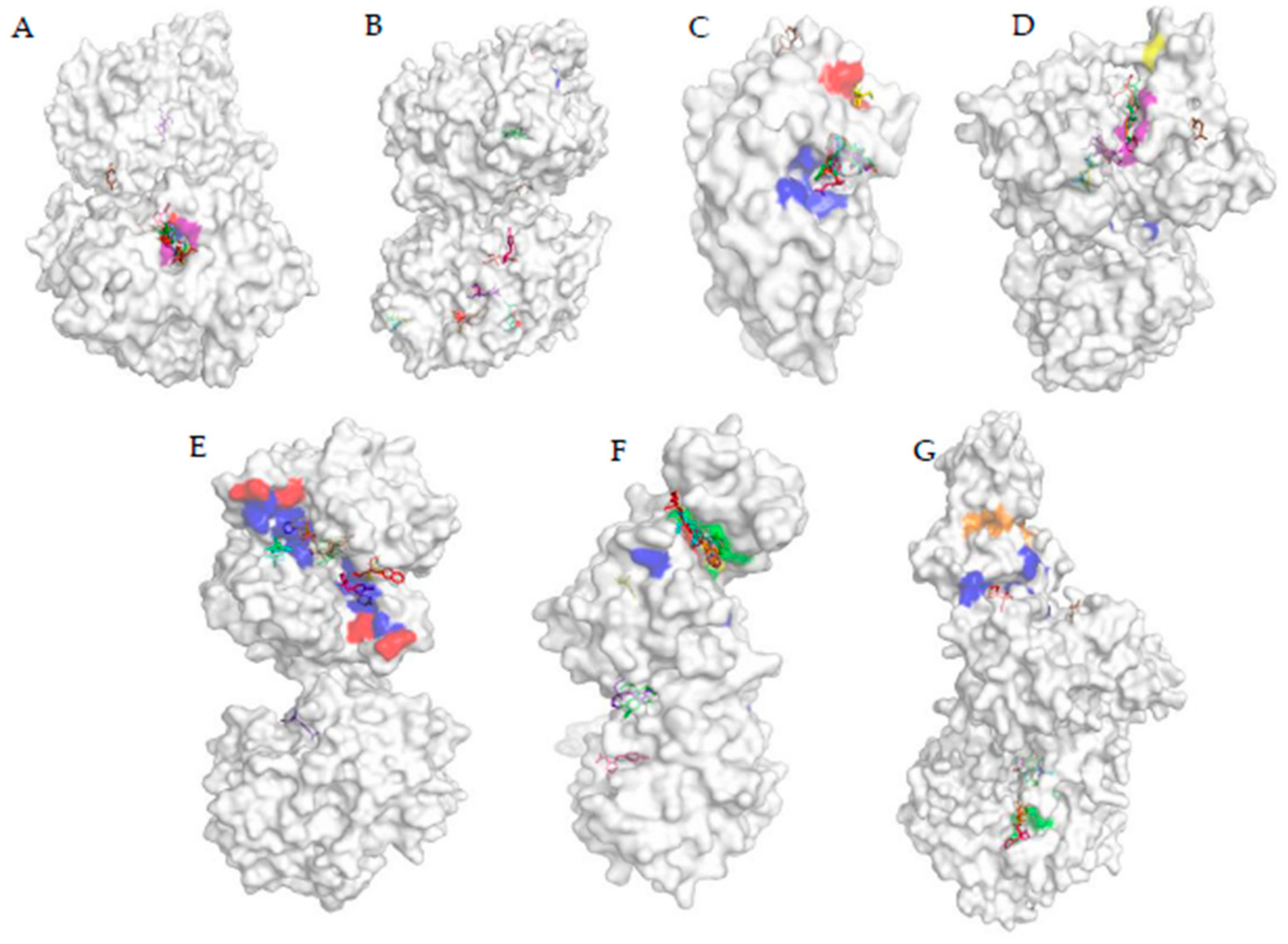
3.2. Protein Active Site Prediction Analysis
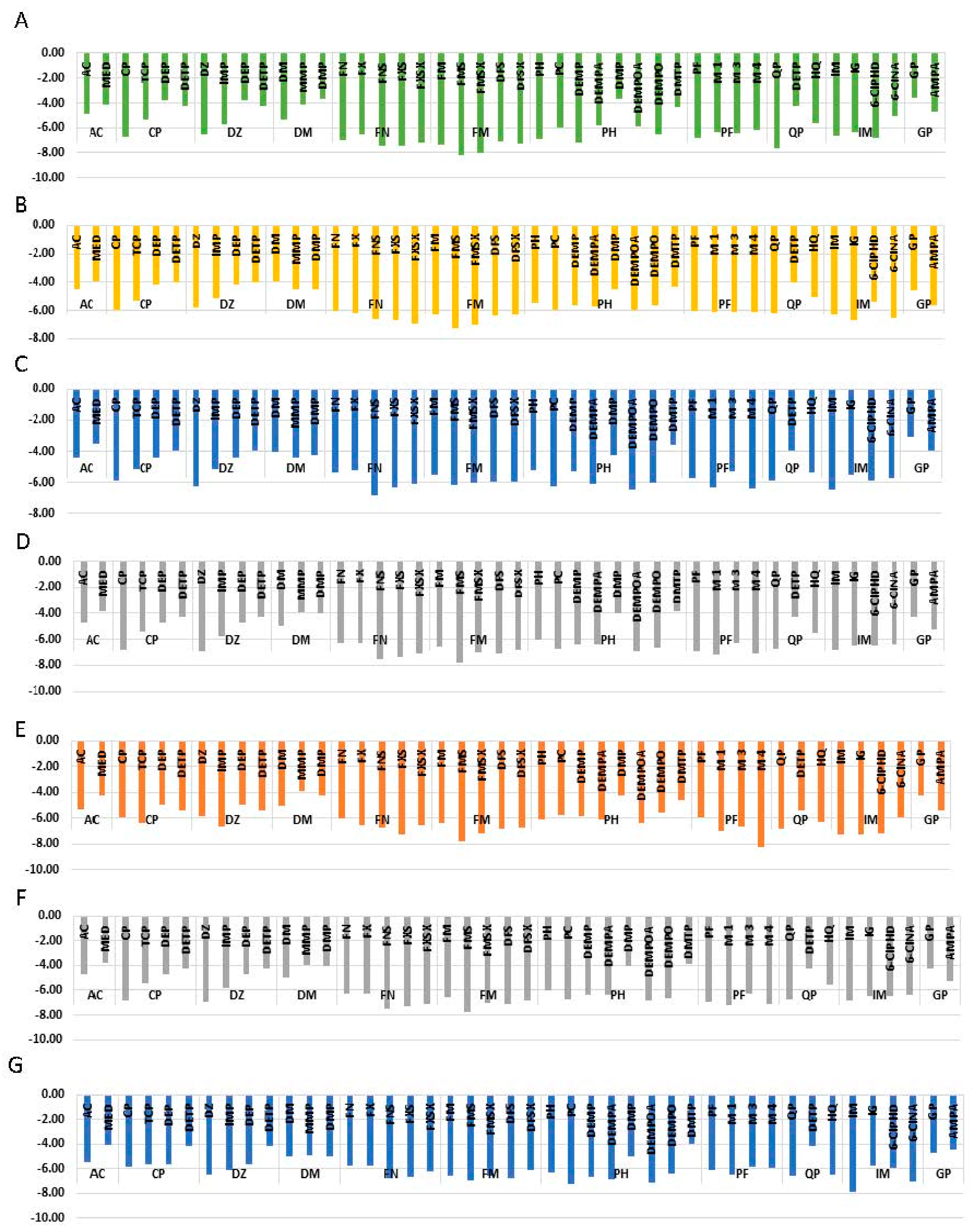
3.3. Molecular Docking
3.4. Validation of the Docking Approach
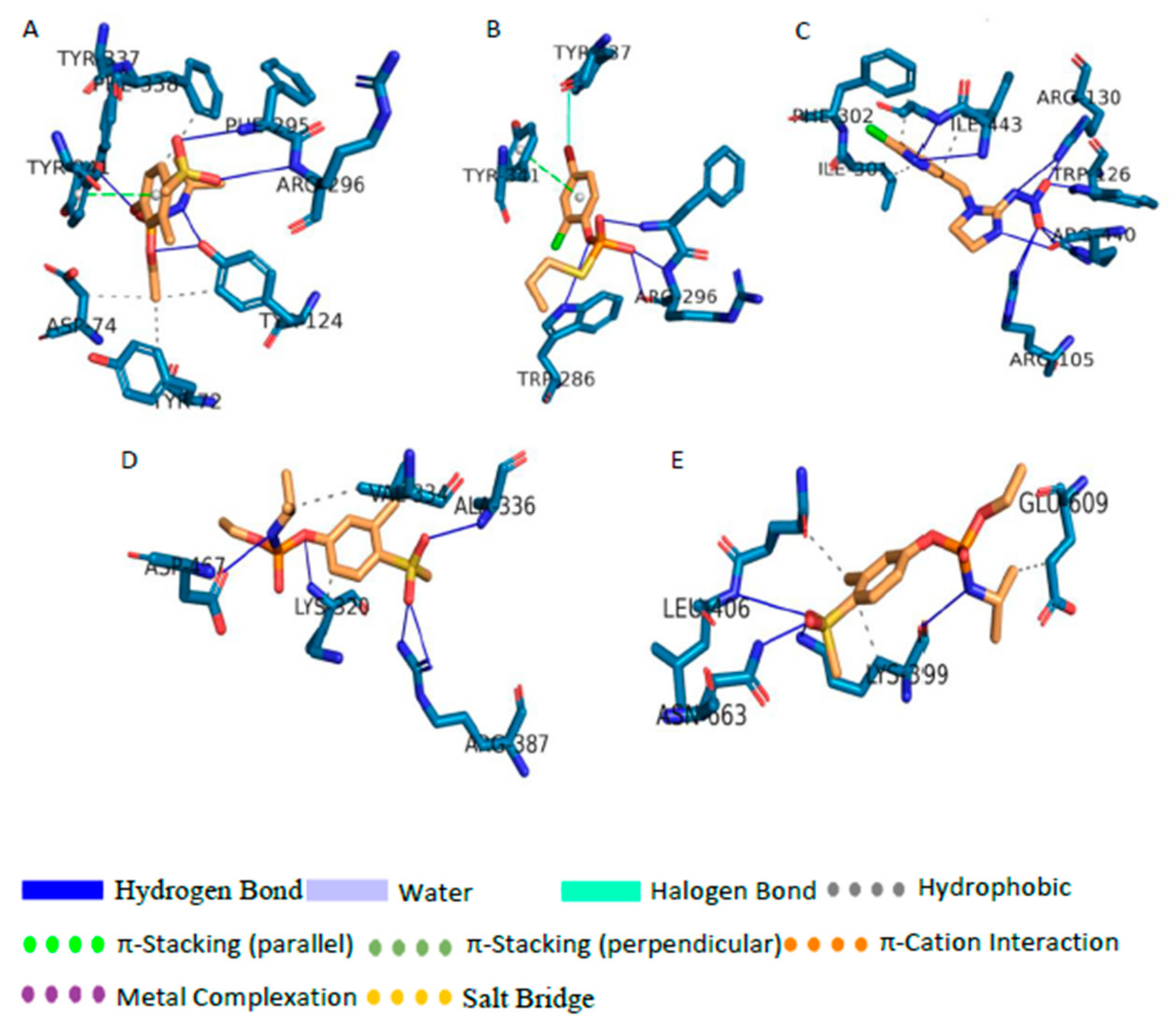
3.5. Analysis of the Docked Location
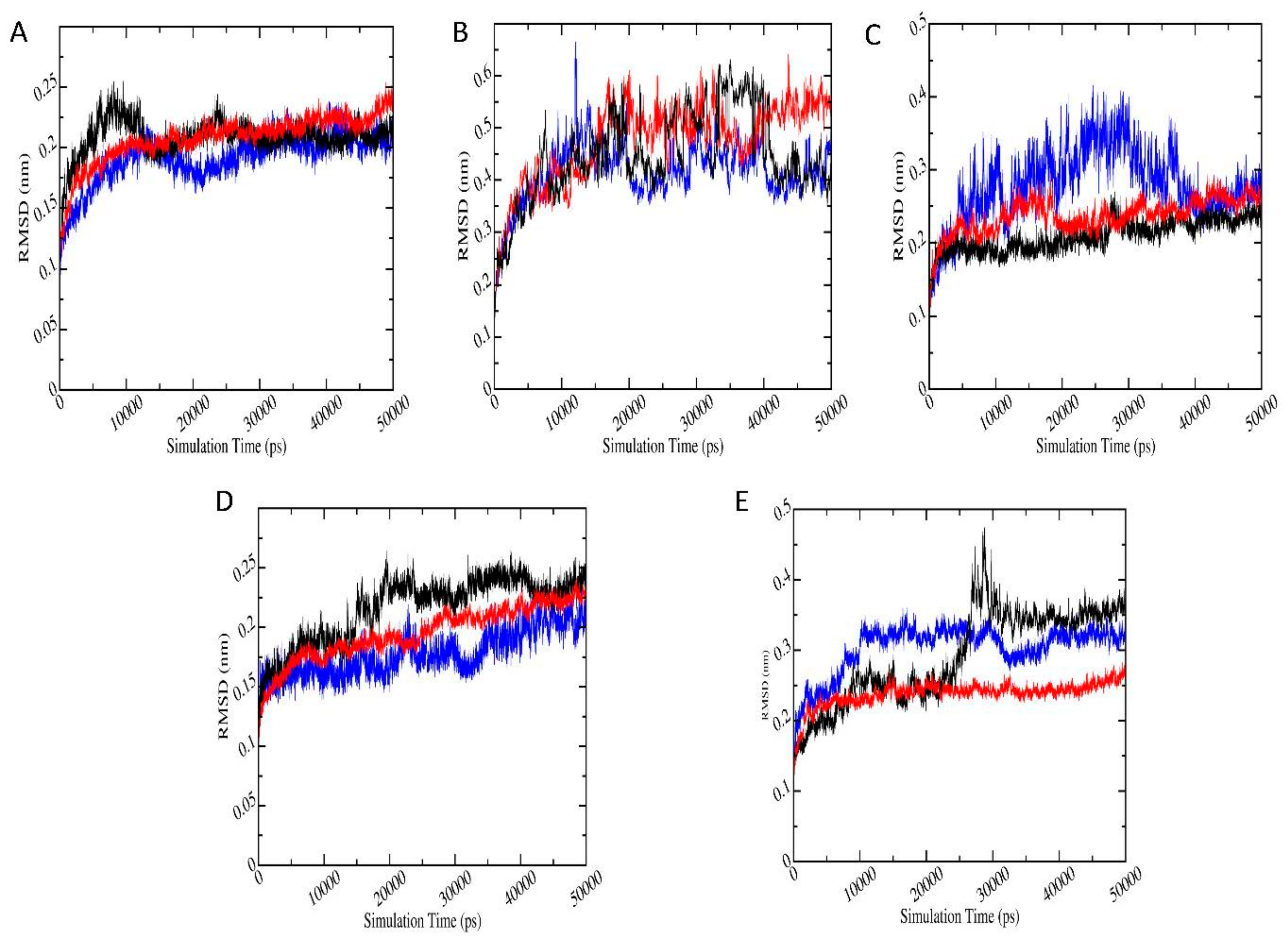
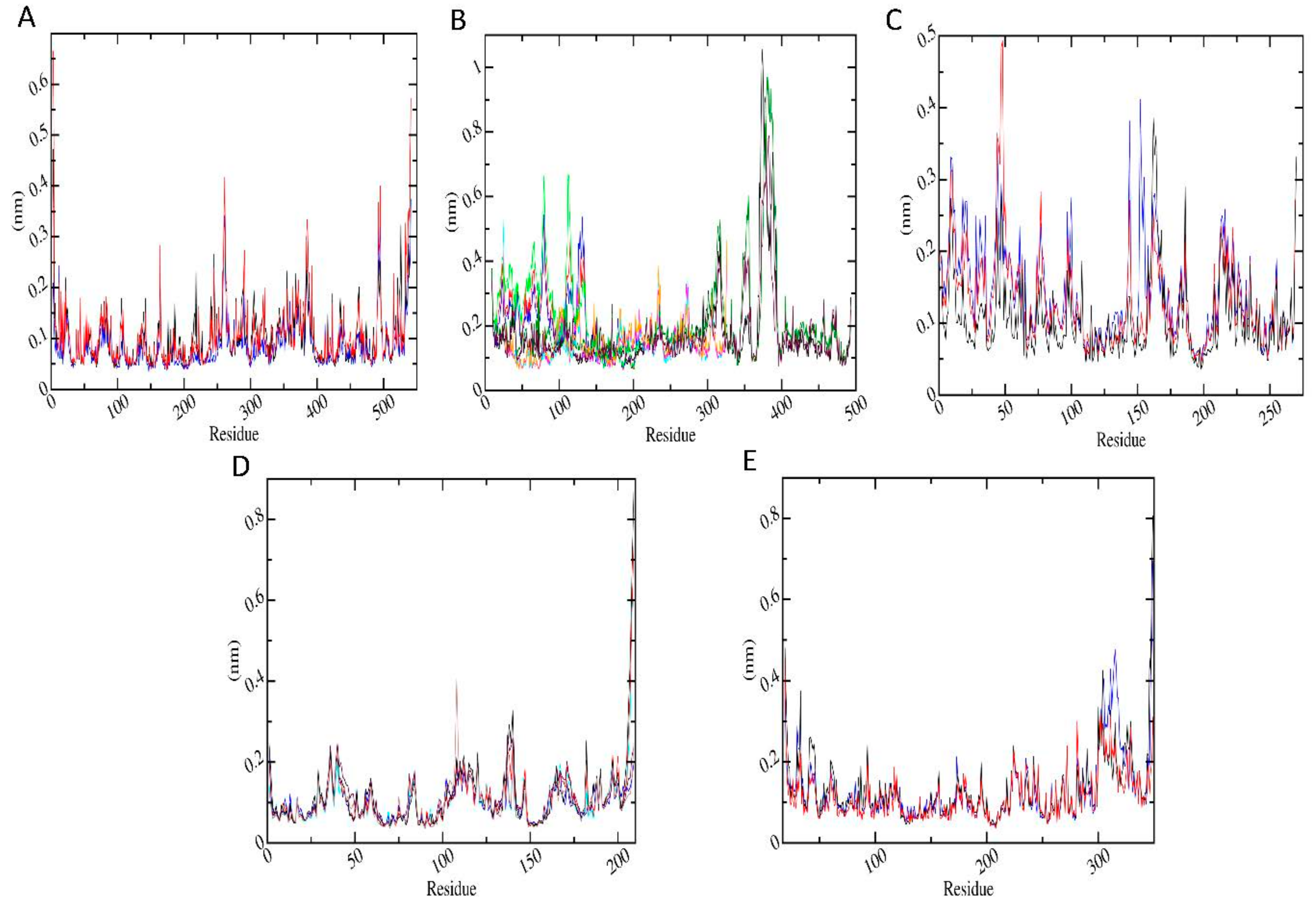
3.6. Molecular Dynamics Simulations
3.6.1. Root Mean Square Deviation (RMSD)
3.6.2. Radius of Gyration (Rg)
3.6.3. Root Mean Square Fluctuation (RMSF)
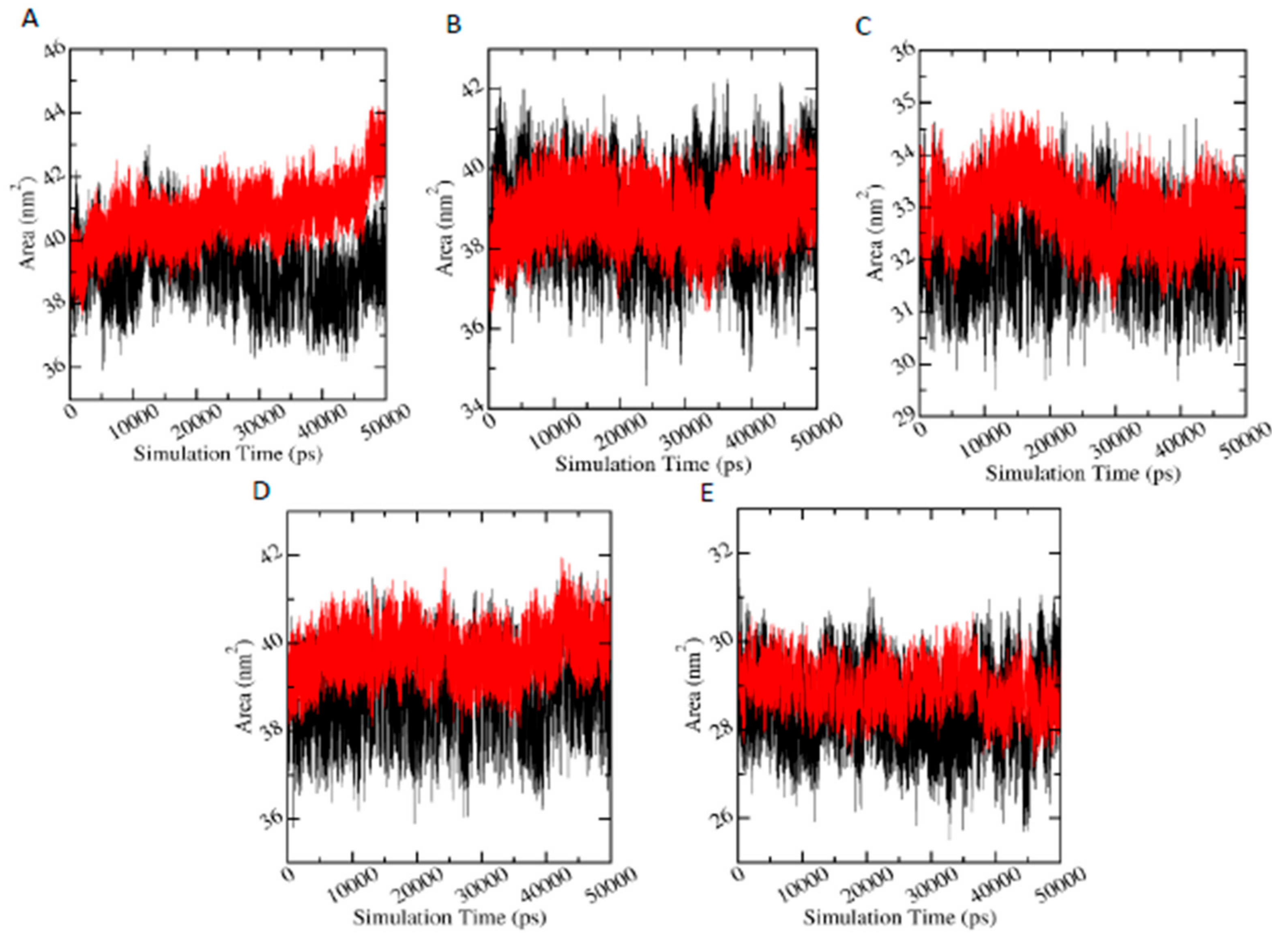
3.6.4. Solvent Accessible Surface Area (SASA)
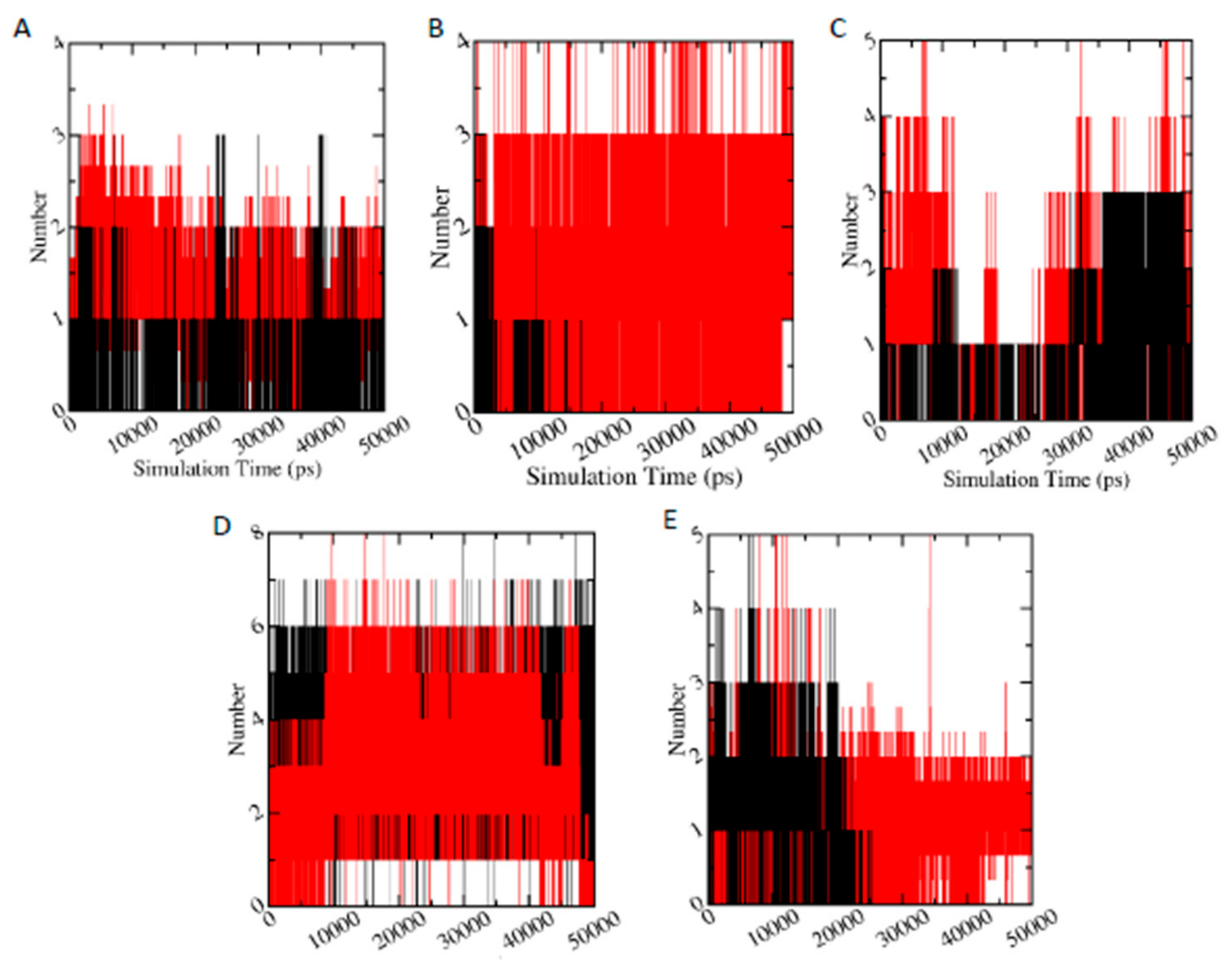
3.6.5. Hydrogen Bond Analysis
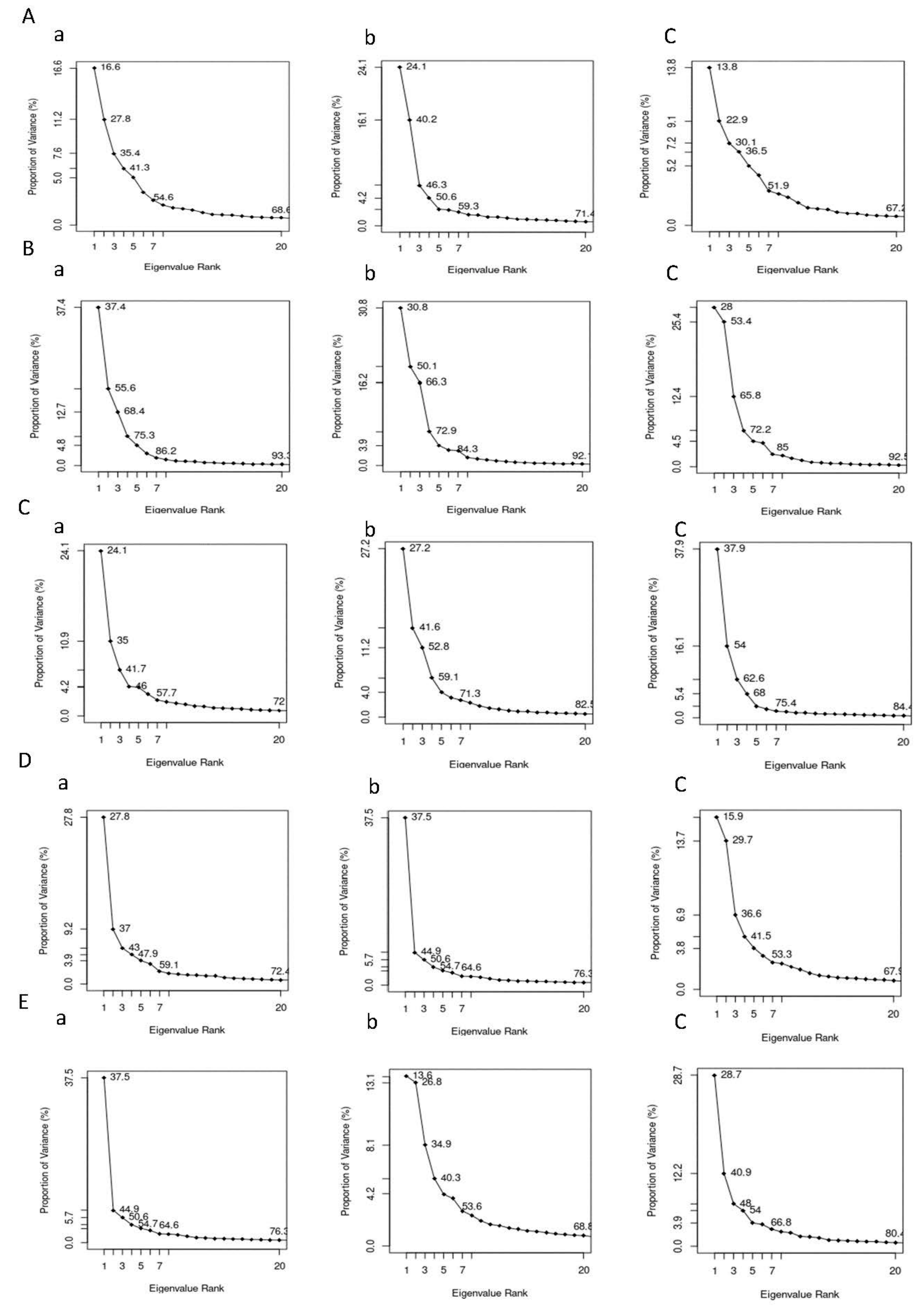
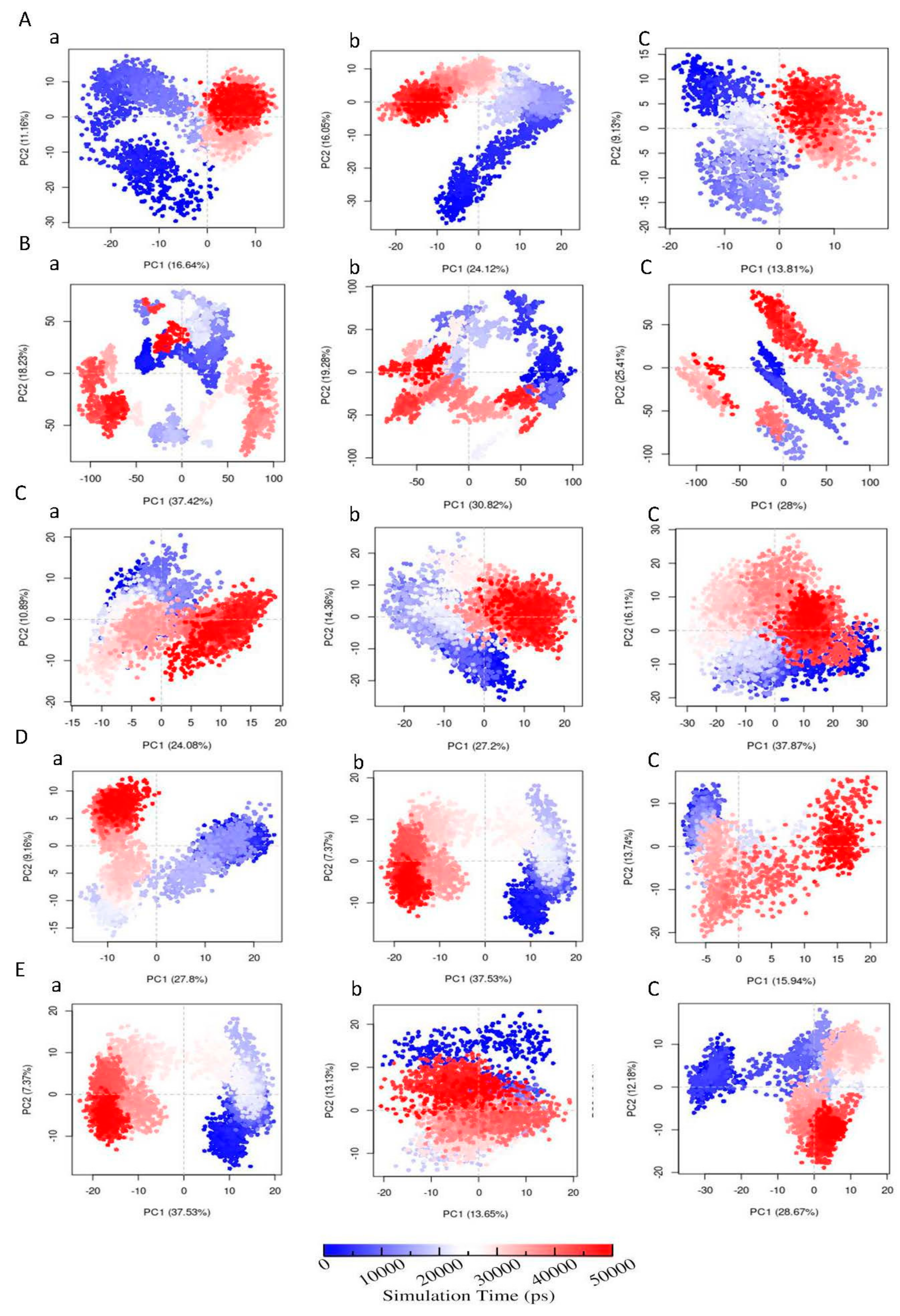
3.6.6. Principal Component Analysis (PCA)
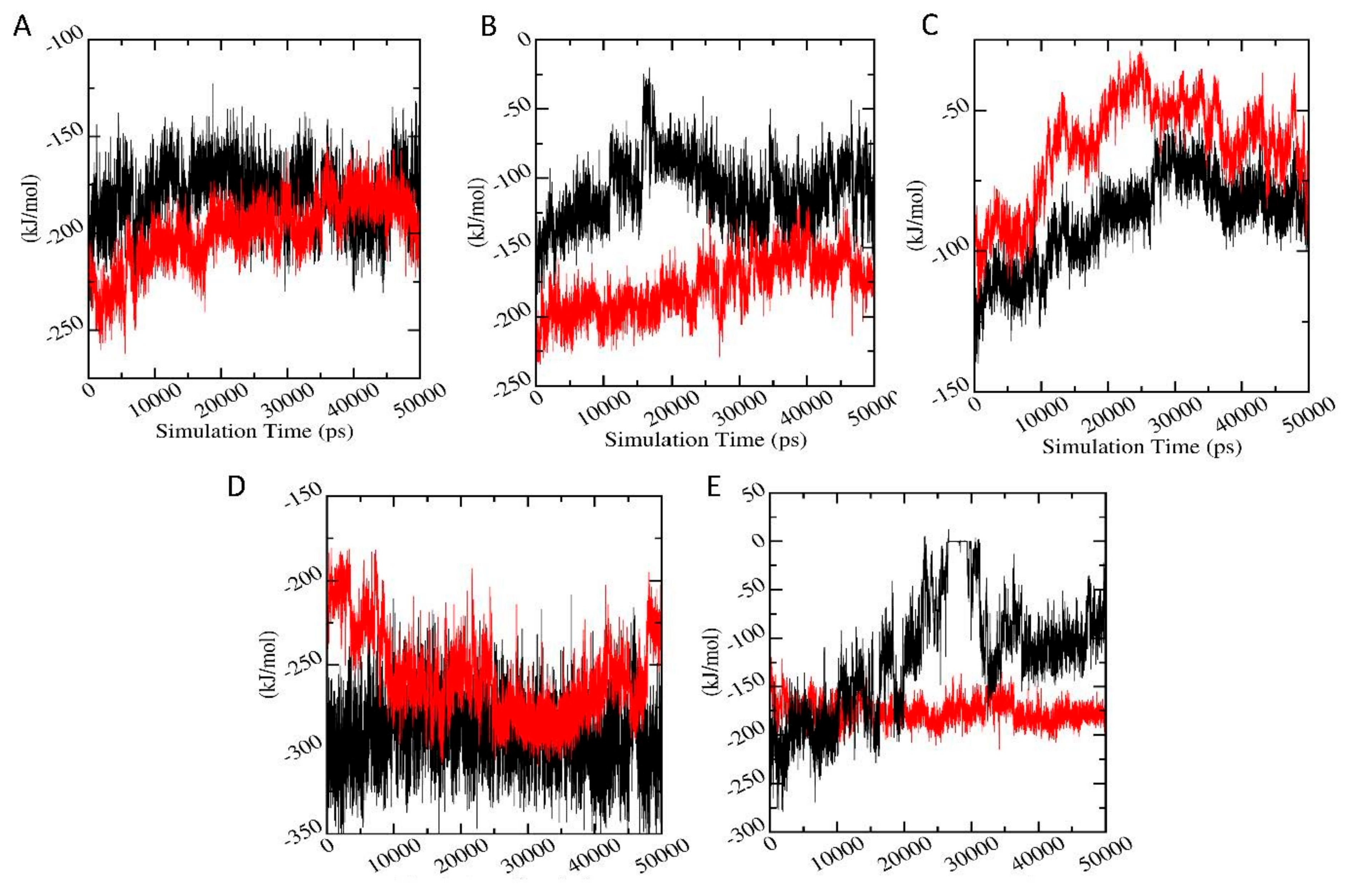
3.6.7. Interaction Energy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wasung, M.E.; Chawla, L.S.; Madero, M. Biomarkers of renal function, which and when? Clin. Chim. Acta 2015, 438, 350–357. [Google Scholar] [CrossRef]
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.-M.; Yang, C.-W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef]
- Weaver, V.M.; Fadrowski, J.J.; Jaar, B.G. Global dimensions of chronic kidney disease of unknown etiology (CKDu): A modern era environmental and/or occupational nephropathy? BMC Nephrol. 2015, 16, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Lebov, J.F.; Valladares, E.; Peña, R.; Peña, E.M.; Sanoff, S.L.; Cisneros, E.C.; Colindres, R.E.; Morgan, D.R.; Hogan, S.L. A Population-Based Study of Prevalence and Risk Factors of Chronic Kidney Disease in León, Nicaragua. Can. J. Kidney Health Dis. 2015, 2, 6–20. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan-Gordo, C.; Shivashankar, R.; Anand, S.; Ghosh, S.; Glaser, J.; Gupta, R.; Jakobsson, K.; Kondal, D.; Krishnan, A.; Mohan, S. Prevalence of and risk factors for chronic kidney disease of unknown aetiology in India: Secondary data analys is of three population-based cross-sectional studies. BMJ Open 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Jayasekara, K.B.; Dissanayake, D.M.; Sivakanesan, R.; Ranasinghe, A.; Karunarathna, R.H.; Kumara, G.W.G.P. Epidemiology of chronic kidney disease, with special emphasis on chronic kidney disease of uncertain etiology, in the north central region of SriLanka. J. Epidemiol. 2015, 25, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Bello, A.K.; Levin, A.; Tonelli, M.; Okpechi, I.G.; Feehally, J.; Harris, D.; Jindal, K.; Salako, B.L.; Rateb, A.; Osman, M.A. Assessment of global kidney health care status. JAMA 2017, 317, 1864–1881. [Google Scholar] [CrossRef]
- Weiner, D.E.; McClean, M.D.; Kaufman, J.S.; Brooks, D.R. The central American epidemic of CKD. Clin. J. Am. Soc. Nephrol. 2013, 8, 504–511. [Google Scholar] [CrossRef]
- Wijkström, J.; González-Quiroz, M.; Hernandez, M.; Trujillo, Z.; Hultenby, K.; Ring, A.; Söderberg, M.; Aragón, A.; Elinder, C.-G.; Wernerson, A. Renal Morphology, Clinical Findings, and Progression Rate in Mesoamerican Nephropathy. Am. J. Kidney Dis. 2017, 69, 626–636. [Google Scholar] [CrossRef]
- Glaser, J.; Hansson, E.; Weiss, I.; Wesseling, C.; Jakobsson, K.; Ekström, U.; Apelqvist, J.; Lucas, R.; Monge, E.A.; Peraza, S. Preventing kidney injury among sugar cane workers: Promising evidence from enhanced work place interventions. Occup. Environ. Med. 2020, 77, 527–534. [Google Scholar] [CrossRef]
- Redmon, J.H.; Elledge, M.F.; Womack, D.S.; Wickremashinghe, R.; Wanigasuriya, K.P.; Peiris-John, R.J.; Lunyera, J.; Smith, K.; Raymer, J.H.; Levine, K.E. Additional perspectives on chronic kidney disease of unknown aetiology (CKDu) in Sri Lanka—lessons learned from the WHO CKDu population prevalence study. BMC Nephrol. 2014, 15, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fitria, L.; Prihartono, N.A.; Ramdhan, D.H.; Wahyono, T.Y.M.; Kongtip, P.; Woskie, S. Environmental and occupational risk factors associated with chronic kidney disease of unknown etiology in West Javanese rice farmers, Indonesia. Int. J. Environ. Res. Publichealth 2020, 17, 4521. [Google Scholar] [CrossRef] [PubMed]
- Small, D.M.; Coombes, J.S.; Bennett, N.; Johnson, D.W.; Gobe, G.C. Oxidative stress, anti-oxidant therapies and chronic kidney disease. Nephrology 2012, 17, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2019, 34, 975–991. [Google Scholar] [CrossRef]
- Sayanthooran, S.; Magana-Arachchi, D.N.; Gunerathne, L.; Abeysekera, T.D.; Sooriyapathirana, S.S. Upregulation of oxida-tive stress related genes in a chronic kidney disease attributed to specific geographical locations of Sri Lanka. Bio Med. Res. Int. 2016, 2016, 1–10. [Google Scholar]
- Sayanthooran, S.; Magana-Arachchi, D.N.; Gunerathne, L.; Abeysekera, T. Potential diagnostic bio markers for chronic kidney disease of unknown etiology (CKDu) in Sri Lanka: A pilot study. BMC Nephrol. 2017, 18, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Deeba, F.; Raza, I.; Muhammad, N.; Rahman, H.; UrRehman, Z.; Azizullah, A.; Khattak, B.; Ullah, F.; Daud, M. Chlorpyrifos and lamb dacyhalothrin-induced oxidative stress in human erythrocytes: In vitro studies. Toxicol. Ind. Health 2017, 33, 297–307. [Google Scholar] [CrossRef]
- Wimalawansa, S.J. Public health interventions for chronic diseases: Cost–benefit modelizations for eradicating chronic kidney disease of multi factorial origin (CKD mfo/CKDu) from tropical countries. Heliyon 2019, 5, 1–11. [Google Scholar] [CrossRef]
- Rose, R.L.; Tang, J.; Choi, J.; Cao, Y.; Usmani, A.; Cherrington, N.; Hodgson, E. Pesticide metabolism in humans, Including polymorphisms. Scand. J. Work. Environ. Health 2005, 156–163. [Google Scholar]
- Rajani, R.; Pastor-Soler, N.M.; Hallows, K.R. Role of AMP-activated protein kinase in kidney tubular transport, metabolism, and disease. Curr. Opin. Nephrol. Hypertens. 2017, 26, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Musashi, M.; Ota, S.; Shiroshita, N. The role of protein kinase C isoforms in cell proliferation and a poptosis. Int. J. Hematol. 2000, 72, 12–19. [Google Scholar]
- Li, J.; Gobe, G. Protein kinase Cactivation and its role in kidney disease. Nephrology 2006, 11, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, K.R.; Johnson, E.C.; Cooney, S.K.; Anderberg, R.J.; Johnson, E.K.; Clifton, G.D.; Meek, R.L. Amino acids in juremesangial cells by advanced glycationend products, oxidative stress and protein kinase C. Kidney Int. 2005, 67, 953–968. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Soomro, I.; Sun, Y.; Li, Z.; Diggs, L.; Hatzivassiliou, G.; Thomas, A.G.; Rais, R.; Slusher, B.S.; Somlo, S.; Skolnik, E.Y. Glutamine metabolism viaglutaminase 1inautosomal-dominantpolycystic kidney disease. Nephrol. Dial. Transplant. 2018, 33, 1343–1353. [Google Scholar] [CrossRef]
- Tesch, G.; Ma, F.Y.; Nikolic-Paterson, D.J. ASK1: A new therapeutic target for kidney disease. Am. J. Physiol. Physiol. 2016, 311, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Grynberg, K.; Ma, F.Y.; Nikolic-Paterson, D.J. The JNK signaling path way in renal fibrosis. Front. Physiol. 2017, 8, 829–841. [Google Scholar] [CrossRef]
- Liles, J.T.; Corkey, B.K.; Notte, G.T.; Budas, G.R.; Lansdon, E.B.; Hinojosa-Kirschenbaum, F.; Badal, S.S.; Lee, M.; Schultz, B.E.; Wise, S.; et al. ASK1 contributes to fibrosis and dysfunction in models of kidney disease. J. Clin. Investig. 2018, 128, 4485–4500. [Google Scholar] [CrossRef]
- Peiris-John, R.; Wanigasuriya, J.; Wickremasinghe, A.; Dissanayake, W.; Hittarage, A. Exposure toacetylcholinester-ase-inhibiting pesticides and chronic renal failure. Ceylon Med. J. 2006, 51, 42–43. [Google Scholar] [CrossRef]
- Prall, Y.G.; Gambhir, K.K.; Cruz, I.A.; Blassingale, J.; Ampy, F.R. Acetyl cholinesterase activity in chronic renal failure. Life Sci. 2000, 66, 835–845. [Google Scholar] [CrossRef]
- Hu, R.; Huang, X.; Huang, J.; Li, Y.; Zhang, C.; Yin, Y.; Chen, Z.; Jin, Y.; Cai, J.; Cui, F. Long- and Short-Term Health Effects of Pesticide Exposure: A Cohort Study from China. PLoS ONE 2015, 10, e0128766. [Google Scholar] [CrossRef] [PubMed]
- Garfitt, S.; Jones, K.; Mason, H.; Cocker, J. Exposure to the organo phosphatediaz in on: Data from a human volunteer study with oral and dermal doses. Toxicol. Lett. 2002, 134, 105–113. [Google Scholar] [CrossRef]
- Liska, D.; Lyon, M.; Jones, D.S. Detoxification and Bio Transformationalim Balances; Explore: New York, NY, USA, 2006; Volume 2, p. 122. [Google Scholar]
- Levengood, J.M.; Beasley, V.R. Principles of eco toxicology. In Veterinary Toxicology; Elsevier Ltd.: Amsterdam, The Netherlands, 2007; pp. 689–708. [Google Scholar]
- Costa, L.G. Toxic effects of pesticides. In Casarett and Doull’s Toxicology: The Basic Science of Poisons; McGraw-Hill: New York, NY, USA, 2008; Volume 8, pp. 883–930. [Google Scholar]
- Barr, D.B.; Needham, L.L. Analytical methods for biological monitoring of exposure to pesticides: A review. J. Chromatogr. B 2002, 778, 5–29. [Google Scholar] [CrossRef]
- Cloyd, R.A. Pesticide Metabolites; Agricultural Experiment Station and Cooperative Extension Service; Kansas State University: Kansas, KS, USA, 2012. [Google Scholar]
- Sandrini, J.Z.; Rola, R.C.; Lopes, F.M.; Buffon, H.F.; Freitas, M.M.; Martins, C.d.M.G.; da Rosa, C.E. Effects of glyphosate on cholinesterase activity of the mussel Perna perna and the fish Dani or erio and Jenynsia mult identata: In vitro studies. Aquat. Toxicol. 2013, 130, 171–173. [Google Scholar] [CrossRef] [PubMed]
- Belpoggi, F.; Soffritti, M.; Guarino, M.; Lambertini, L.; Cevolani, D.; Maltoni, C. Resultsoflong-term experimental studies on the carcino genicity of ethylene-bis-dithiocarbamate(Mancozeb)inrats. Ann. N. Y. Acad. Sci. 2002, 982, 123–136. [Google Scholar] [CrossRef]
- Amoros, I.; Connon, R.; Garelick, H.; Alonso, J.L.; Carrasco, J. An assessment of the toxicity of some pesticides and the irme-tabolites affecting a natural aquatic environment using the Microtox™ system. Water Sci. Technol. 2000, 42, 19–24. [Google Scholar] [CrossRef]
- Riediger, S.; Behrends, A.; Croll, B.; Vega-Naredo, I.; Hänig, N.; Poeggeler, B.; Böker, J.; Grube, S.; Gipp, J.; Coto-Montes, A. Toxicity of the quinal phosmetabolite 2-hydroxy quinoxaline: Growth inhibition, induction of oxidative stress and genotoxicity in test organisms. Environ. Toxicol. Int. J. 2007, 22, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Epa, U. Special Report on Environmental Endocrine Disruption: An Effects Assessment and Analysis Office of Research and Development; REPA/630/R-96/012; National Service Center for Environmental Publications (NSCEP): Washington, DC, USA, 1997. [Google Scholar]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization and analysis platform. J. Cheminform. 2012, 4, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16Rev. C. 01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. A binitio calculation of vibrational absorption and circular dichroism spectra using density functional for cefields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Yan, Y.; Zhou, X.E.; Novick, S.; Shaw, S.J.; Li, Y.; Brunzelle, J.S.; Hitoshi, Y.; Griffin, P.R.; Xu, H.E.; Melcher, K. Structures of AMP-activated protein kinase bound to novel pharmacological activators in phosphorylated, non-phosphorylated, and nucleotide-free states. J. Biol. Chem. 2019, 294, 953–967. [Google Scholar] [CrossRef]
- Grodsky, N.; Li, Y.; Bouzida, D.; Love, R.; Jensen, J.; Nodes, B.; Nonomiya, J.; Grant, S. Structure of the catalytic domain of human protein kinase CβIIc omplexed with a bi sindolylmaleimide inhibitor. Biochemistry 2006, 45, 13970–13981. [Google Scholar] [CrossRef] [PubMed]
- Thangavelu, K.; Pan, C.Q.; Karlberg, T.; Balaji, G.; Uttamchandani, M.; Suresh, V.; Schüler, H.; Low, B.C.; Sivaraman, J. Structural basis for the all osteric inhibitory mechanism of human kidney-type glutaminase (KGA) and its regulation byR af-Mek-Erk signaling in cancer cell metabolism. Proc. Natl. Acad. Sci. USA 2012, 109, 7705–7710. [Google Scholar] [CrossRef] [PubMed]
- Terao, Y.; Suzuki, H.; Yoshikawa, M.; Yashiro, H.; Takekawa, S.; Fujitani, Y.; Okada, K.; Inoue, Y.; Yamamoto, Y.; Nakagawa, H. Design and biological evaluation of imidazo [1,2-a] pyridinesas novel and potent ASK1 inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 7326–7329. [Google Scholar] [CrossRef] [PubMed]
- Hsu, F.L.; Bae, S.Y.; McGuire, J.; Anderson, D.R.; Bester, S.M.; Height, J.J.; Pegan, S.D.; Walz, A.J. Synthesis and Molecular Properties of Nerve Agent Reactivator HLö-7Dimethane sulfonate. ACS Med. Chem. Lett. 2019, 10, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Sevrioukova, I.F.; Poulos, T.L. Structure and mechanism of the complex between cytochrome P4503A4 and ritonavir. Proc. Natl. Acad. Sci. USA 2010, 107, 18422–18427. [Google Scholar] [CrossRef]
- Prade, L.; Huber, R.; Manoharan, T.H.; Fahl, W.E.; Reuter, W. Structures of class piglutathioneS-transferase from human placenta in complex with substrate, transition-state analogueand inhibitor. Structure 1997, 5, 1287–1295. [Google Scholar] [CrossRef]
- Madden, T. The BLAST sequence analysis tool. In The NCBI Handbook, 2nd ed.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2013. [Google Scholar]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Eisenberg, D.; Lüthy, R.; Bowie, J.U. VERIFY3D: Assessment of protein models with three-dimensional profiles. In Methods in Enzymology; Elsevier BV: Amsterdam, The Netherlands, 1997; Volume 277, pp. 396–404. [Google Scholar]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of non bonded atomic interactions. Protein. Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- MacArthur, M.W.; Laskowski, R.A.; Thornton, J.M. Knowledge-based validation of protein structure coordinates derived by X-ray crystallography and NMR spectroscopy. Curr. Opin. Struct. Biol. 1994, 4, 731–737. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic. Acids. Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed]
- Moraes, J.P.; Pappa, G.L.; Pires, D.E.; Izidoro, S.C. GASS-WEB:a web server for identifying enzyme activesites based on genetic algorithms. Nucleic. Acids. Res. 2017, 45, 315–W319. [Google Scholar] [CrossRef] [PubMed]
- Gibrat, J.-F.; Madej, T.; Bryant, S.H. Surprising similarities in structure comparison. Curr. Opin. Struct. Biol. 1996, 6, 377–385. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Tomizawa, M.; Casida, J.E. Desnitro-imidacloprid activates the extracellular signal-regulated kinase cascade via the nicotinic receptor and intracellular calcium mobilization in N1E-115 cells. Toxicol. Appl. Pharm. 2002, 184, 180–186. [Google Scholar] [CrossRef] [PubMed]
- GROMACS, Version 2016; The Royal Institute of Technology and Uppsala University: Uppsala, Sweden, 2017.
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I. CHARMM general force field: A force field for drug-like molecules compatible with the CHARM Mall-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.C.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simu-lating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald-an N. Log(N)Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Arfken, G. Mathematical Methods for Physicists, 3rd ed.; Academic Press: Orlando, FL, USA, 1985. [Google Scholar]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; Vangunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular-Dynamics with Coupling to an Ex-ternal Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. Lig Plot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP:fully automated protein–ligand interaction profiler. Nucleic. Acids. Res. 2015, 43, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. NonDrug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Duan, L.; Liu, X.; Zhang, J.Z. Interaction Entropy: A New Paradigm for Highly Efficient and Reliable Computation of Protein–Ligand Binding Free Energy. J. Am. Chem. Soc. 2016, 138, 5722–5728. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.J.; Rodrigues, A.P.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S. Bio 3d: An Rpackage for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef]
- Tuck, S.; Graham-Lorence, S.; Peterson, J.; De Montellano, P.O. Active sites of the cytochrome p450cam (CYP101) F87W and F87A mutants. Evidence for significant structural reorganization without alteration of catalytic regiospecificity. J. Biol. Chem. 1993, 268, 269–275. [Google Scholar] [CrossRef]
- Thangavelu, K.; Chong, Q.Y.; Low, B.C.; Sivaraman, J. Structural basis for the active site inhibition mechanism of human kidney-typeglutaminase (KGA). Sci. Rep. 2014, 4, 3827–3834. [Google Scholar] [CrossRef] [PubMed]
- Tanoli, S.T.; Ramzan, M.; Hassan, A.; Sadiq, A.; Jan, M.S.; Khan, F.A.; Ullah, F.; Ahmad, H.; Bibi, M.; Mahmood, T. Design, synthesis and bioevaluation of tricyclicfusedring system as dual binding site acetylcholinesterase inhibitors. Bioorg. Chem. 2019, 83, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Bunkoczi, G.; Salah, E.; Filippakopoulos, P.; Fedorov, O.; Müller, S.; Sobott, F.; Parker, S.A.; Zhang, H.; Min, W.; Turk, B.E. Structural and functional characterization of the human proteinkinase ASK1. Structure 2007, 15, 1215–1226. [Google Scholar] [CrossRef]
- Gould, C.M.; Antal, C.E.; Reyes, G.; Kunkel, M.T.; Adams, R.A.; Ziyar, A.; Riveros, T.; Newton, A.C. Active site inhibitors protect proteinkinase C from dephosphorylation and stabilize its mature form. J. Biol. Chem. 2011, 286, 28922–28930. [Google Scholar] [CrossRef]
- Leonard, T.A.; Różycki, B.; Saidi, L.F.; Hummer, G.; Hurley, J.H. Crystals tructure and all osteric activation of proteinkinase CβII. Cell 2011, 144, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Oligschlaeger, Y.; Miglianico, M.; Chanda, D.; Scholz, R.; Thali, R.F.; Tuerk, R.; Stapleton, D.I.; Gooley, P.R.; Neumann, D. The recruitment of AMP-activated protein kinasetoglycogenis regulated by autophosphorylation. J. Biol. Chem. 2015, 290, 11715–11728. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef]
- Verschueren, K.; Cobbaut, M.; Demaerel, J.; Saadah, L.; Voet, A.R.; Van Lint, J.; DeBorggraeve, W.M. Discovery of a potent protein kinase Dinhibitor: Insights in the binding mode of pyrazolo [3,4-d] pyrimidine analogues. MedChemComm 2017, 8, 640–646. [Google Scholar] [CrossRef]
- Quesada-Soriano, I.; Parker, L.J.; Primavera, A.; Wielens, J.; Holien, J.K.; Casas-Solvas, J.M.; Vargas-Berenguel, A.; Aguilera, A.M.; Nuccetelli, M.; Mazzetti, A.P. Diuretic drug binding to human glutathionetransferase P1-1: Potentialrole of Cys-101 revealed in the double mutant C47S/Y108V. J. Mol. Recognit. 2011, 24, 220–234. [Google Scholar] [CrossRef]
- Lobanov, M.Y.; Bogatyreva, N.S.; Galzitskaya, O.V. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Jensen, P.K.; Wujcik, C.E.; McGuire, M.K.; McGuire, M.A. Validation of reliable and selective methods for direct determination of glyphosate and aminomethylphosphonicacidin milk and urine using LC-MS/MS. J. Environ. Sci. Health Part B 2016, 51, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Colovic, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetyl choline sterase Inhibitors: Pharmacology and Toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed]
- Davydov, D.R.; Halpert, J.R. Allosteric P450 mechanisms: Multiple bindingsites, multiple conformers or both? Expert. Opin. Drug Metab. Toxicol. 2008, 4, 1523–1535. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Pesticide | Metabolite of the Pesticide |
|---|---|---|
| AChE | FM | FMS |
| AMPK | FN | FNS |
| ASK1 | FM | FMS |
| GST | IM | 6-CIPHD |
| PKC | IM | 6-CIPHD |
| Protein | Chemical Identity | Action | Binding Residues From Literature | Binding Energy (kcal/mol) | Theoretical Binding Residues |
|---|---|---|---|---|---|
| AMPK | 5-{[6-chloro-5-(2′-hydroxy[1,1′-biphenyl]-4-yl)-1H-benzimidazol-2-yl]oxy}-N-hydroxy-2-methylbenzamide | Activator | ARG83B, ASN50A, VAL113B, ASP108B, GLY30A, LY33A, ILE48A, VAL13A, LEU20A, VAL81B [46] | −9.52 | ARG83B, ASN50A, VAL113B, ASP108B, GLY30A, LY33A, ILE48A, VAL13A, LEU20A, |
| PKC | 3-{1-[3-(dimethylamino)propyl]-2-methyl-1h-indol-3-yl}-4-(2-methyl-1h-indol-3-yl)-1h-pyrrole-2,5-dione | Inhibitor | GLU421A, THR404A, VAL356A, LEU348A, ASP484A, ALA483A, PHE353A [47] | −8.42 | GLU421A, THR404A, VAL356A, LEU348A, ASP484A, ALA483A, PHE353A |
| GLS | 5,5′-(sulfanediyldiethane-2,1-diyl)bis(1,3,4-thiadiazol-2-amine) | Inhibitor | LEU323A, TYR394A [48] | −9.37 | LEU323A, TYR394A |
| ASK1 | 4-tert-butyl-N-[6-(1H-imidazol-1-yl)imidazo[1,2-a]pyridin-2-yl]benzamide | Inhibitor | LYS709A, PRO758A, VAL649A, LEU810A, ALA707A, VAL757A [49] | −8.25 | LYS709A, PRO758A, VAL649A, ALA707A, VAL757A |
| AChE | 1-[({2,4-bis[(e)-(hydroxyimino)methyl]pyridinium-1-yl}methoxy)methyl]-4-carbamoylpyridinium | Reactivator | TYR337A, PHE338A, TYR341A, TRP286A, VAL282A, ASP74A, SER125A, ASN87A, TYR72A, TYR124A [50] | −7.98 | TYR337A, PHE338A, TRP286A, VAL282A, SER125A, ASN87A, TYR72A, TYR124A |
| Protein | Rg of Crystal Structure of Protein (nm)Metabolite | Average Rg and Standard Deviation of Protein Ligand Complex During MD Simulation | Average Rg and Standard Deviation of Protein during MD Simulation | ||
|---|---|---|---|---|---|
| Metabolite | Parent Pesticide | Metabolite | Parent Pesticide | ||
| AChE | 2.266 | 2.308 (±0.041) | 2.308 (±0.083) | 2.312 (±0.001) | 2.312 (±0.001) |
| ASK1 | 1.901 | 1.957 (±0.013) | 1.918 (±0.008) | 1.939 (±0.001) | 1.924 (±0.001) |
| PKC | 2.0541 | 2.075 (±0.006) | 2.081 (±0.001) | 2.079 (±0.019) | 2.082 (±0.013) |
| GST | 2.104 | 2.127 (±0.005) | 2.113 (±0.007) | 2.132 (±0.017) | 2.117 (±0.067) |
| AMPK | 3.427 | 3.452 (±0.016) | 3.478 (±0.020) | 3.452 (±0.022) | 3.475 (±0.022) |
| Complex | VdE Energy (kJ/mol) | Elec. Energy (kJ/mol) | Polar Solvation Energy (kJ/mol) | SASA Energy (kJ/mol) | Binding Energy (kJ/mol) | Entropy of Binding TΔS (kJ/mol) |
|---|---|---|---|---|---|---|
| AChE-FM | −180.89 ± 8.92 | −16.43 ± 6.58 | 115.53 ± 10.37 | −18.77 ± 0.65 | −100.56 ± 12.50 | −14.36 |
| AChE-FMS | −175.27 ± 9.22 | −13.34 ± 6.56 | 94.07 ± 18.03 | −18.81 ± 1.04 | −113.34 ± 15.42 | −15.84 |
| ASK1-FM | −116.32 ± 9.10 | −35.00 ± 6.67 | 92.26 ± 7.07 | −14.44 ± 0.74 | −73.49 ± 9.24 | −11.44 |
| ASK1-FMS | −116.61 ± 20.747 | −8.22 ± 1.265 | 78.60 ± 17.334 | −14.00 ± 2.015 | −60.23 ± 14.03 | −31.2 |
| AMPK-FN | −95.75 ± 14.32 | −13.72 ± 9.25 | 58.96 ± 13.21 | 10.70 ± 0.64 | −61.20 ± 13.31 | −15.59 |
| AMPK-FNS | −118.99 ± 6.80 | −21.62 ± 1.56 | 83.24 ± 20.22 | −13.35 ± 0.73 | −70.72 ± 14.7 | −25.30 |
| PKC-IM | −77.04 ± 12.46 | −29.36 ± 3.61 | 75.44 ± 13.32 | −8.679 ± 1.0 | −39.64 ± 15.40 | −42.809 |
| PKC-6CIPHD | −109.58 ± 12.92 | −21.77 ± 9.54 | 91.09 ± 26.82 | −12.78 ± 1.15 | −53.03 ± 24.60 | −14.22 |
| GST-IM | −108.88 ± 9.38 | −113.57 ± 12.44 | 254.65 ± 17.67 | −13.27 ± 0.51 | 18.92 ± 7.71 | −17.449 |
| GST-6CIPHD | −137.52 ± 6.45 | −137.52 ± 6.45 | 231.87 ± 23.23 | −13.83 ± 0.73 | −21.40 ± 10.36 | −17.443 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajapaksha, H.; Pandithavidana, D.R.; Dahanayake, J.N. Demystifying Chronic Kidney Disease of Unknown Etiology (CKDu): Computational Interaction Analysis of Pesticides and Metabolites with Vital Renal Enzymes. Biomolecules 2021, 11, 261. https://doi.org/10.3390/biom11020261
Rajapaksha H, Pandithavidana DR, Dahanayake JN. Demystifying Chronic Kidney Disease of Unknown Etiology (CKDu): Computational Interaction Analysis of Pesticides and Metabolites with Vital Renal Enzymes. Biomolecules. 2021; 11(2):261. https://doi.org/10.3390/biom11020261
Chicago/Turabian StyleRajapaksha, Harindu, Dinesh R. Pandithavidana, and Jayangika N. Dahanayake. 2021. "Demystifying Chronic Kidney Disease of Unknown Etiology (CKDu): Computational Interaction Analysis of Pesticides and Metabolites with Vital Renal Enzymes" Biomolecules 11, no. 2: 261. https://doi.org/10.3390/biom11020261
APA StyleRajapaksha, H., Pandithavidana, D. R., & Dahanayake, J. N. (2021). Demystifying Chronic Kidney Disease of Unknown Etiology (CKDu): Computational Interaction Analysis of Pesticides and Metabolites with Vital Renal Enzymes. Biomolecules, 11(2), 261. https://doi.org/10.3390/biom11020261