
The Role of Interferons in the Pathogenesis of Sjögren’s Syndrome and Future Therapeutic Perspectives

 , ,
, ,
Abstract
1. Introduction
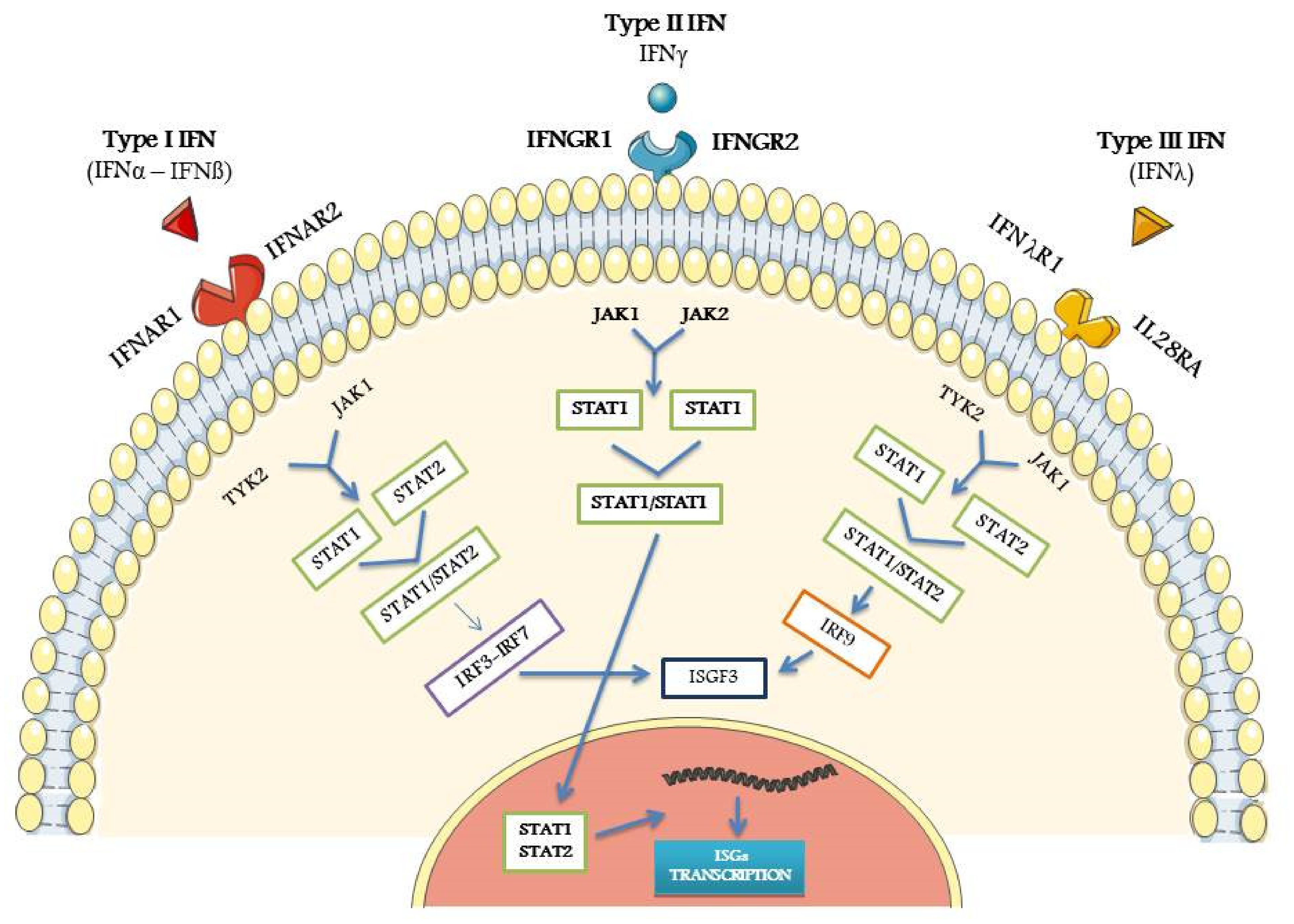
2. Biological Properties of IFNs
3. Detection of IFN-Related Activity
4. Role of IFNs Signature in the Pathogenesis of pSS in Humans
5. IFN Signature, from Human Studies to Animal Models
6. Perspectives of IFN-Modulating Therapies in pSS
6.1. Therapies Preventing the Activation of TLRs
6.2. Therapies Modulating IFN Pathway Activation in pDCs
6.3. Therapy against IFNα and Related Receptors
6.4. Therapies Contrasting JAK Activation and the Subsequent IFN Production
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moutsopoulos, H.M. Sjögren’s syndrome: A forty-year scientific journey. J. Autoimmun. 2014, 51, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Vitali, C.; Del Papa, N. Pain in primary Sjögren’s syndrome. Best Pract. Res. Clin. Rheumatol. 2015, 29, 63–70. [Google Scholar] [CrossRef]
- Nakamura, H.; Shimizu, T.; Kawakami, A. Role of viral infections in the pathogenesis of Sjögren’s syndrome: Different characteristics of Epstein-Barr virus and HTLV-1. J. Clin. Med. 2020, 9, 1459. [Google Scholar] [CrossRef]
- Utomo, S.W.; Putri, J.F. Infections as risk factor of Sjögren’s syndrome. Open Access Rheumatol. 2020, 12, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Vitali, C. Immunopathologic differences of Sjögren’s syndrome versus sicca syndrome in HCV and HIV infection. Arthritis Res. Ther. 2011, 13, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Takahashi, Y.; Yamamoto-Fukuda, T.; Horai, Y.; Nakashima, Y.; Arima, K.; Nakamura, T.; Koji, T.; Kawakami, A. Direct infection of primary salivary gland epithelial cells by human T lymphotropic virus type I in patients with Sjögren’s syndrome. Arthritis Rheumatol. 2015, 67, 1096–1106. [Google Scholar] [CrossRef] [PubMed]
- Brodziak, A.; Ziółko, E.; Muc-Wierzgoń, M.; Nowakowska-Zajdel, E.; Kokot, T.; Klakla, K. The role of human endogenous retroviruses in the pathogenesis of autoimmune diseases. Med. Sci. Monit. 2012, 18, 80–88. [Google Scholar]
- Moyes, D.L.; Martin, A.; Sawcer, S.; Temperton, N.; Worthington, J.; Griffiths, D.J.; Venables, P.J. The distribution of the endogenous retroviruses HERV-K113 and HERV-K115 in health and disease. Genomics 2005, 86, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Lessard, C.J.; Li, H.; Adrianto, I.; Ice, J.A.; Rasmussen, A.; Grundahl, K.M.; Kelly, J.A.; Dozmorov, M.G.; Miceli-Richard, C.; Bowman, S.; et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjögren’s syndrome. Nat. Genet. 2013, 45, 1284–1292. [Google Scholar] [CrossRef]
- Song, I.W.; Chen, H.C.; Lin, Y.F.; Yang, J.H.; Chang, C.C.; Chou, C.T.; Lee, M.M.; Chou, Y.C.; Chen, C.H.; Chen, Y.T.; et al. Identification of susceptibility gene associated with female primary Sjögren’s syndrome in Han Chinese by genome-wide association study. Hum. Genet. 2016, 135, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Miceli-Richard, C.; Comets, E.; Loiseau, P.; Puechal, X.; Hachulla, E.; Mariette, X. Association of an IRF5 gene functional polymorphism with Sjögren’s syndrome. Arthritis Rheum. 2007, 56, 3989–3994. [Google Scholar] [CrossRef] [PubMed]
- Miceli-Richard, C.; Gestermann, N.; Ittah, M.; Comets, E.; Loiseau, P.; Puechal, X.; Hachulla, E.; Gottenberg, J.-E.; Lebon, P.; Becquemont, L.; et al. The CGGGG insertion/deletion polymorphism of the IRF5 promoter is a strong risk factor for primary Sjögren’s syndrome. Arthritis Rheum. 2009, 60, 1991–1997. [Google Scholar] [CrossRef]
- Gestermann, N.; Mekinian, A.; Comets, E.; Loiseau, P.; Puechal, X.; Hachulla, E.; Gottenberg, J.E.; Mariette, X.; Miceli-Richard, C. STAT4 is a confirmed genetic risk factor for Sjögren’s syndrome and could be involved in type 1 interferon pathway signaling. Genes Immun. 2010, 11, 432–438. [Google Scholar] [CrossRef]
- Marketos, N.; Cinoku, I.; Rapti, A.; Mavragani, C.P. Type I interferon signature in Sjögren’s syndrome: Pathophysiological and clinical implications. Clin. Exp. Rheumatol. 2019, 37, 185–191. [Google Scholar] [PubMed]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signaling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Meyer, O. Interferons and autoimmune disorders. Joint Bone Spine. 2009, 76, 464–473. [Google Scholar] [CrossRef]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef]
- Muskardin, T.L.W.; Niewold, T.B. Type I interferon in rheumatic diseases. Nat. Rev. Rheumatol. 2018, 14, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 1, 519–525. [Google Scholar] [CrossRef]
- Le Bon, A.; Schiavoni, G.; D’Agostino, G.; Gresser, I.; Belardinelli, F.; Tough, D.F. Type I interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity 2001, 14, 461–470. [Google Scholar] [CrossRef]
- Hervas-Stubbs, S.; Perez-Garcia, J.L.; Rouzaut, A.; Sanmame, M.F.; Le Bon, A.; Melero, I. Direct effects of type I interferons on cells of the immune system. Clin. Cancer Res. 2011, 17, 2619–2627. [Google Scholar] [CrossRef]
- Chen, K.; Liu, J.; Cao, X. Regulation of type I interferon signaling in immunity and inflammation: A comprehensive review. J. Autoimmun. 2017, 83, 1–11. [Google Scholar] [CrossRef]
- Witas, R.; Gupta, S.; Nguyen, C.Q. Contributions of major cell populations to Sjögren’s Syndrome. J. Clin. Med. 2020, 9, 3057. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Mann-Nuttel, R.; Schulze, A.; RichterI, L.; Alferink, J.; Scheu, S. Sources of type I interferons in infectious immunity: Plasmacytoid dendritic cells not always in the driver’s seat. Front. Immunol. 2019, 10, 778–798. [Google Scholar] [CrossRef] [PubMed]
- Aculumbre, S.; Raieli, S.; Hoffmann, C.; Chelbi, R. Plasmacytoid pre-dendritic cells (pDC): From molecular pathways to function and disease association. Semin. Cell. Dev. Biol. 2019, 86, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003, 4, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-γ: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef]
- Sheppard, P.; Kindsvogel, W.; Xu, W.; Henderson, K.; Schlutsmeyer, S.; Whitmore, T.E.; Kuestner, R.; Garrigues, U.; Birks, C.; Roraback, J.; et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 2003, 4, 63–68. [Google Scholar] [CrossRef]
- Hu, X.; Ivashkiv, L.B. Cross-regulation of signaling pathways by interferon-gamma: Implications for immune responses and autoimmune diseases. Immunity 2009, 31, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Schoenborn, J.R.; Wilson, C.B. Regulation of Interferon-γ during innate and adaptive immune responses. Adv. Immunol. 2007, 96, 41–101. [Google Scholar] [PubMed]
- Wang, F.; Zhang, S.; Jeon, R.; Vuckovic, I.; Jiang, X.; Lerman, A.; Folmes, C.D.; Dzeja, P.D.; Herrmann, J. Interferon gamma induces reversible metabolic reprogramming of M1 macrophages to sustain cell viability and pro-inflammatory activity. EBioMed. 2018, 30, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.G.; Mariani, L.; Radbruch, A.; Höfer, T. Sequential polarization and imprinting of type 1 T helper lymphocytes by interferon-γ and interleukin-12. Immunity 2009, 30, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Berger, A. Th1 and Th2 responses: What are they? BMJ 2000, 321, 424. [Google Scholar] [CrossRef] [PubMed]
- Karabiyik, A.; Peck, A.B.; Nguyen, C.Q. The important role of T cells and receptor expression in Sjögren’s syndrome. Scand. J. Immunol. 2013, 78, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Wack, A.; Terczynska-Dyla, E.; Hartmann, R. Guarding the frontiers: The biology of type III interferons. Nat. Immunol. 2015, 16, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Lamot, L.; Niemietz, I.; Brown, K.L. Methods for type I interferon detection and their relevance for clinical utility and improved understanding of rheumatic diseases. Clin. Exp. Rheumatol. 2019, 37, 1077–1083. [Google Scholar]
- Wilson, D.H.; Rissin, D.M.; Kan, C.W.; Fournier, D.R.; Piech, T.; Campbell, T.G.; Meyer, R.E.; Fishburn, M.W.; Cabrera, C.; Patel, P.P.; et al. The Simoa HD-1 analyzer: A novel fully automated digital immunoassay analyzer with single- molecule sensitivity and multiplexing. J. Lab. Autom. 2016, 21, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Kirou, K.A.; Lee, C.; George, S.; Louca, K.; Papagiannis, I.G.; Peterson, M.G.E.; Ly, N.; Woodward, R.N.; Fry, K.E.; Lau, A.Y.-H.; et al. Coordinate overexpression of interferon-α-induced genes in systemic lupus erythematosus. Arthritis Rheum. 2004, 50, 3958–3967. [Google Scholar] [CrossRef]
- Tan, F.K.; Zhou, X.; Maayes, M.D.; Gourh, P.; Guo, X.; Marcum, C.; Jin, L.; Arnett, F.C., Jr. Signatures of differentially regulated interferon gene expression and vasculotrophism in the peripheral blood cells of systemic sclerosis patients. Rheumatology 2006, 45, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Gottenberg, J.E.; Cagnard, N.; Lucchesi, C.; Letourneur, F.; Mistou, S.; Lazure, T.; Jacques, S.; Ba, N.; Ittah, M.; Lepajolec, C.; et al. Activation of IFN pathways and plasmacytoid dendritic cell recruitment in target organs of primary Sjögren’s syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 2770–2775. [Google Scholar] [CrossRef]
- Emamian, E.S.; Leon, J.M.; Lessard, C.J.; Grandits, M.; Baechler, E.C.; Gaffney, P.M.; Segal, B.; Rhodus, N.L.; Moser, K.L. Peripheral blood gene expression profiling in Sjögren’s syndrome. Genes Immun. 2009, 10, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Brkic, Z.; Maria, N.I.; van Helden-Meeuwsen, C.G.; van de Merwe, J.P.; van Daele, P.L.; Dalm, V.A.; Wildenberg, M.E.; Beumer, W.; Drexhage, H.A.; Versnel, M.A. Prevalence of interferon type I signature in CD14 monocytes of patients with Sjögren’s syndrome and association with disease activity and BAFF gene expression. Ann. Rheum. Dis. 2013, 72, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Hjelmervik, T.O.R.; Petersen, K.; Jonassen, I.; Jonsson, R.; Bolstad, A.I. Gene expression profiling of minor salivary glands clearly distinguishes primary Sjögren’s syndrome patients from healthy control subjects. Arthritis Rheum. 2005, 52, 1534–1544. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Wu, H.; Grossman, J.M.; Hanvivadhanakul, P.; Fitzgerald, J.D.; Park, G.S.; Dong, X.; Chen, W.; Kim, M.H.; Weng, H.H.; et al. Association of increased interferon-inducible gene expression with disease activity and lupus nephritis in patients with systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 2951–2962. [Google Scholar] [CrossRef]
- Hall, J.C.; Casciola-Rosen, L.; Berger, A.E.; Kapsogeorgou, E.K.; Cheadle, C.; Tzioufas, A.G.; Baer, A.N.; Rosen, A. Precise probes of type II interferon activity define the origin of interferon signatures in target tissues in rheumatic diseases. Proc. Natl. Acad. Sci. USA 2012, 109, 17609–17614. [Google Scholar] [CrossRef]
- Hall, J.C.; Baer, A.N.; Shah, A.A.; Criswell, L.A.; Shiboski, C.H.; Rosen, A.; Casciola-Rosen, L. Molecular subsetting of interferon pathways in Sjögren’s Syndrome. Arthritis Rheum. 2015, 67, 2437–2446. [Google Scholar] [CrossRef]
- Nezos, A.; Gravani, F.; Tassidou, A.; Kapsogeorgou, K.E.; Voulgarelis, M.; Koutsilieris, M.; Crow, M.K.; Mavragani, C.P. Type I and II interferon signatures in Sjögren’s syndrome pathogenesis: Contributions in distinct clinical phenotypes and Sjögren’s related lymphomagenesis. J. Autoimmun. 2015, 63, 47–58. [Google Scholar] [CrossRef]
- Kimoto, O.; Sawada, J.; Shimoyama, K.; Suzuki, D.; Nakamura, S.; Hayashi, H.; Ogawa, N. Activation of the interferon pathway in peripheral blood of patients with Sjögren’s syndrome. J. Rheumatol. 2011, 38, 310–316. [Google Scholar] [CrossRef]
- Wildemberg, M.E.; Van Helden-Meeuwsen, C.G.; Van De Merwe, J.P.; Drexhage, H.A.; Versnell, M.A. Systemic increase in type I interferon activity in Sjögren’s syndrome: A putative role for plasmacytoid dendritic cells. Eur. J. Immunol. 2008, 38, 2024–2033. [Google Scholar] [CrossRef] [PubMed]
- Imgenberg-Kreuz, J.; Sandling, J.K.; Bjork, A.; Nordlund, J.; Kvarnström, M.; Eloranta, M.L.; Rönnblom, L.; Wahren-Herlenius, M.; Syvänen, A.C.; Nordmark., G. Transcription profiling of peripheral B cells in antibody-positive primary Sjögren’s syndrome reveals upregulated expression of CX3CR1 and a type I and type II interferon signature. Scand. J. Immunol. 2018, 87, 1–15. [Google Scholar] [CrossRef]
- Chiche, L.; Jourde-Chiche, N.; Whalen, E.; Presnell, S.; Gersuk, V.; Dang, K.; Anguiano, E.; Quinn, C.; Burtey, S.; Berland, Y.; et al. Modular transcriptional repertoire analyses of adults with systemic lupus erythematosus reveal distinct type I and type II interferon signatures. Arthritis Rheumatol. 2014, 66, 1583–1595. [Google Scholar] [CrossRef] [PubMed]
- Bodewes, I.L.A.; Al-Ali, S.; van Helden-Meeuwsen, C.G.; Maria, N.I.; Tarn, J.; Lendrem, D.W.; Schreurs, M.W.J.; Steenwijk, E.C.; van Daele, P.L.A.; Both, T.; et al. Systemic interferon type I and type II signatures in primary Sjögren’s syndrome reveal differences in biological disease activity. Rheumatology 2018, 57, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Seror, R.; Ravaud, P.; Bowman, S.J.; Baron, G.; Tzioufas, A.; Theander, E.; Gottenberg, J.E.; Bootsma, H.; Mariette, X.; Vitali, C. EULAR Sjögren’s syndrome disease activity index: Development of a consensus systemic disease activity index for primary Sjögren’s syndrome. Ann. Rheum. Dis. 2010, 69, 1103–1109. [Google Scholar] [CrossRef] [PubMed]
- Seror, R.; Meiners, P.; Baron, G.; Bootsma, H.; Bowman, S.J.; Vitali, C.; Gottenberg, J.E.; Theander, E.; Tzioufas, A.; De Vita, S.; et al. Development of the ClinESSDAI: A clinical score without biological domain. A tool for biological studies. Ann. Rheum. Dis. 2016, 75, 1945–1950. [Google Scholar] [CrossRef]
- Seror, R.; Ravaud, P.; Mariette, X.; Bootsma, H.; Theander, E.; Hansen, A.; Ramos-Casals, M.; Dörner, T.; Bombardieri, S.; Hachulla, E.; et al. EULAR Sjögren’s task force. EULAR Sjögren’s syndrome patient reported index (ESSPRI): Development of a consensus patient index for primary Sjögren’s syndrome. Ann. Rheum. Dis. 2011, 70, 968–972. [Google Scholar] [CrossRef] [PubMed]
- Del Papa, N.; Vitali, C.; Lorini, M.; Carbonelli, V.; Maglione, W.; Pignataro, F.; Minniti, A.; Montano, N.; Caporali, R. AB0125 expression of interferon type I- and type II-induced genes in patients with Sjögren’s syndrome with and without extraglandular involvement. Ann. Rheum. Dis. 2020, 79, 1362–1363. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.W.; Feng, G.S. Relationship between interferon-γ, indoleamine 2,3-dioxygenase, and tryptophan catabolism. FASEB J. 1991, 5, 2516–2522. [Google Scholar] [CrossRef]
- Baban, B.; Chandler, P.R.; Sharma, M.D.; Pihkala, J.; Koni, P.A.; Munn, D.H.; Mellor, A.L. IDO activates regulatory T cells and blocks their conversion into Th17-like T Cells. J. Immunol. 2009, 183, 2475–2483. [Google Scholar] [CrossRef]
- Lenany, N.; Berta, L.; Kovalcs, L.; Balog, A.; Toldi, G. The role of B7 family costimulatory molecules and indoleamine 2,3-dioxygenase in primary Sjögren’s syndrome and systemic sclerosis. Immunol. Res. 2017, 65, 622–629. [Google Scholar]
- Maria, N.I.; van Helden-Meeuwsen, C.G.; Brkic, Z.; Paulissen, S.M.; Steenwijk, E.C.; Dalm, V.A.; van Daele, P.L.; Martin van Hagen, P.; Kroese, F.G.; van Roon, J.A.; et al. Increased Tregs associated with elevated indoleamine-2,3-dioxygenase activity and an imbalanced kynurenine pathway in IFN positive primary Sjögren’s syndrome. Arthritis Rheum. 2016, 68, 1688–1699. [Google Scholar] [CrossRef] [PubMed]
- Mellor, A.L.; Lemos, H.; Huang, L. Indoleamine 2,3-dioxygenase and tolerance: Where are we now? Front. Immunol. 2017, 8, 1360–1366. [Google Scholar] [CrossRef]
- Liu, M.; Wang, X.; Wang, L.; Ma, X.; Gong, Z.; Zhang, S.; Li, Y. Targeting the IDO1 pathway in cancer: From bench to bedside. J. Hematol. Oncol. 2018, 11, 100–112. [Google Scholar] [CrossRef]
- Szodoray, P.; Papp, G.; Horvath, I.F.; Barath, S.; Sipka, S.; Nakken, B.; Zeher, M. Cells with regulatory function of the innate and adaptive immune system in primary Sjögren’s syndrome. Clin. Exp. Immunol. 2009, 157, 343–349. [Google Scholar] [CrossRef]
- Sarigul, M.; Yazisiz, V.; Bassorgun, C.I.; Ulker, M.; Avci, A.B.; Erbasan, F.; Gelen, T.; Gorczynski, R.M.; Terzioglu, E. The numbers of Foxp3 + Treg cells are positively correlated with higher grade of infiltration at the salivary glands in primary Sjögren’s syndrome. Lupus 2010, 19, 138–145. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, F.R.; Fantucci, M.Z.; Adriano, L.; Valim, V.; Cunha, T.M.; Louzada-Junior, P.; Rocha, E.M. Neurological and inflammatory manifestations in Sjögren’s syndrome: The role of the kynurenine metabolic pathway. Int. J. Mol. Sci. 2018, 19, 3953. [Google Scholar] [CrossRef]
- Jovanovic, F.; Candido, K.D.; Knezevic, N.N. The role of the kynurenine signaling pathway in different chronic pain conditions and potential use of therapeutic agents. Int. J. Mol. Sci. 2020, 21, 6045. [Google Scholar] [CrossRef]
- Howard Tripp, N.; Tarn, J.; Natasari, A.; Gillespie, C.; Mitchell, S.; Hackett, K.L.; Bowman, S.J.; Price, E.; Pease, C.T.; Emery, P.; et al. Fatigue in primary Sjögren’s syndrome is associated with lower levels of proinflammatory cytokines. RMD Open 2016, 2, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Imgenberg-Kreuz, J.; Sandling, J.K.; Almlöf, J.C.; Nordlund, J.; Signér, L.; Norheim, K.B.; Omdal, R.; Rönnblom, L.; Eloranta, M.L.; Syvänen, A.C.; et al. Genome-wide DNA methylation analysis in multiple tissues in primary Sjögren’s syndrome reveals regulatory effects at interferon-induced genes. Ann. Rheum. Dis. 2016, 75, 2029–2036. [Google Scholar] [CrossRef]
- Karageorgas, T.; Fragioudaki, S.; Nezos, A.; Karaiskos, D.; Moutsopoulos, H.M.; Mavragani, C.P. Fatigue in primary Sjögren’s syndrome: Clinical, laboratory, psychometric, and biologic associations. Arthritis Care Res. 2016, 68, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Bodewes, I.L.A.; van der Spek, P.J.; Leon, L.G.; Wijkhuijs, A.J.M.; van Helden-Meeuwsen, C.G.; Tas, L.; Schreurs, M.W.J.; van Daele, P.L.A.; Katsikis, P.D.; Versnel, M.A. Fatigue in Sjögren’s Syndrome: A search for biomarkers and treatment targets. Front. Immunol. 2019, 10, 312–322. [Google Scholar] [CrossRef]
- Apostolou, E.; Kapsogeorgou, E.K.; Konsta, O.D.; Giotakis, I.; Saridaki, M.I.; Andreakos, E.; Tzioufas, A.G. Expression of type III interferons (IFNλs) and their receptor in Sjögren’s syndrome. Clin. Exp. Immunol. 2016, 186, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Thorlacius, G.E.; Wahren-Herlenius, M.; Ronnblom, L. An update on the role of type I interferons in systemic lupus erythematosus and Sjögren’s syndrome. Curr. Opin. Rheumatol. 2018, 30, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Vitali, C.; Dolcino, M.; Del Papa, N.; Minniti, A.; Pignataro, F.; Maglione, W.; Lunardi, C.; Puccetti, A. Gene expression profiles in primary Sjögren’s syndrome with and without systemic manifestations. ACR Open Rheumatol. 2019, 1, 603–613. [Google Scholar] [CrossRef]
- Delaleu, N.; Nguyen, C.Q.; Peck, A.B.; Jonsson, R. Sjögren’s syndrome: Studying the disease in mice. Arthritis Res. Ther. 2011, 13, 217–232. [Google Scholar] [CrossRef] [PubMed]
- Maria, N.I.; Vogelsang, P.; Versnel, M.A. The clinical relevance of animal models in Sjögren’s syndrome: The interferon signature from mouse to man. Arthritis Res. Ther. 2015, 17, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Lodde, B.M.; Mineshiba, F.; Kok, M.R.; Wang, J.; Zheng, C.; Schmidt, M.; Cotrim, A.P.; Kriete, M.; Tak, P.P.; Baum, B.J. NOD mouse model for Sjögren’s syndrome: Lack of longitudinal stability. Oral Dis. 2006, 12, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Szczerba, B.M.; Rybakowska, P.D.; Dey, P.; Payerhin, K.M.; Peck, A.B.; Bagavant, H.; Deshmukh, U.S. Type I interferon receptor deficiency prevents murine Sjogren’s syndrome. J. Dent. Res. 2013, 92, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Nandula, S.R.; Dey, P.; Corbin, K.L.; Nunemaker, C.S.; Bagavant, H.; Deshmukh, U.S. Salivary gland hypofunction induced by activation of innate immunity is dependent on type I interferon signaling. J. Oral. Pathol. Med. 2012, 42, 66–72. [Google Scholar] [CrossRef]
- Cha, S.; Brayer, J.; Gao, J.; Brown, V.; Killedar, S.; Yasunari, U.; Peck, A.B. A dual role for interferon-gamma in the pathogenesis of Sjogren’s syndrome-like autoimmune exocrinopathy in the nonobese diabetic mouse. Scand. J. Immunol. 2004, 60, 552–565. [Google Scholar] [CrossRef]
- Yin, H.; Vosters, J.L.; Roescher, N.; D’Souza, A.; Kurien, B.T.; Tak, P.P.; Chiorini, J.A. Location of immunization and interferon-γ are central to induction of salivary gland dysfunction in Ro60 peptide immunized model of Sjögren’s syndrome. PLoS ONE 2011, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Desnues, B.; Macedo, A.B.; Roussel-Queval, A.; Bonnardel, J.; Henri, S.; Demaria, O.; Alexopoulou, L. TLR8 on dendritic cells and TLR9 on B cells restrain TLR7-mediated spontaneous autoimmunity in C57BL/6 mice. Proc. Natl. Acad. Sci. USA 2014, 111, 1497–1502. [Google Scholar] [CrossRef] [PubMed]
- Kosenda, K.; Ichii, O.; Otsuka, S.; Hashimoto, Y.; Kon, Y. BXSB/MpJ-Yaa mice develop autoimmune dacryoadenitis with the appearance of inflammatory cell marker messenger RNAs in the lacrimal fluid. Clin. Exp. Ophthalmol. 2013, 41, 788–797. [Google Scholar] [CrossRef]
- Posada, J.; Valadkhan, S.; Burge, D.; Davies, K.; Tarn, J.; Casement, J.; Jobling, K.; Gallagher, P.; Wilson, D.; Barone, F.; et al. Improvement of severe fatigue following nuclease therapy in patients with primary Sjögren’s Syndrome: A randomized clinical trial. Arthritis Rheumatol. 2020, 73, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Furie, R.; Werth, V.P.; Merola, J.F.; Stevenson, L.; Reynolds, T.L.; Naik, H.; Wang, W.; Christmann, R.; Gardet, A.; Pellerin, A.; et al. Monoclonal antibody targeting BDCA2 ameliorates skin lesions in systemic lupus erythematosus. J. Clin. Invest. 2019, 129, 1359–1371. [Google Scholar] [CrossRef]
- Bodewes, I.L.A.; Huijser, E.; van Helden-Meeuwsen, C.G.; Tas, L.; Huizinga, R.; Dalm, V.A.S.H.; van Hagen, P.M.; Groot, N.; Kamphuis, S.; van Daele, P.L.A.; et al. TBK1: A key regulator and potential treatment target for interferon positive Sjögren’s syndrome, systemic lupus erythematosus and systemic sclerosis. J. Autoimmun. 2018, 91, 97–102. [Google Scholar] [CrossRef]
- Kalunian, K.C.; Merrill, J.T.; Maciuca, R.; McBride, J.M.; Townsend, M.J.; Wei, X.; Davis, J.C., Jr.; Kennedy, W.P. A phase II study of the efficacy and safety of rontalizumab (rhuMAb interferon-α) in patients with systemic lupus erythematosus (ROSE). Ann. Rheum. Dis. 2015, 75, 196–202. [Google Scholar] [CrossRef]
- Khamashta, M.; Merrill, J.T.; Werth, V.P.; Furie, R.; Kalunian, K.; Illei, G.G.; Drappa, J.; Wang, L.; Greth, W. Sifalimumab, an anti-interferon-α monoclonal antibody, in moderate to severe systemic lupus erythematosus: A randomised, double-blind, placebo-controlled study. Ann. Rheum. Dis. 2016, 75, 1909–1916. [Google Scholar] [CrossRef]
- Boedigheimer, M.J.; Martin, D.A.; Amoura, Z.; Sánchez-Guerrero, J.; Romero-Diaz, J.; Kivitz, A.; Aranow, C.; Chan, T.M.; Chong, Y.B.; Chiu, K.; et al. Safety, pharmacokinetics and pharmacodynamics of AMG 811, an anti-interferon-γ monoclonal antibody, in SLE subjects without or with lupus nephritis. Lupus Sci. Med. 2017, 4, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Song, G.G. Anifrolumab for the treatment of active systemic lupus erythematosus: A meta-analysis of randomized controlled trials. Zeitschrift fur Rheumatologie 2020. [Google Scholar] [CrossRef] [PubMed]
- Sacre, K.; Criswell, L.A.; McCune, J.M. Hydroxychloroquine is associated with impaired interferon-alpha and tumor necrosis factor-alpha production by plasmacytoid dendritic cells in systemic lupus erythematosus. Arthritis Res. Ther. 2012, 14, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Gottenberg, J.E.; Ravaud, P.; Puéchal, X.; Le Guern, V.; Sibilia, J.; Goeb, V.; Larroche, C.; Dubost, J.J.; Rist, S.; Saraux, A.; et al. Effects of hydroxychloroquine on symptomatic improvement in primary Sjögren syndrome: The JOQUER randomized clinical trial. J. Am. Med. Assoc. 2014, 312, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Burge, D.J.; Eisenman, J.; Byrnes-Blake, K.; Smolak, P.; Lau, K.; Cohen, S.B.; Kivitz, A.J.; Levin, R.; Martin, R.W.; Sherrer, Y.; et al. Safety, pharmacokinetics, and pharmacodynamics of RSLV-132, an RNase-Fc fusion protein in systemic lupus erythematosus: A randomized, double-blind, placebo-controlled study. Lupus 2017, 26, 825–834. [Google Scholar] [CrossRef]
- Cao, W.; Bover, L. Signaling and ligand interaction of ILT7: Receptor-mediated regulatory mechanisms for plasmacytoid dendritic cells. Immunol. Rev. 2010, 234, 163–176. [Google Scholar] [CrossRef]
- Tavano, B.; Boasso, A. Effect of immunoglobin-like transcript 7 cross-linking on plasmacytoid dendritic cells differentiation into antigen-presenting cells. PLoS ONE 2014, 9, 1–9. [Google Scholar] [CrossRef][Green Version]
- Dzionek, A.; Sohma, Y.; Nagafune, J.; Cella, M.; Colonna, M.; Facchetti, F.; Günther, G.; Johnston, I.; Lanzavecchia, A.; Nagasaka, T.; et al. BDCA-2, a novel plasmacytoid dendritic cell-specific type II C-type lectin, mediates antigen capture and is a potent inhibitor of interferon alpha/beta induction. J. Exp. Med. 2001, 194, 1823–1834. [Google Scholar] [CrossRef] [PubMed]
- Chatham, W.W.; Furie, R.; Saxena, A.; Brohawn, P.; Schwetje, E.; Abreu, G.; Tummala, R. Long-term safety and efficacy of anifrolumab in adults with systemic lupus erythematosus: Results of a phase 2 open-label extension study. Arthritis Rheumatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Damsky, W.; Peterson, D.; Ramseier, J.; Al-Bawardy, B.; Chun, H.; Proctor, D.; Strand, V.; Flavell, R.A.; King, B. The emerging role of Janus kinase inhibitors in the treatment of autoimmune and inflammatory diseases. J. Allergy Clin. Immunol. 2020, 28, 1–13. [Google Scholar] [CrossRef]
- Lee, J.; Lee, J.; Kwok, S.K.; Baek, S.; Jang, S.G.; Hong, S.M.; Min, J.W.; Choi, S.S.; Lee, J.; Cho, M.L.; et al. JAK-1 inhibition suppresses interferon-induced BAFF production in human salivary gland: Potential therapeutic strategy for primary Sjögren’s syndrome. Arthritis Rheumatol. 2018, 70, 2057–2066. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Gene | MX1 | IFIT1 | IFT3 | IFI44 | IDO1 | GBP1 | MIG | IP-10 | P2RY14 |
|---|---|---|---|---|---|---|---|---|---|
| pSS-EGMs | 85.9 | 38.5 | 24.4 | 40.4 | 25.1 | 8.3 | 4.5 | 1.5 | 5.5 |
| pSS-WP | 4.2 | 1.7 | 2.0 | 4.8 | 4.1 | 1.2 | 0.6 | 0.3 | 1.3 |
| HC | 2.1 | 1.6 | 1.1 | 1.5 | 1.4 | 1.3 | 1.5 | 1.3 | 1.4 |
| Agent Code/Name | Biological Property | Mechanism of Action | Trial/Study | Status | Results | Reference |
|---|---|---|---|---|---|---|
| RSLV132 | IgG1 fused with RNAse | degradation of circulating RNA | phase II in pSS | completed | reduction of fatigue | [84] |
| MEDI7734 | mAb stimulating ILT7 | dowregulation of TLR7 and TLR9 | phase I in different autoimmune diseases | completed | no results available | clin.trials.gov: NCT02780674 |
| BIIB059 | mAb against BDCA2 antigen on pDCs | internalization of BDCA2 and inhibition of type I IFN release | phase I in SLE | completed | reduction of SLE-related skin lesions and IFN expression | [85] |
| BX795 | Inhibition of TBK1 | downregulation of IRF3 and IFR7 | in vitro study on PBMCs from different autoimmune diseases | completed | reduced expression of ISGs | [86] |
| rontalizumab | mAb against IFN | inactivaction of IFN | phase II in SLE | completed | reduction of DA and clinical flares | [87] |
| sifalimumab | mAb against IFN | inactivaction of IFN | phase II in SLE | completed | reduction of DA | [88] |
| AMG811 | mAb against IFN | inactivaction of IFN | phase Ib in SLE | completed | acceptable safety no clinical effects | [89] |
| anifrolumab | mAb against IFNAR | inhibition of IFN receptor | 3 RCTs in SLE | completed | efficacy only in some disease manifestations | [90] |
| tofacitinib | JAK 3 inhibitor | suppression of ISGs transcription | phase Ib-IIa in pSS | ongoing | no results available | clin.trials.gov: NCT04496960 |
| filgotinib | JAK 1 inhibitor | suppression of ISGs transcription | phase II in active pSS | ongoing | no results available | clin.trials.gov: NCT03100942 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Del Papa, N.; Minniti, A.; Lorini, M.; Carbonelli, V.; Maglione, W.; Pignataro, F.; Montano, N.; Caporali, R.; Vitali, C. The Role of Interferons in the Pathogenesis of Sjögren’s Syndrome and Future Therapeutic Perspectives. Biomolecules 2021, 11, 251. https://doi.org/10.3390/biom11020251
Del Papa N, Minniti A, Lorini M, Carbonelli V, Maglione W, Pignataro F, Montano N, Caporali R, Vitali C. The Role of Interferons in the Pathogenesis of Sjögren’s Syndrome and Future Therapeutic Perspectives. Biomolecules. 2021; 11(2):251. https://doi.org/10.3390/biom11020251
Chicago/Turabian StyleDel Papa, Nicoletta, Antonina Minniti, Maurizio Lorini, Vincenzo Carbonelli, Wanda Maglione, Francesca Pignataro, Nicola Montano, Roberto Caporali, and Claudio Vitali. 2021. "The Role of Interferons in the Pathogenesis of Sjögren’s Syndrome and Future Therapeutic Perspectives" Biomolecules 11, no. 2: 251. https://doi.org/10.3390/biom11020251
APA StyleDel Papa, N., Minniti, A., Lorini, M., Carbonelli, V., Maglione, W., Pignataro, F., Montano, N., Caporali, R., & Vitali, C. (2021). The Role of Interferons in the Pathogenesis of Sjögren’s Syndrome and Future Therapeutic Perspectives. Biomolecules, 11(2), 251. https://doi.org/10.3390/biom11020251