
Signaling Nodes Associated with Endoplasmic Reticulum Stress during NAFLD Progression


Abstract
1. Introduction
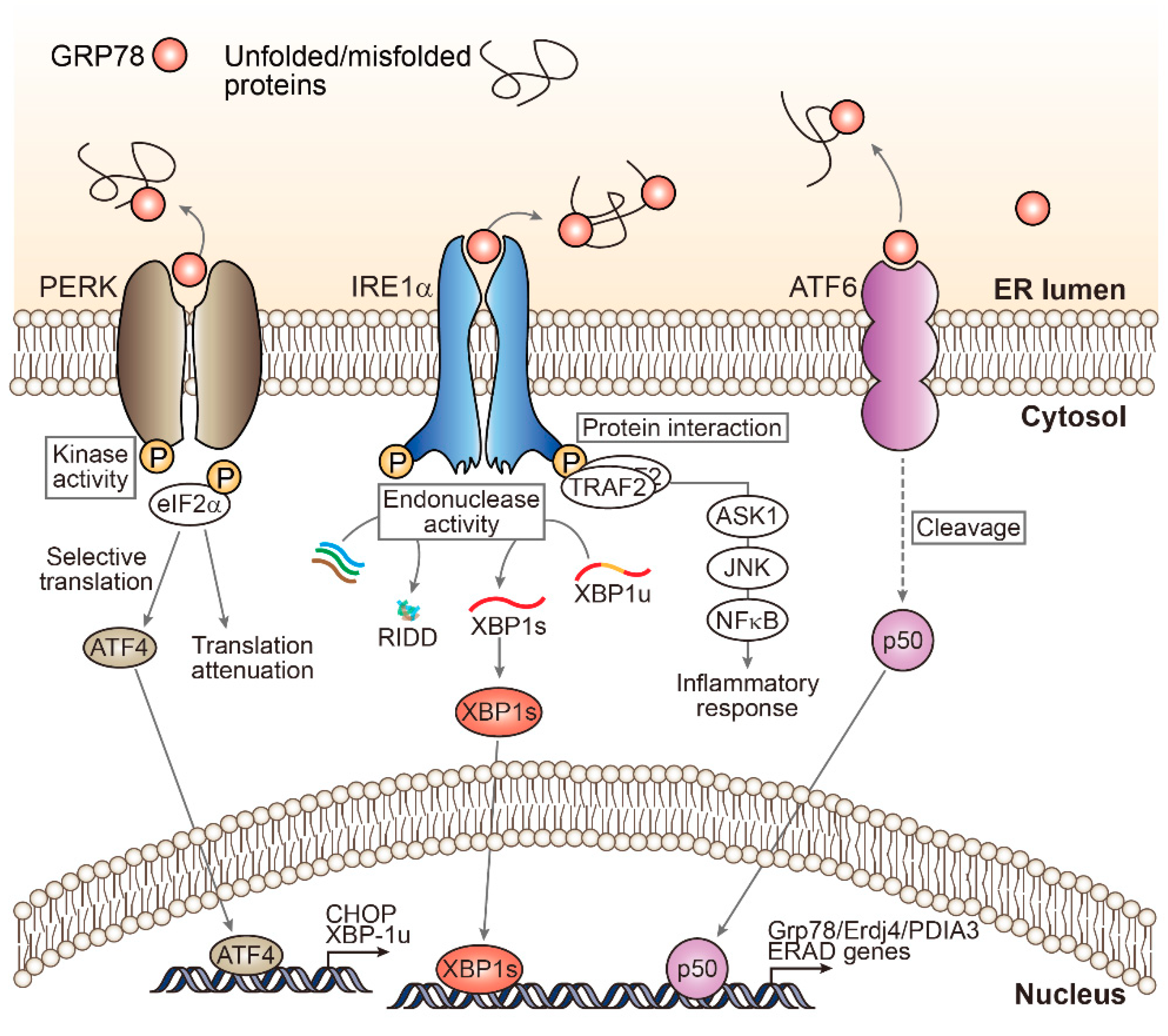
2. Unfolded Protein Response Signaling: A Myriad of ER Stress Effectors
3. Clinical Association of ER Stress in NAFLD Patients
4. Experimental Models Depicting ER Stress as the Key Trigger of NAFLD
5. Cell-Type Dependent Effects of ER Stress in Liver
5.1. Hepatocytes
5.1.1. BAX/BAK
5.1.2. ASK1
5.1.3. PHLDA3
5.1.4. DUSP5
5.1.5. FXR
5.2. Hepatic Stellate Cells
5.2.1. SMAD2
5.2.2. JNK1/2
5.2.3. NOX1/NOX4
5.3. Kupffer Cells
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Hetz, C.; Chevet, E.; Harding, H.P. Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 2013, 12, 703–719. [Google Scholar] [CrossRef]
- Lin, J.H.; Walter, P.; Yen, T.S. Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol. 2008, 3, 399–425. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, K.R.; Chaudhary, M.; Kim, H.R.; Chae, H.J. Endoplasmic Reticulum (ER) Stress Response Failure in Diseases. Trends Cell Biol. 2020, 30, 672–675. [Google Scholar] [CrossRef] [PubMed]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef]
- Ghemrawi, R.; Battaglia-Hsu, S.F.; Arnold, C. Endoplasmic Reticulum Stress in Metabolic Disorders. Cells 2018, 7, 63. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, L.; Tabas, I. Role of endoplasmic reticulum stress in metabolic disease and other disorders. Annu. Rev. Med. 2012, 63, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, D.; Wootz, H.; Korhonen, L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006, 13, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.K.; Chae, S.W.; Kim, H.R.; Chae, H.J. Endoplasmic reticulum stress and cancer. J. Cancer Prev. 2014, 19, 75–88. [Google Scholar] [CrossRef]
- Watt, M.J.; Miotto, P.M.; De Nardo, W.; Montgomery, M.K. The Liver as an Endocrine Organ-Linking NAFLD and Insulin Resistance. Endocr. Rev. 2019, 40, 1367–1393. [Google Scholar] [CrossRef]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef] [PubMed]
- Dara, L.; Ji, C.; Kaplowitz, N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology 2011, 53, 1752–1763. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Konerman, M.A.; Jones, J.C.; Harrison, S.A. Pharmacotherapy for NASH: Current and emerging. J. Hepatol. 2018, 68, 362–375. [Google Scholar] [CrossRef]
- Wattacheril, J.; Issa, D.; Sanyal, A. Nonalcoholic Steatohepatitis (NASH) and Hepatic Fibrosis: Emerging Therapies. Annu. Rev. Pharm. Toxicol. 2018, 58, 649–662. [Google Scholar] [CrossRef]
- Rinella, M.E. Nonalcoholic fatty liver disease: A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Peng, C.; Stewart, A.G.; Woodman, O.L.; Ritchie, R.H.; Qin, C.X. Non-Alcoholic Steatohepatitis: A Review of Its Mechanism, Models and Medical Treatments. Front. Pharm. 2020, 11, 603926. [Google Scholar] [CrossRef]
- Pagliassotti, M.J. Endoplasmic reticulum stress in nonalcoholic fatty liver disease. Annu. Rev. Nutr. 2012, 32, 17–33. [Google Scholar] [CrossRef]
- Karagöz, G.E.; Aragón, T.; Acosta-Alvear, D. Recent advances in signal integration mechanisms in the unfolded protein response. F1000Research 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Martinon, F.; Rodriguez, D.; Glimcher, L.H. The unfolded protein response: Integrating stress signals through the stress sensor IRE1α. Physiol. Rev. 2011, 91, 1219–1243. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2018, 9, 3083. [Google Scholar] [CrossRef]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef]
- Matsumoto, H.; Miyazaki, S.; Matsuyama, S.; Takeda, M.; Kawano, M.; Nakagawa, H.; Nishimura, K.; Matsuo, S. Selection of autophagy or apoptosis in cells exposed to ER-stress depends on ATF4 expression pattern with or without CHOP expression. Biol. Open 2013, 2, 1084–1090. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Reimold, A.M.; Etkin, A.; Clauss, I.; Perkins, A.; Friend, D.S.; Zhang, J.; Horton, H.F.; Scott, A.; Orkin, S.H.; Byrne, M.C.; et al. An essential role in liver development for transcription factor XBP-1. Genes Dev. 2000, 14, 152–157. [Google Scholar]
- Zhang, K.; Wong, H.N.; Song, B.; Miller, C.N.; Scheuner, D.; Kaufman, R.J. The unfolded protein response sensor IRE1alpha is required at 2 distinct steps in B cell lymphopoiesis. J. Clin. Investig. 2005, 115, 268–281. [Google Scholar] [CrossRef]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef]
- Rashid, H.O.; Kim, H.K.; Junjappa, R.; Kim, H.R.; Chae, H.J. Endoplasmic reticulum stress in the regulation of liver diseases: Involvement of Regulated IRE1α and β-dependent decay and miRNA. J. Gastroenterol. Hepatol. 2017, 32, 981–991. [Google Scholar] [CrossRef]
- Yoshida, H.; Haze, K.; Yanagi, H.; Yura, T.; Mori, K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 1998, 273, 33741–33749. [Google Scholar] [CrossRef]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, A.; Mayoral, R.; Agra, N.; Valdecantos, M.P.; Pardo, V.; Miquilena-Colina, M.E.; Vargas-Castrillon, J.; Lo Iacono, O.; Corazzari, M.; Fimia, G.M.; et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014, 5, e1179. [Google Scholar] [CrossRef] [PubMed]
- Lebeaupin, C.; Proics, E.; de Bieville, C.H.; Rousseau, D.; Bonnafous, S.; Patouraux, S.; Adam, G.; Lavallard, V.J.; Rovere, C.; Le Thuc, O.; et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015, 6, e1879. [Google Scholar] [CrossRef]
- Toriguchi, K.; Hatano, E.; Tanabe, K.; Takemoto, K.; Nakamura, K.; Koyama, Y.; Seo, S.; Taura, K.; Uemoto, S. Attenuation of steatohepatitis, fibrosis, and carcinogenesis in mice fed a methionine-choline deficient diet by CCAAT/enhancer-binding protein homologous protein deficiency. J. Gastroenterol. Hepatol. 2014, 29, 1109–1118. [Google Scholar] [CrossRef]
- Puri, P.; Mirshahi, F.; Cheung, O.; Natarajan, R.; Maher, J.W.; Kellum, J.M.; Sanyal, A.J. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Farrell, G.; Schattenberg, J.M.; Leclercq, I.; Yeh, M.M.; Goldin, R.; Teoh, N.; Schuppan, D. Mouse Models of Nonalcoholic Steatohepatitis: Toward Optimization of Their Relevance to Human Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2241–2257. [Google Scholar] [CrossRef]
- Lau, J.K.; Zhang, X.; Yu, J. Animal models of non-alcoholic fatty liver disease: Current perspectives and recent advances. J. Pathol. 2017, 241, 36–44. [Google Scholar] [CrossRef]
- Ibrahim, S.H.; Hirsova, P.; Malhi, H.; Gores, G.J. Animal Models of Nonalcoholic Steatohepatitis: Eat, Delete, and Inflame. Dig. Dis. Sci. 2016, 61, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Savard, C.; Tartaglione, E.V.; Kuver, R.; Haigh, W.G.; Farrell, G.C.; Subramanian, S.; Chait, A.; Yeh, M.M.; Quinn, L.S.; Ioannou, G.N. Synergistic interaction of dietary cholesterol and dietary fat in inducing experimental steatohepatitis. Hepatology 2013, 57, 81–92. [Google Scholar] [CrossRef]
- Fujii, M.; Shibazaki, Y.; Wakamatsu, K.; Honda, Y.; Kawauchi, Y.; Suzuki, K.; Arumugam, S.; Watanabe, K.; Ichida, T.; Asakura, H.; et al. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med. Mol. Morphol. 2013, 46, 141–152. [Google Scholar] [CrossRef]
- Tsuchida, T.; Lee, Y.A.; Fujiwara, N.; Ybanez, M.; Allen, B.; Martins, S.; Fiel, M.I.; Goossens, N.; Chou, H.I.; Hoshida, Y.; et al. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J. Hepatol. 2018, 69, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Asgharpour, A.; Cazanave, S.C.; Pacana, T.; Seneshaw, M.; Vincent, R.; Banini, B.A.; Kumar, D.P.; Daita, K.; Min, H.K.; Mirshahi, F.; et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J. Hepatol. 2016, 65, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Henkel, A.S.; Dewey, A.M.; Anderson, K.A.; Olivares, S.; Green, R.M. Reducing endoplasmic reticulum stress does not improve steatohepatitis in mice fed a methionine- and choline-deficient diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G54–G59. [Google Scholar] [CrossRef]
- Bashiri, A.; Nesan, D.; Tavallaee, G.; Sue-Chue-Lam, I.; Chien, K.; Maguire, G.F.; Naples, M.; Zhang, J.; Magomedova, L.; Adeli, K.; et al. Cellular cholesterol accumulation modulates high fat high sucrose (HFHS) diet-induced ER stress and hepatic inflammasome activation in the development of non-alcoholic steatohepatitis. Biochim. Biophys. Acta 2016, 1861, 594–605. [Google Scholar] [CrossRef]
- Kandel-Kfir, M.; Almog, T.; Shaish, A.; Shlomai, G.; Anafi, L.; Avivi, C.; Barshack, I.; Grosskopf, I.; Harats, D.; Kamari, Y. Interleukin-1alpha deficiency attenuates endoplasmic reticulum stress-induced liver damage and CHOP expression in mice. J. Hepatol. 2015, 63, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Choi, Y.; Godlewski, G.; Ha, S.K.; Banerjee, A.; Jang, S.; Song, B.J. Cytochrome P450-2E1 promotes fast food-mediated hepatic fibrosis. Sci. Rep. 2017, 7, 39764. [Google Scholar] [CrossRef]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Gorgun, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef]
- Ben Mosbah, I.; Alfany-Fernandez, I.; Martel, C.; Zaouali, M.A.; Bintanel-Morcillo, M.; Rimola, A.; Rodes, J.; Brenner, C.; Rosello-Catafau, J.; Peralta, C. Endoplasmic reticulum stress inhibition protects steatotic and non-steatotic livers in partial hepatectomy under ischemia-reperfusion. Cell Death Dis. 2010, 1, e52. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef]
- Peña-Blanco, A.; García-Sáez, A.J. Bax, Bak and beyond—Mitochondrial performance in apoptosis. FEBS J. 2018, 285, 416–431. [Google Scholar] [CrossRef]
- Akazawa, Y.; Cazanave, S.; Mott, J.L.; Elmi, N.; Bronk, S.F.; Kohno, S.; Charlton, M.R.; Gores, G.J. Palmitoleate attenuates palmitate-induced Bim and PUMA up-regulation and hepatocyte lipoapoptosis. J. Hepatol. 2010, 52, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Bernasconi, P.; Fisher, J.; Lee, A.H.; Bassik, M.C.; Antonsson, B.; Brandt, G.S.; Iwakoshi, N.N.; Schinzel, A.; Glimcher, L.H.; et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science 2006, 312, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Lisbona, F.; Rojas-Rivera, D.; Thielen, P.; Zamorano, S.; Todd, D.; Martinon, F.; Glavic, A.; Kress, C.; Lin, J.H.; Walter, P.; et al. BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1alpha. Mol. Cell 2009, 33, 679–691. [Google Scholar] [CrossRef]
- Bailly-Maitre, B.; Belgardt, B.F.; Jordan, S.D.; Coornaert, B.; von Freyend, M.J.; Kleinridders, A.; Mauer, J.; Cuddy, M.; Kress, C.L.; Willmes, D.; et al. Hepatic Bax inhibitor-1 inhibits IRE1alpha and protects from obesity-associated insulin resistance and glucose intolerance. J. Biol. Chem. 2010, 285, 6198–6207. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Brahmbhatt, H.; Mergenthaler, P.; Zhang, Z.; Sang, J.; Daude, M.; Ehlert, F.G.R.; Diederich, W.E.; Wong, E.; Zhu, W.; et al. A Small-Molecule Inhibitor of Bax and Bak Oligomerization Prevents Genotoxic Cell Death and Promotes Neuroprotection. Cell Chem. Biol. 2017, 24, 493–506.e5. [Google Scholar] [CrossRef]
- Garner, T.P.; Amgalan, D.; Reyna, D.E.; Li, S.; Kitsis, R.N.; Gavathiotis, E. Small-molecule allosteric inhibitors of BAX. Nat. Chem. Biol. 2019, 15, 322–330. [Google Scholar] [CrossRef]
- Jensen, K.; WuWong, D.J.; Wong, S.; Matsuyama, M.; Matsuyama, S. Pharmacological inhibition of Bax-induced cell death: Bax-inhibiting peptides and small compounds inhibiting Bax. Exp. Biol. Med. 2019, 244, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Nishitoh, H.; Matsuzawa, A.; Tobiume, K.; Saegusa, K.; Takeda, K.; Inoue, K.; Hori, S.; Kakizuka, A.; Ichijo, H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002, 16, 1345–1355. [Google Scholar] [CrossRef]
- Takeda, K.; Matsuzawa, A.; Nishitoh, H.; Ichijo, H. Roles of MAPKKK ASK1 in stress-induced cell death. Cell Struct. Funct. 2003, 28, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Imarisio, C.; Alchera, E.; Bangalore Revanna, C.; Valente, G.; Follenzi, A.; Trisolini, E.; Boldorini, R.; Carini, R. Oxidative and ER stress-dependent ASK1 activation in steatotic hepatocytes and Kupffer cells sensitizes mice fatty liver to ischemia/reperfusion injury. Free Radic. Biol. Med. 2017, 112, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Lawitz, E.; Mantry, P.S.; Jayakumar, S.; Caldwell, S.H.; Arnold, H.; Diehl, A.M.; Djedjos, C.S.; Han, L.; Myers, R.P.; et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology 2018, 67, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Schuster-Gaul, S.; Geisler, L.J.; McGeough, M.D.; Johnson, C.D.; Zagorska, A.; Li, L.; Wree, A.; Barry, V.; Mikaelian, I.; Jih, L.J.; et al. ASK1 inhibition reduces cell death and hepatic fibrosis in an Nlrp3 mutant liver injury model. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Challa, T.D.; Wueest, S.; Lucchini, F.C.; Dedual, M.; Modica, S.; Borsigova, M.; Wolfrum, C.; Blüher, M.; Konrad, D. Liver ASK1 protects from non-alcoholic fatty liver disease and fibrosis. EMBO Mol. Med. 2019, 11, e10124. [Google Scholar] [CrossRef] [PubMed]
- Kawase, T.; Ohki, R.; Shibata, T.; Tsutsumi, S.; Kamimura, N.; Inazawa, J.; Ohta, T.; Ichikawa, H.; Aburatani, H.; Tashiro, F.; et al. PH domain-only protein PHLDA3 is a p53-regulated repressor of Akt. Cell 2009, 136, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Ohki, R.; Saito, K.; Chen, Y.; Kawase, T.; Hiraoka, N.; Saigawa, R.; Minegishi, M.; Aita, Y.; Yanai, G.; Shimizu, H.; et al. PHLDA3 is a novel tumor suppressor of pancreatic neuroendocrine tumors. Proc. Natl. Acad. Sci. USA 2014, 111, E2404–E2413. [Google Scholar] [CrossRef] [PubMed]
- Muroi, H.; Nakajima, M.; Satomura, H.; Takahashi, M.; Yamaguchi, S.; Sasaki, K.; Yokobori, T.; Miyazaki, T.; Kuwano, H.; Kato, H. Low PHLDA3 expression in oesophageal squamous cell carcinomas is associated with poor prognosis. Anticancer Res. 2015, 35, 949–954. [Google Scholar]
- Takikawa, M.; Ohki, R. A vicious partnership between AKT and PHLDA3 to facilitate neuroendocrine tumors. Cancer Sci. 2017, 108, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Han, C.Y.; Lim, S.W.; Koo, J.H.; Kim, W.; Kim, S.G. PHLDA3 overexpression in hepatocytes by endoplasmic reticulum stress via IRE1-Xbp1s pathway expedites liver injury. Gut 2016, 65, 1377–1388. [Google Scholar] [CrossRef]
- Lee, C.G.; Kang, Y.J.; Kim, H.S.; Moon, A.; Kim, S.G. Phlda3, a urine-detectable protein, causes p53 accumulation in renal tubular cells injured by cisplatin. Cell Biol. Toxicol. 2015, 31, 121–130. [Google Scholar] [CrossRef]
- Sakata, N.; Yamaguchi, Y.; Chen, Y.; Shimoda, M.; Yoshimatsu, G.; Unno, M.; Sumi, S.; Ohki, R. Pleckstrin homology-like domain family A, member 3 (PHLDA3) deficiency improves islets engraftment through the suppression of hypoxic damage. PLoS ONE 2017, 12, e0187927. [Google Scholar] [CrossRef] [PubMed]
- Bensellam, M.; Chan, J.Y.; Lee, K.; Joglekar, M.V.; Hardikar, A.A.; Loudovaris, T.; Thomas, H.E.; Jonas, J.C.; Laybutt, D.R. Phlda3 regulates beta cell survival during stress. Sci. Rep. 2019, 9, 12827. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Tan, T.H. DUSPs, to MAP kinases and beyond. Cell Biosci. 2012, 2, 24. [Google Scholar] [CrossRef]
- Chen, H.F.; Chuang, H.C.; Tan, T.H. Regulation of Dual-Specificity Phosphatase (DUSP) Ubiquitination and Protein Stability. Int. J. Mol. Sci. 2019, 20, 2668. [Google Scholar] [CrossRef] [PubMed]
- Lang, R.; Raffi, F.A.M. Dual-Specificity Phosphatases in Immunity and Infection: An Update. Int. J. Mol. Sci. 2019, 20, 2710. [Google Scholar] [CrossRef]
- Jo, H.J.; Yang, J.W.; Park, J.H.; Choi, E.S.; Lim, C.S.; Lee, S.; Han, C.Y. Endoplasmic Reticulum Stress Increases DUSP5 Expression via PERK-CHOP Pathway, Leading to Hepatocyte Death. Int. J. Mol. Sci. 2019, 20, 4369. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.Y.; Lee, H.; Hubbert, M.L.; Edwards, P.A.; Zhang, Y. FXR, a multipurpose nuclear receptor. Trends Biochem. Sci. 2006, 31, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Han, C.Y. Update on FXR Biology: Promising Therapeutic Target? Int. J. Mol. Sci. 2018, 19, 2069. [Google Scholar] [CrossRef]
- Xiong, X.; Wang, X.; Lu, Y.; Wang, E.; Zhang, Z.; Yang, J.; Zhang, H.; Li, X. Hepatic steatosis exacerbated by endoplasmic reticulum stress-mediated downregulation of FXR in aging mice. J. Hepatol. 2014, 60, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Han, C.Y.; Rho, H.S.; Kim, A.; Kim, T.H.; Jang, K.; Jun, D.W.; Kim, J.W.; Kim, B.; Kim, S.G. FXR Inhibits Endoplasmic Reticulum Stress-Induced NLRP3 Inflammasome in Hepatocytes and Ameliorates Liver Injury. Cell Rep. 2018, 24, 2985–2999. [Google Scholar] [CrossRef]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.A.; Kowdley, K.V. Obeticholic acid for the treatment of nonalcoholic steatohepatitis. Expert Rev. Gastroenterol. Hepatol. 2020, 14, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Mederacke, I.; Hsu, C.C.; Troeger, J.S.; Huebener, P.; Mu, X.; Dapito, D.H.; Pradere, J.P.; Schwabe, R.F. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 2013, 4, 2823. [Google Scholar] [CrossRef]
- Koo, J.H.; Lee, H.J.; Kim, W.; Kim, S.G. Endoplasmic Reticulum Stress in Hepatic Stellate Cells Promotes Liver Fibrosis via PERK-Mediated Degradation of HNRNPA1 and Up-regulation of SMAD2. Gastroenterology 2016, 150, 181–193.e8. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Gea, V.; Hilscher, M.; Rozenfeld, R.; Lim, M.P.; Nieto, N.; Werner, S.; Devi, L.A.; Friedman, S.L. Endoplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J. Hepatol. 2013, 59, 98–104. [Google Scholar] [CrossRef]
- Kim, R.S.; Hasegawa, D.; Goossens, N.; Tsuchida, T.; Athwal, V.; Sun, X.; Robinson, C.L.; Bhattacharya, D.; Chou, H.I.; Zhang, D.Y.; et al. The XBP1 Arm of the Unfolded Protein Response Induces Fibrogenic Activity in Hepatic Stellate Cells Through Autophagy. Sci. Rep. 2016, 6, 39342. [Google Scholar] [CrossRef] [PubMed]
- Mannaerts, I.; Thoen, L.F.R.; Eysackers, N.; Cubero, F.J.; Batista Leite, S.; Coldham, I.; Colle, I.; Trautwein, C.; van Grunsven, L.A. Unfolded protein response is an early, non-critical event during hepatic stellate cell activation. Cell Death Dis. 2019, 10, 98. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; Brenner, D.A.; Karin, M. A liver full of JNK: Signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology 2012, 143, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Demay, Y.; Perochon, J.; Szuplewski, S.; Mignotte, B.; Gaumer, S. The PERK pathway independently triggers apoptosis and a Rac1/Slpr/JNK/Dilp8 signaling favoring tissue homeostasis in a chronic ER stress Drosophila model. Cell Death Dis. 2014, 5, e1452. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhou, X.; Cai, J.; Zhao, F.; Cao, T.; Ning, L.; Luo, C.; Xiao, X.; Liu, S. Recombinant Treponema pallidum protein Tp0768 promotes proinflammatory cytokine secretion of macrophages through ER stress and ROS/NF-kappaB pathway. Appl. Microbiol. Biotechnol. 2021, 105, 353–366. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Kluwe, J.; Pradere, J.P.; Gwak, G.Y.; Mencin, A.; De Minicis, S.; Osterreicher, C.H.; Colmenero, J.; Bataller, R.; Schwabe, R.F. Modulation of hepatic fibrosis by c-Jun-N-terminal kinase inhibition. Gastroenterology 2010, 138, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Hatting, M.; Nevzorova, Y.A.; Peng, J.; Hu, W.; Boekschoten, M.V.; Roskams, T.; Muller, M.; Gassler, N.; Liedtke, C.; et al. Jnk1 in murine hepatic stellate cells is a crucial mediator of liver fibrogenesis. Gut 2014, 63, 1159–1172. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Qiu, T.; Yao, X.; Jiang, L.; Wang, N.; Jiang, J.; Jia, X.; Wei, S.; Zhang, J.; Zhu, Y.; et al. IRE1alpha/NOX4 signaling pathway mediates ROS-dependent activation of hepatic stellate cells in NaAsO2-induced liver fibrosis. J. Cell Physiol. 2021, 236, 1469–1480. [Google Scholar] [CrossRef]
- Lan, T.; Kisseleva, T.; Brenner, D.A. Deficiency of NOX1 or NOX4 Prevents Liver Inflammation and Fibrosis in Mice through Inhibition of Hepatic Stellate Cell Activation. PLoS ONE 2015, 10, e0129743. [Google Scholar] [CrossRef]
- Gaggini, F.; Laleu, B.; Orchard, M.; Fioraso-Cartier, L.; Cagnon, L.; Houngninou-Molango, S.; Gradia, A.; Duboux, G.; Merlot, C.; Heitz, F.; et al. Design, synthesis and biological activity of original pyrazolo-pyrido-diazepine, -pyrazine and -oxazine dione derivatives as novel dual Nox4/Nox1 inhibitors. Bioorg. Med. Chem. 2011, 19, 6989–6999. [Google Scholar] [CrossRef]
- Laleu, B.; Gaggini, F.; Orchard, M.; Fioraso-Cartier, L.; Cagnon, L.; Houngninou-Molango, S.; Gradia, A.; Duboux, G.; Merlot, C.; Heitz, F.; et al. First in class, potent, and orally bioavailable NADPH oxidase isoform 4 (Nox4) inhibitors for the treatment of idiopathic pulmonary fibrosis. J. Med. Chem. 2010, 53, 7715–7730. [Google Scholar] [CrossRef]
- Wenfeng, Z.; Yakun, W.; Di, M.; Jianping, G.; Chuanxin, W.; Chun, H. Kupffer cells: Increasingly significant role in nonalcoholic fatty liver disease. Ann. Hepatol. 2014, 13, 489–495. [Google Scholar] [CrossRef]
- Yang, F.; Liu, Y.; Ren, H.; Zhou, G.; Yuan, X.; Shi, X. ER-stress regulates macrophage polarization through pancreatic EIF-2alpha kinase. Cell Immunol. 2019, 336, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Carey, E.J.; Ali, A.H.; Lindor, K.D. Primary biliary cirrhosis. Lancet 2015, 386, 1565–1575. [Google Scholar] [CrossRef]
- Hetz, C.; Axten, J.M.; Patterson, J.B. Pharmacological targeting of the unfolded protein response for disease intervention. Nat. Chem. Biol. 2019, 15, 764–775. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Model | Altered ER Stress Marker | Human NAFLD Reflection | Effect of UPR Intervention |
|---|---|---|---|
| MCD diet | p-eIF2α↑ CHOP↑ GRP78↑ | NASH and fibrosis, without steatosis | TUDCA administration (500 mg/kg/day) prevented liver injury CHOP KO reduced NASH progression |
| High-fat diet (20 weeks) | p-PERK↑ CHOP↑ | Steatosis, NASH, and mild fibrosis | |
| High-fat, high-sucrose diet | XBP1s↑ CHOP↑ ER free cholesterol/phospholipid ratio↑ | Steatosis and mild NASH and fibrosis | |
| Atherogenic diet | CHOP↑ | Steatosis, NASH, and fibrosis | |
| High-cholesterol diet | ER morphological disruption p-PERK↑ | Steatosis, NASH, and mild fibrosis | |
| ALIOS model | CHOP↑ | Steatosis, NASH, and fibrosis | |
| DIAMOND mice | CHOP↑ | Steatosis, NASH, and fibrosis | |
| ob/ob (in combination with LPS) | GRP78↑ XBP1s↑ p-eIF2α↑ | Steatosis, NASH, and fibrosis | TUDCA administration (500 mg/kg/day) prevented steatosis, hepatocyte death and inflammation |
| MUP-uPA (in combination with HFD) | GRP78↑ p-eIF2α↑ CHOP↑ p-IRE1α↑ p-JNK1/2 | Steatosis, NASH, and fibrosis | TUDCA administration (250 mg/kg/day) reduced steatosis and hepatocyte death |
| Chronic CCl4 | GRP78↑ p-PERK↑ CHOP↑ | NASH and fibrosis, without steatosis | Targeted lentiviral delivery of GRP78 in hepatic stellate cells reduces fibrosis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koo, J.H.; Han, C.Y. Signaling Nodes Associated with Endoplasmic Reticulum Stress during NAFLD Progression. Biomolecules 2021, 11, 242. https://doi.org/10.3390/biom11020242
Koo JH, Han CY. Signaling Nodes Associated with Endoplasmic Reticulum Stress during NAFLD Progression. Biomolecules. 2021; 11(2):242. https://doi.org/10.3390/biom11020242
Chicago/Turabian StyleKoo, Ja Hyun, and Chang Yeob Han. 2021. "Signaling Nodes Associated with Endoplasmic Reticulum Stress during NAFLD Progression" Biomolecules 11, no. 2: 242. https://doi.org/10.3390/biom11020242
APA StyleKoo, J. H., & Han, C. Y. (2021). Signaling Nodes Associated with Endoplasmic Reticulum Stress during NAFLD Progression. Biomolecules, 11(2), 242. https://doi.org/10.3390/biom11020242