
ER Stress and Unfolded Protein Response in Leukemia: Friend, Foe, or Both?

 , , and
, , and
Abstract
1. Introduction
2. The Unfolded Protein Response
2.1. The Translational Pathway: Activation of the PERK Kinase
2.2. The Transcriptional Pathway: Activation of ATF6α and IRE1α
3. Hematopoiesis and Leukemias
3.1. Hematopoiesis
3.2. Acute Myeloid Leukemia (AML)
3.3. Acute Lymphoblastic Leukemia (ALL)
3.4. Chronic Myeloid Leukemia (CML)
3.5. Chronic Lymphocytic Leukemia (CLL)
4. Endoplasmic Reticulum Stress Induction in Hematopoietic and Leukemic Cells
4.1. ER Stress Activation in HSCs
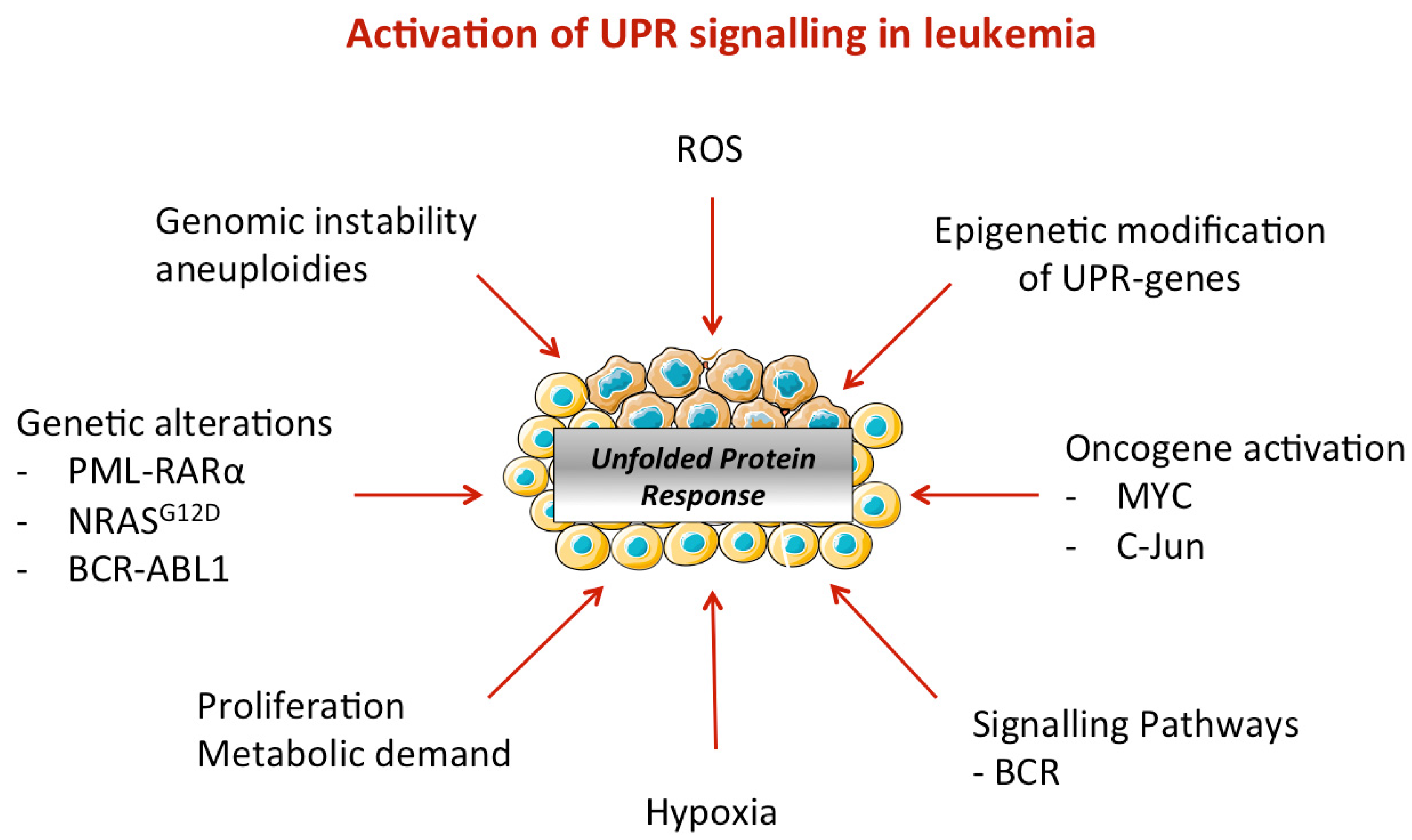
4.2. ER Stress Activation in Leukemic Cells
4.3. UPR Modulation: A Double-Edged Sword to Fight Against Leukemia
5. Conclusions
Funding
Conflicts of Interest
References
- Alberts, B.; Johnson, A.; Lewis, J.; Morgan, D.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 6th ed.; Garland Science: New York, NY, USA, 2014. [Google Scholar]
- Araki, K.; Nagata, K. Protein folding and quality control in the ER. Cold Spring Harb. Perspect. Biol. 2011, 3, a007526. [Google Scholar] [CrossRef] [PubMed]
- Eletto, D.; Chevet, E.; Argon, Y.; Appenzeller-Herzog, C. Redox controls UPR to control redox. J. Cell Sci. 2014, 127, 3649–3658. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Brodsky, J.L. Protein quality control in the secretory pathway. J. Cell Biol. 2019, 218, 3171–3187. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Xia, B.; Wang, S.; Xu, J. Folded or Degraded in Endoplasmic Reticulum. Adv. Exp. Med. Biol. 2020, 1248, 265–294. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Moore, K.A.; Hollien, J. The unfolded protein response in secretory cell function. Annu. Rev. Genet. 2012, 46, 165–183. [Google Scholar] [CrossRef]
- Kaufman, R.J. Stress signaling from the lumen of the endoplasmic reticulum: Coordination of gene transcriptional and translational controls. Genes. Dev. 1999, 13, 1211–1233. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Sicari, D.; Igbaria, A.; Chevet, E. Control of Protein Homeostasis in the Early Secretory Pathway: Current Status and Challenges. Cells 2019, 8, 1347. [Google Scholar] [CrossRef]
- Maurel, M.; McGrath, E.P.; Mnich, K.; Healy, S.; Chevet, E.; Samali, A. Controlling the unfolded protein response-mediated life and death decisions in cancer. Semin. Cancer Biol. 2015, 33, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Chevet, E. Theme Series—UPR in cancer. Semin. Cancer Biol. 2015, 33, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Hosoi, T.; Ozawa, K. Endoplasmic reticulum stress in disease: Mechanisms and therapeutic opportunities. Clin. Sci. 2009, 118, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Axten, J.M. Pharmacological targeting of the unfolded protein response for disease intervention. Nat. Chem. Biol. 2019, 15, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Chevet, E.; Harding, H.P. Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 2013, 12, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic Reticulum Stress, Unfolded Protein Response, and Cancer Cell Fate. Nat. Chem. Biol. 2017, 7, 78. [Google Scholar] [CrossRef]
- Oakes, S.A. Endoplasmic Reticulum Stress Signaling in Cancer Cells. Am. J. Pathol. 2020, 190, 934–946. [Google Scholar] [CrossRef]
- Oakes, S.A. Endoplasmic reticulum proteostasis: A key checkpoint in cancer. Am. J. Physiol. Cell Physiol. 2017, 312, C93–C102. [Google Scholar] [CrossRef]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef]
- Chevet, E.; Hetz, C.; Samali, A. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov. 2015, 5, 586–597. [Google Scholar] [CrossRef]
- Korennykh, A.; Walter, P. Structural basis of the unfolded protein response. Annu. Rev. Cell Dev. Biol. 2012, 28, 251–277. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef]
- Gardner, B.M.; Walter, P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 2011, 333, 1891–1894. [Google Scholar] [CrossRef] [PubMed]
- Karagoz, G.E.; Acosta-Alvear, D.; Nguyen, H.T.; Lee, C.P.; Chu, F.; Walter, P. An unfolded protein-induced conformational switch activates mammalian IRE1. eLife 2017, 6, e30700. [Google Scholar] [CrossRef] [PubMed]
- Bakunts, A.; Orsi, A.; Vitale, M.; Cattaneo, A.; Lari, F.; Tade, L.; Sitia, R.; Raimondi, A.; Bachi, A.; van Anken, E. Ratiometric sensing of BiP-client versus BiP levels by the unfolded protein response determines its signaling amplitude. eLife 2017, 6, e27518. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Garcia-Bonilla, L.; Hu, J.; Harding, H.P.; Ron, D. Activation-dependent substrate recruitment by the eukaryotic translation initiation factor 2 kinase PERK. J. Cell Biol. 2006, 172, 201–209. [Google Scholar] [CrossRef]
- DuRose, J.B.; Scheuner, D.; Kaufman, R.J.; Rothblum, L.I.; Niwa, M. Phosphorylation of eukaryotic translation initiation factor 2alpha coordinates rRNA transcription and translation inhibition during endoplasmic reticulum stress. Mol. Cell. Biol. 2009, 29, 4295–4307. [Google Scholar] [CrossRef] [PubMed]
- Koumenis, C.; Naczki, C.; Koritzinsky, M.; Rastani, S.; Diehl, A.; Sonenberg, N.; Koromilas, A.; Wouters, B.G. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol. Cell. Biol. 2002, 22, 7405–7416. [Google Scholar] [CrossRef] [PubMed]
- Wek, R.C.; Cavener, D.R. Translational control and the unfolded protein response. Antioxid. Redox Signal. 2007, 9, 2357–2371. [Google Scholar] [CrossRef] [PubMed]
- Jaud, M.; Philippe, C.; Di Bella, D.; Tang, W.; Pyronnet, S.; Laurell, H.; Mazzolini, L.; Rouault-Pierre, K.; Touriol, C. Translational Regulations in Response to Endoplasmic Reticulum Stress in Cancers. Cells 2020, 9, 540. [Google Scholar] [CrossRef] [PubMed]
- Vattem, K.M.; Wek, R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269–11274. [Google Scholar] [CrossRef] [PubMed]
- Palam, L.R.; Baird, T.D.; Wek, R.C. Phosphorylation of eIF2 facilitates ribosomal bypass of an inhibitory upstream ORF to enhance CHOP translation. J. Biol. Chem. 2011, 286, 10939–10949. [Google Scholar] [CrossRef]
- Bonnet-Magnaval, F.; Philippe, C.; Van Den Berghe, L.; Prats, H.; Touriol, C.; Lacazette, E. Hypoxia and ER stress promote Staufen1 expression through an alternative translation mechanism. Cancers 2016, 479, 365–371. [Google Scholar] [CrossRef]
- Jaud, M.; Philippe, C.; Van Den Berghe, L.; Ségura, C.; Mazzolini, L. The PERK Branch of the Unfolded Protein Response Promotes DLL4 Expression by Activating an Alternative Translation Mechanism. Cancers 2019, 11, 142. [Google Scholar] [CrossRef]
- Philippe, C.; Dubrac, A.; Quelen, C.; Desquesnes, A.; Van Den Berghe, L.; Ségura, C.; Filleron, T.; Pyronnet, S.; Prats, H.; Brousset, P.; et al. PERK mediates the IRES-dependent translational activation of mRNAs encoding angiogenic growth factors after ischemic stress. Sci. Signal. 2016, 9, ra44. [Google Scholar] [CrossRef]
- Fernandez, J.; Bode, B.; Koromilas, A.; Diehl, J.A.; Krukovets, I.; Snider, M.D.; Hatzoglou, M. Translation mediated by the internal ribosome entry site of the cat-1 mRNA is regulated by glucose availability in a PERK kinase-dependent manner. J. Biol. Chem. 2002, 277, 11780–11787. [Google Scholar] [CrossRef]
- Reid, D.W.; Tay, A.S.; Sundaram, J.R.; Lee, I.C.; Chen, Q.; George, S.E.; Nicchitta, C.V.; Shenolikar, S. Complementary Roles of GADD34- and CReP-Containing Eukaryotic Initiation Factor 2α Phosphatases during the Unfolded Protein Response. Mol. Cell. Biol. 2016, 36, 1868–1880. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Hendershot, L.M. Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J. Biol. Chem. 2003, 278, 34864–34873. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.Y.; Cevallos, R.C.; Jan, E. An upstream open reading frame regulates translation of GADD34 during cellular stresses that induce eIF2alpha phosphorylation. J. Biol. Chem. 2009, 284, 6661–6673. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Brewer, J.W.; Diehl, J.A.; Hendershot, L.M. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J. Mol. Biol. 2002, 318, 1351–1365. [Google Scholar] [CrossRef]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2018, 9, 3083. [Google Scholar] [CrossRef] [PubMed]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes. Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Diehl, J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 20108–20117. [Google Scholar] [CrossRef]
- Bobrovnikova-Marjon, E.; Diehl, J.A. Coping with stress: ATF6alpha takes the stage. Dev. Cell 2007, 13, 322–324. [Google Scholar] [CrossRef][Green Version]
- Hillary, R.F.; FitzGerald, U. A lifetime of stress: ATF6 in development and homeostasis. J. Biomed. Sci. 2018, 25, 48. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Chemaly, E.R.; Troncone, L.; Lebeche, D. SERCA control of cell death and survival. Cell Calcium 2018, 69, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Bommiasamy, H.; Back, S.H.; Fagone, P.; Lee, K.; Meshinchi, S.; Vink, E.; Sriburi, R.; Frank, M.; Jackowski, S.; Kaufman, R.J.; et al. ATF6alpha induces XBP1-independent expansion of the endoplasmic reticulum. J. Cell Sci. 2009, 122, 1626–1636. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Takahara, K.; Oyadomari, S.; Okada, T.; Sato, T.; Harada, A.; Mori, K. Induction of liver steatosis and lipid droplet formation in ATF6alpha-knockout mice burdened with pharmacological endoplasmic reticulum stress. Mol. Biol. Cell 2010, 21, 2975–2986. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Rutkowski, D.T.; Dubois, M.; Swathirajan, J.; Saunders, T.; Wang, J.; Song, B.; Yau, G.D.; Kaufman, R.J. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell 2007, 13, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Guan, D.; Xu, Y.; Yang, M.; Wang, H.; Wang, X.; Shen, Z. N-acetyl cysteine and penicillamine induce apoptosis via the ER stress response-signaling pathway. Mol. Carcinog. 2010, 49, 68–74. [Google Scholar] [CrossRef]
- Yoshida, H.; Okada, T.; Haze, K.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol. 2000, 20, 6755–6767. [Google Scholar] [CrossRef]
- Yang, H.; Niemeijer, M.; van de Water, B.; Beltman, J.B. ATF6 Is a Critical Determinant of CHOP Dynamics during the Unfolded Protein Response. iScience 2020, 23, 100860. [Google Scholar] [CrossRef]
- Thuerauf, D.J.; Morrison, L.E.; Hoover, H.; Glembotski, C.C. Coordination of ATF6-mediated transcription and ATF6 degradation by a domain that is shared with the viral transcription factor, VP16. J. Biol. Chem. 2002, 277, 20734–20739. [Google Scholar] [CrossRef]
- Urano, F.; Bertolotti, A.; Ron, D. IRE1 and efferent signaling from the endoplasmic reticulum. J. Cell Sci. 2000, 113 Pt 21, 3697–3702. [Google Scholar]
- Tirasophon, W.; Welihinda, A.A.; Kaufman, R.J. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes. Dev. 1998, 12, 1812–1824. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Z.; Harding, H.P.; Zhang, Y.; Jolicoeur, E.M.; Kuroda, M.; Ron, D. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 1998, 17, 5708–5717. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Wang, X.; Novoa, I.; Jungreis, R.; Schlessinger, K.; Cho, J.H.; West, A.B.; Ron, D. Increased sensitivity to dextran sodium sulfate colitis in IRE1beta-deficient mice. J. Clin. Investig. 2001, 107, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Martino, M.B.; Jones, L.; Brighton, B.; Ehre, C.; Abdulah, L.; Davis, C.W.; Ron, D.; O’Neal, W.K.; Ribeiro, C.M. The ER stress transducer IRE1beta is required for airway epithelial mucin production. Mucosal Immunol. 2013, 6, 639–654. [Google Scholar] [CrossRef] [PubMed]
- Shamu, C.E.; Walter, P. Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 1996, 15, 3028–3039. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Schröder, M.; Kaufman, R.J. Ligand-independent dimerization activates the stress response kinases IRE1 and PERK in the lumen of the endoplasmic reticulum. J. Biol. Chem. 2000, 275, 24881–24885. [Google Scholar] [CrossRef] [PubMed]
- Peschek, J.; Acosta-Alvear, D.; Mendez, A.S.; Walter, P. A conformational RNA zipper promotes intron ejection during non-conventional XBP1 mRNA splicing. EMBO Rep. 2015, 16, 1688–1698. [Google Scholar] [CrossRef]
- Lu, Y.; Liang, F.X.; Wang, X. A synthetic biology approach identifies the mammalian UPR RNA ligase RtcB. Mol. Cell 2014, 55, 758–770. [Google Scholar] [CrossRef]
- Acosta-Alvear, D.; Zhou, Y.; Blais, A.; Tsikitis, M.; Lents, N.H.; Arias, C.; Lennon, C.J.; Kluger, Y.; Dynlacht, B.D. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 2007, 27, 53–66. [Google Scholar] [CrossRef]
- Guo, F.; Lin, E.A.; Liu, P.; Lin, J.; Liu, C. XBP1U inhibits the XBP1S-mediated upregulation of the iNOS gene expression in mammalian ER stress response. Cell. Signal. 2010, 22, 1818–1828. [Google Scholar] [CrossRef] [PubMed]
- Byrd, A.E.; Brewer, J.W. Intricately Regulated: A Cellular Toolbox for Fine-Tuning XBP1 Expression and Activity. Cells 2012, 1, 738–753. [Google Scholar] [CrossRef] [PubMed]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Maurel, M.; Chevet, E.; Tavernier, J.; Gerlo, S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 2014, 39, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.P.; Wang, L.; Han, D.; Wang, E.S.; Huskey, N.E.; Lim, L.; Truitt, M.; McManus, M.T.; Ruggero, D.; Goga, A.; et al. IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 2012, 338, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Pluquet, O.; Dejeans, N.; Bouchecareilh, M.; Lhomond, S.; Pineau, R.; Higa, A.; Delugin, M.; Combe, C.; Loriot, S.; Cubel, G.; et al. Posttranscriptional regulation of PER1 underlies the oncogenic function of IREalpha. Cancer Res. 2013, 73, 4732–4743. [Google Scholar] [CrossRef] [PubMed]
- Dejeans, N.; Pluquet, O.; Lhomond, S.; Grise, F.; Bouchecareilh, M.; Juin, A.; Meynard-Cadars, M.; Bidaud-Meynard, A.; Gentil, C.; Moreau, V.; et al. Autocrine control of glioma cells adhesion and migration through IRE1alpha-mediated cleavage of SPARC mRNA. J. Cell Sci. 2012, 125, 4278–4287. [Google Scholar] [CrossRef]
- Bright, M.D.; Itzhak, D.N.; Wardell, C.P.; Morgan, G.J.; Davies, F.E. Cleavage of BLOC1S1 mRNA by IRE1 Is Sequence Specific, Temporally Separate from XBP1 Splicing, and Dispensable for Cell Viability under Acute Endoplasmic Reticulum Stress. Mol. Cell. Biol. 2015, 35, 2186–2202. [Google Scholar] [CrossRef]
- Han, D.; Lerner, A.G.; Vande Walle, L.; Upton, J.P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef]
- So, J.S.; Hur, K.Y.; Tarrio, M.; Ruda, V.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Lichtman, A.H.; Iwawaki, T.; Glimcher, L.H.; et al. Silencing of lipid metabolism genes through IRE1alpha-mediated mRNA decay lowers plasma lipids in mice. Cell Metab. 2012, 16, 487–499. [Google Scholar] [CrossRef]
- Moore, K.; Hollien, J. Ire1-mediated decay in mammalian cells relies on mRNA sequence, structure, and translational status. Mol. Biol. Cell 2015, 26, 2873–2884. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Lawrence, D.A.; Marsters, S.; Acosta-Alvear, D.; Kimmig, P.; Mendez, A.S.; Paton, A.W.; Paton, J.C.; Walter, P.; Ashkenazi, A. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014, 345, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Glimcher, L.H. Fine-tuning of the unfolded protein response: Assembling the IRE1alpha interactome. Mol. Cell 2009, 35, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, D.; Rojas-Rivera, D.; Rodriguez, D.A.; Groenendyk, J.; Kohler, A.; Lebeaupin, C.; Ito, S.; Urra, H.; Carreras-Sureda, A.; Hazari, Y.; et al. Interactome Screening Identifies the ER Luminal Chaperone Hsp47 as a Regulator of the Unfolded Protein Response Transducer IRE1alpha. Mol. Cell 2018, 69, 238–252.e237. [Google Scholar] [CrossRef] [PubMed]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, S.; Xia, J.; Liu, F. Hematopoietic Hierarchy—An Updated Roadmap. Trends Cell Biol. 2018, 28, 976–986. [Google Scholar] [CrossRef]
- Chao, M.P.; Seita, J.; Weissman, I.L. Establishment of a normal hematopoietic and leukemia stem cell hierarchy. Cold Spring Harb. Symp. Quant. Biol. 2008, 73, 439–449. [Google Scholar] [CrossRef]
- Warr, M.R.; Pietras, E.M.; Passegué, E. Mechanisms controlling hematopoietic stem cell functions during normal hematopoiesis and hematological malignancies. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011, 3, 681–701. [Google Scholar] [CrossRef]
- Noetzli, L.J.; French, S.L.; Machlus, K.R. New Insights into the Differentiation of Megakaryocytes from Hematopoietic Progenitors. Arter. Thromb. Vasc. Biol. 2019, 39, 1288–1300. [Google Scholar] [CrossRef]
- Akashi, K.; Traver, D.; Miyamoto, T.; Weissman, I.L. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature 2000, 404, 193–197. [Google Scholar] [CrossRef]
- Passegué, E.; Jamieson, C.H.; Ailles, L.E.; Weissman, I.L. Normal and leukemic hematopoiesis: Are leukemias a stem cell disorder or a reacquisition of stem cell characteristics? Proc. Natl. Acad. Sci. USA 2003, 100 (Suppl. 1), 11842–11849. [Google Scholar] [CrossRef]
- Sawyers, C.L.; Denny, C.T.; Witte, O.N. Leukemia and the disruption of normal hematopoiesis. Cell 1991, 64, 337–350. [Google Scholar] [CrossRef]
- De Kouchkovsky, I.; Abdul-Hay, M. Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J. 2016, 6, e441. [Google Scholar] [CrossRef] [PubMed]
- Grove, C.S.; Vassiliou, G.S. Acute myeloid leukaemia: A paradigm for the clonal evolution of cancer? Dis. Models Mech. 2014, 7, 941–951. [Google Scholar] [CrossRef]
- Klco, J.M.; Spencer, D.H.; Miller, C.A.; Griffith, M.; Lamprecht, T.L.; O’Laughlin, M.; Fronick, C.; Magrini, V.; Demeter, R.T.; Fulton, R.S.; et al. Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell 2014, 25, 379–392. [Google Scholar] [CrossRef]
- Hackl, H.; Astanina, K.; Wieser, R. Molecular and genetic alterations associated with therapy resistance and relapse of acute myeloid leukemia. J. Hematol. Oncol. 2017, 10, 51. [Google Scholar] [CrossRef]
- Lagunas-Rangel, F.A.; Chávez-Valencia, V.; Gómez-Guijosa, M.; Cortes-Penagos, C. Acute Myeloid Leukemia-Genetic Alterations and Their Clinical Prognosis. Int. J. Hematol. Oncol. Stem Cell Res. 2017, 11, 328–339. [Google Scholar]
- Goldman, S.L.; Hassan, C.; Khunte, M.; Soldatenko, A.; Jong, Y.; Afshinnekoo, E.; Mason, C.E. Epigenetic Modifications in Acute Myeloid Leukemia: Prognosis, Treatment, and Heterogeneity. Front. Genet. 2019, 10, 133. [Google Scholar] [CrossRef]
- Pourrajab, F.; Zare-Khormizi, M.R.; Hashemi, A.S.; Hekmatimoghaddam, S. Genetic Characterization and Risk Stratification of Acute Myeloid Leukemia. Cancer Manag. Res. 2020, 12, 2231–2253. [Google Scholar] [CrossRef]
- Benedetti, L.; Levin, A.A.; Scicchitano, B.M.; Grignani, F.; Allenby, G.; Diverio, D.; Lo Coco, F.; Avvisati, G.; Ruthardt, M.; Adamo, S.; et al. Characterization of the retinoid binding properties of the major fusion products present in acute promyelocytic leukemia cells. Blood 1997, 90, 1175–1185. [Google Scholar] [CrossRef]
- Visani, G.; Buonamici, S.; Malagola, M.; Isidori, A.; Piccaluga, P.P.; Martinelli, G.; Ottaviani, E.; Grafone, T.; Baccarani, M.; Tura, S. Pulsed ATRA as single therapy restores long-term remission in PML-RARalpha-positive acute promyelocytic leukemia patients: Real time quantification of minimal residual disease. A pilot study. Leukemia 2001, 15, 1696–1700. [Google Scholar] [CrossRef] [PubMed]
- Cicconi, L.; Divona, M.; Ciardi, C.; Ottone, T.; Ferrantini, A.; Lavorgna, S.; Alfonso, V.; Paoloni, F.; Piciocchi, A.; Avvisati, G.; et al. PML-RARα kinetics and impact of FLT3-ITD mutations in newly diagnosed acute promyelocytic leukaemia treated with ATRA and ATO or ATRA and chemotherapy. Leukemia 2016, 30, 1987–1992. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, G.; Ottaviani, E.; Visani, G.; Testoni, N.; Montefusco, V.; Tura, S. Long-term disease-free acute promyelocytic leukemia patients really can be cured at molecular level. Haematologica 1998, 83, 860–863. [Google Scholar] [PubMed]
- Haferlach, T.; Bacher, U.; Haferlach, C.; Kern, W.; Schnittger, S. Insight into the molecular pathogenesis of myeloid malignancies. Curr. Opin. Hematol. 2007, 14, 90–97. [Google Scholar] [CrossRef]
- Haferlach, T.; Kohlmann, A.; Bacher, U.; Schnittger, S.; Haferlach, C.; Kern, W. Gene expression profiling for the diagnosis of acute leukaemia. Br. J. Cancer 2007, 96, 535–540. [Google Scholar] [CrossRef]
- Prada-Arismendy, J.; Arroyave, J.C.; Röthlisberger, S. Molecular biomarkers in acute myeloid leukemia. Blood Rev. 2017, 31, 63–76. [Google Scholar] [CrossRef]
- Inaba, H.; Greaves, M.; Mullighan, C.G. Acute lymphoblastic leukaemia. Lancet 2013, 381, 1943–1955. [Google Scholar] [CrossRef]
- Teitell, M.A.; Pandolfi, P.P. Molecular genetics of acute lymphoblastic leukemia. Annu. Rev. Pathol. 2009, 4, 175–198. [Google Scholar] [CrossRef]
- Armstrong, S.A.; Look, A.T. Molecular genetics of acute lymphoblastic leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 6306–6315. [Google Scholar] [CrossRef]
- Moorman, A.V. New and emerging prognostic and predictive genetic biomarkers in B-cell precursor acute lymphoblastic leukemia. Haematologica 2016, 101, 407–416. [Google Scholar] [CrossRef]
- Palmi, C.; Savino, A.M.; Silvestri, D.; Bronzini, I.; Cario, G.; Paganin, M.; Buldini, B.; Galbiati, M.; Muckenthaler, M.U.; Bugarin, C.; et al. CRLF2 over-expression is a poor prognostic marker in children with high risk T-cell acute lymphoblastic leukemia. Oncotarget 2016, 7, 59260–59272. [Google Scholar] [CrossRef] [PubMed]
- Malinowska-Ozdowy, K.; Frech, C.; Schönegger, A.; Eckert, C.; Cazzaniga, G.; Stanulla, M.; zur Stadt, U.; Mecklenbräuker, A.; Schuster, M.; Kneidinger, D.; et al. KRAS and CREBBP mutations: A relapse-linked malicious liaison in childhood high hyperdiploid acute lymphoblastic leukemia. Leukemia 2015, 29, 1656–1667. [Google Scholar] [CrossRef] [PubMed]
- Van Vlierberghe, P.; Ferrando, A. The molecular basis of T cell acute lymphoblastic leukemia. J. Clin. Investig. 2012, 122, 3398–3406. [Google Scholar] [CrossRef] [PubMed]
- Malard, F.; Mohty, M. Acute lymphoblastic leukaemia. Lancet 2020, 395, 1146–1162. [Google Scholar] [CrossRef]
- Brown, L.M.; Lonsdale, A.; Zhu, A.; Davidson, N.M.; Schmidt, B.; Hawkins, A.; Wallach, E.; Martin, M.; Mechinaud, F.M.; Khaw, S.L.; et al. The application of RNA sequencing for the diagnosis and genomic classification of pediatric acute lymphoblastic leukemia. Blood Adv. 2020, 4, 930–942. [Google Scholar] [CrossRef] [PubMed]
- Waanders, E.; Gu, Z.; Dobson, S.M.; Antić, Ž.; Crawford, J.C.; Ma, X.; Edmonson, M.N.; Payne-Turner, D.; van der Vorst, M.; Jongmans, M.C.J.; et al. Mutational landscape and patterns of clonal evolution in relapsed pediatric acute lymphoblastic leukemia. Blood Cancer Discov. 2020, 1, 96–111. [Google Scholar] [CrossRef]
- Apperley, J.F. Chronic myeloid leukaemia. Lancet 2015, 385, 1447–1459. [Google Scholar] [CrossRef]
- Siveen, K.S.; Prabhu, K.S.; Achkar, I.W.; Kuttikrishnan, S.; Shyam, S.; Khan, A.Q.; Merhi, M.; Dermime, S.; Uddin, S. Role of Non Receptor Tyrosine Kinases in Hematological Malignances and its Targeting by Natural Products. Mol. Cancer 2018, 17, 31. [Google Scholar] [CrossRef]
- Steegmann, J.L.; Baccarani, M.; Breccia, M.; Casado, L.F.; García-Gutiérrez, V.; Hochhaus, A.; Kim, D.W.; Kim, T.D.; Khoury, H.J.; Le Coutre, P.; et al. European LeukemiaNet recommendations for the management and avoidance of adverse events of treatment in chronic myeloid leukaemia. Leukemia 2016, 30, 1648–1671. [Google Scholar] [CrossRef]
- Westerweel, P.E.; Te Boekhorst, P.A.W.; Levin, M.D.; Cornelissen, J.J. New Approaches and Treatment Combinations for the Management of Chronic Myeloid Leukemia. Front. Oncol. 2019, 9, 665. [Google Scholar] [CrossRef]
- Kipps, T.J.; Stevenson, F.K.; Wu, C.J.; Croce, C.M.; Packham, G.; Wierda, W.G.; O’Brien, S.; Gribben, J.; Rai, K. Chronic lymphocytic leukaemia. Nat. Rev. Dis. Prim. 2017, 3, 16096. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, T.J.; Davis, Z.; Gardiner, A.; Oscier, D.G.; Stevenson, F.K. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999, 94, 1848–1854. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Vicente, A.E.; Díaz, M.G.; Hernández-Rivas, J.M. Chronic lymphocytic leukemia: A clinical and molecular heterogenous disease. Cancer Genet. 2013, 206, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Puiggros, A.; Blanco, G. Genetic abnormalities in chronic lymphocytic leukemia: Where we are and where we go. Biomed Res. Int. 2014, 2014, 435983. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, Y.; Schulte, C.; Tiemann, M.; Bullerdiek, J. Chronic lymphocytic leukemia-associated chromosomal abnormalities and miRNA deregulation. Appl. Clin. Genet. 2012, 5, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Nabhan, C.; Raca, G.; Wang, Y.L. Predicting Prognosis in Chronic Lymphocytic Leukemia in the Contemporary Era. Jama Oncol. 2015, 1, 965–974. [Google Scholar] [CrossRef]
- Kipps, T.J. Chronic lymphocytic leukemia. Curr. Opini. Hematol. 2000, 7, 223–234. [Google Scholar] [CrossRef]
- Döhner, H.; Stilgenbauer, S.; Benner, A.; Leupolt, E.; Kröber, A.; Bullinger, L.; Döhner, K.; Bentz, M.; Lichter, P. Genomic aberrations and survival in chronic lymphocytic leukemia. N. Engl. J. Med. 2000, 343, 1910–1916. [Google Scholar] [CrossRef]
- Haferlach, C.; Dicker, F.; Schnittger, S.; Kern, W.; Haferlach, T. Comprehensive genetic characterization of CLL: A study on 506 cases analysed with chromosome banding analysis, interphase FISH, IgV(H) status and immunophenotyping. Leukemia 2007, 21, 2442–2451. [Google Scholar] [CrossRef]
- Zent, C.S.; Burack, W.R. Mutations in chronic lymphocytic leukemia and how they affect therapy choice: Focus on NOTCH1, SF3B1, and TP53. Hematol. Am. Soc. Hematol. Educ. Program. 2014, 2014, 119–124. [Google Scholar] [CrossRef]
- Kipps, T.J.; Choi, M.Y. Targeted Therapy in Chronic Lymphocytic Leukemia. Cancer J. 2019, 25, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A. Treatment of Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2020, 383, 460–473. [Google Scholar] [CrossRef]
- Doulatov, S.; Notta, F.; Laurenti, E.; Dick, J.E. Hematopoiesis: A human perspective. Cell Stem Cell 2012, 10, 120–136. [Google Scholar] [CrossRef]
- Vedi, A.; Santoro, A.; Dunant, C.F.; Dick, J.E.; Laurenti, E. Molecular landscapes of human hematopoietic stem cells in health and leukemia. Ann. N. Y. Acad. Sci. 2016, 1370, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Signer, R.A.; Magee, J.A.; Salic, A.; Morrison, S.J. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 2014, 509, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Khateb, A.; Ronai, Z.A. Unfolded Protein Response in Leukemia: From Basic Understanding to Therapeutic Opportunities. Trends Cancer 2020, 6, 960–973. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo San Jose, L.; Sunshine, M.J.; Dillingham, C.H.; Chua, B.A.; Kruta, M.; Hong, Y.; Hatters, D.M.; Signer, R.A.J. Modest Declines in Proteome Quality Impair Hematopoietic Stem Cell Self-Renewal. Cell Rep. 2020, 30, 69–80.e66. [Google Scholar] [CrossRef] [PubMed]
- van Galen, P.; Kreso, A.; Mbong, N.; Kent, D.G.; Fitzmaurice, T.; Chambers, J.E.; Xie, S.; Laurenti, E.; Hermans, K.; Eppert, K.; et al. The unfolded protein response governs integrity of the haematopoietic stem-cell pool during stress. Nature 2014, 510, 268–272. [Google Scholar] [CrossRef]
- van Galen, P.; Mbong, N.; Kreso, A.; Schoof, E.M.; Wagenblast, E.; Ng, S.W.K.; Krivdova, G.; Jin, L.; Nakauchi, H.; Dick, J.E. Integrated Stress Response Activity Marks Stem Cells in Normal Hematopoiesis and Leukemia. Cell Rep. 2018, 25, 1109–1117.e1105. [Google Scholar] [CrossRef]
- Miharada, K.; Sigurdsson, V.; Karlsson, S. Dppa5 improves hematopoietic stem cell activity by reducing endoplasmic reticulum stress. Cell Rep. 2014, 7, 1381–1392. [Google Scholar] [CrossRef]
- Chapple, R.H.; Hu, T.; Tseng, Y.J.; Liu, L.; Kitano, A.; Luu, V.; Hoegenauer, K.A.; Iwawaki, T.; Li, Q.; Nakada, D. ERalpha promotes murine hematopoietic regeneration through the Ire1alpha-mediated unfolded protein response. eLife 2018, 7, e31159. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhao, M.; Jin, X.; Ney, G.; Yang, K.B.; Peng, F.; Cao, J.; Iwawaki, T.; Del Valle, J.; Chen, X.; et al. Adaptive endoplasmic reticulum stress signalling via IRE1alpha-XBP1 preserves self-renewal of haematopoietic and pre-leukaemic stem cells. Nat. Cell Biol. 2019, 21, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.Z.; Garcia-Prat, L.; Voisin, V.; Ferrari, R.; Gan, O.I.; Wagenblast, E.; Kaufmann, K.B.; Zeng, A.G.X.; Takayanagi, S.I.; Patel, I.; et al. Sphingolipid Modulation Activates Proteostasis Programs to Govern Human Hematopoietic Stem Cell Self-Renewal. Cell Stem Cell 2019, 25, 639–653.e637. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Liu, X.; Peng, F.; Zhang, W.; Zheng, L.; Ding, Y.; Gu, T.; Lv, K.; Wang, J.; Ortinau, L.; et al. Protein quality control through endoplasmic reticulum-associated degradation maintains haematopoietic stem cell identity and niche interactions. Nat. Cell Biol. 2020, 22, 1162–1169. [Google Scholar] [CrossRef]
- Rouault-Pierre, K.; Hamilton, A.; Bonnet, D. Effect of hypoxia-inducible factors in normal and leukemic stem cell regulation and their potential therapeutic impact. Expert Opin. Biol. Ther. 2016, 16, 463–476. [Google Scholar] [CrossRef]
- Sigurdsson, V.; Miharada, K. Regulation of unfolded protein response in hematopoietic stem cells. Int. J. Hematol. 2018, 107, 627–633. [Google Scholar] [CrossRef]
- Doultsinos, D.; Avril, T.; Lhomond, S.; Dejeans, N.; Guédat, P.; Chevet, E. Control of the Unfolded Protein Response in Health and Disease. Slas Discov. Adv. Life Sci. R D 2017, 22, 787–800. [Google Scholar] [CrossRef]
- Siwecka, N.; Rozpędek, W.; Pytel, D.; Wawrzynkiewicz, A.; Dziki, A.; Dziki, Ł.; Diehl, J.A.; Majsterek, I. Dual role of Endoplasmic Reticulum Stress-Mediated Unfolded Protein Response Signaling Pathway in Carcinogenesis. Int. J. Mol. Sci. 2019, 20, 4354. [Google Scholar] [CrossRef]
- Martelli, A.M.; Paganelli, F.; Chiarini, F.; Evangelisti, C. The Unfolded Protein Response: A Novel Therapeutic Target in Acute Leukemias. Cancers 2020, 12, 333. [Google Scholar] [CrossRef]
- Kharabi Masouleh, B.; Chevet, E.; Panse, J.; Jost, E.; O’Dwyer, M.; Bruemmendorf, T.H.; Samali, A. Drugging the unfolded protein response in acute leukemias. J. Hematol. Oncol. 2015, 8, 87. [Google Scholar] [CrossRef]
- Schardt, J.A.; Weber, D.; Eyholzer, M.; Mueller, B.U.; Pabst, T. Activation of the unfolded protein response is associated with favorable prognosis in acute myeloid leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 3834–3841. [Google Scholar] [CrossRef]
- Tanimura, A.; Yujiri, T.; Tanaka, Y.; Tanaka, M.; Mitani, N.; Nakamura, Y.; Ariyoshi, K.; Tanizawa, Y. Activation of the unfolded protein response in primary acute myeloid leukemia cells. Int. J. Hematol. 2011, 94, 300–302. [Google Scholar] [CrossRef] [PubMed]
- Krysov, S.; Steele, A.J.; Coelho, V.; Linley, A.; Sanchez Hidalgo, M.; Carter, M.; Potter, K.N.; Kennedy, B.; Duncombe, A.S.; Ashton-Key, M.; et al. Stimulation of surface IgM of chronic lymphocytic leukemia cells induces an unfolded protein response dependent on BTK and SYK. Blood 2014, 124, 3101–3109. [Google Scholar] [CrossRef] [PubMed]
- Kharabi, M.B.; Geng, H.; Hurtz, C.; Chan, L.N.; Logan, A.C.; Chang, M.S.; Huang, C.; Swaminathan, S.; Sun, H.; Paietta, E.; et al. Mechanistic rationale for targeting the unfolded protein response in pre-B acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2014, 111, E2219–E2228. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Martinez, E.; Di Marcantonio, D.; Solanki-Patel, N.; Aghayev, T.; Peri, S.; Ferraro, F.; Skorski, T.; Scholl, C.; Fröhling, S.; et al. JUN is a key transcriptional regulator of the unfolded protein response in acute myeloid leukemia. Leukemia 2017, 31, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Lin, D.C.; Guo, X.; Kharabi, M.B.; Gery, S.; Cao, Q.; Alkan, S.; Ikezoe, T.; Akiba, C.; Paquette, R.; et al. Inhibition of IRE1α-driven pro-survival pathways is a promising therapeutic application in acute myeloid leukemia. Oncotarget 2016, 7, 18736–18749. [Google Scholar] [CrossRef] [PubMed]
- Bujisic, B.; De Gassart, A. Impairment of both IRE1 expression and XBP1 activation is a hallmark of GCB DLBCL and contributes to tumor growth. Blood 2017, 129, 2420–2428. [Google Scholar] [CrossRef] [PubMed]
- Hart, L.S.; Cunningham, J.T.; Datta, T.; Dey, S.; Tameire, F.; Lehman, S.L.; Qiu, B.; Zhang, H.; Cerniglia, G.; Bi, M.; et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J. Clin. Investig. 2012, 122, 4621–4634. [Google Scholar] [CrossRef] [PubMed]
- Brancolini, C.; Iuliano, L. Proteotoxic Stress and Cell Death in Cancer Cells. Cancers 2020, 12, 2385. [Google Scholar] [CrossRef]
- Uckun, F.M.; Qazi, S.; Ozer, Z.; Garner, A.L.; Pitt, J.; Ma, H.; Janda, K.D. Inducing apoptosis in chemotherapy-resistant B-lineage acute lymphoblastic leukaemia cells by targeting HSPA5, a master regulator of the anti-apoptotic unfolded protein response signalling network. Br. J. Haematol. 2011, 153, 741–752. [Google Scholar] [CrossRef]
- Kusio-Kobialka, M.; Podszywalow-Bartnicka, P.; Peidis, P.; Glodkowska-Mrowka, E.; Wolanin, K.; Leszak, G.; Seferynska, I.; Stoklosa, T.; Koromilas, A.E.; Piwocka, K. The PERK-eIF2α phosphorylation arm is a pro-survival pathway of BCR-ABL signaling and confers resistance to imatinib treatment in chronic myeloid leukemia cells. Cell Cycle 2012, 11, 4069–4078. [Google Scholar] [CrossRef] [PubMed]
- Higa, A.; Taouji, S.; Lhomond, S.; Jensen, D.; Fernandez-Zapico, M.E.; Simpson, J.C.; Pasquet, J.M.; Schekman, R.; Chevet, E. Endoplasmic reticulum stress-activated transcription factor ATF6α requires the disulfide isomerase PDIA5 to modulate chemoresistance. Mol. Cell. Biol. 2014, 34, 1839–1849. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.H.; Ranatunga, S.; Kriss, C.L.; Cubitt, C.L.; Tao, J.; Pinilla-Ibarz, J.A.; Del Valle, J.R.; Hu, C.C. Inhibition of ER stress-associated IRE-1/XBP-1 pathway reduces leukemic cell survival. J. Clin. Investig. 2014, 124, 2585–2598. [Google Scholar] [CrossRef] [PubMed]
- Tanimura, A.; Yujiri, T.; Tanaka, Y.; Hatanaka, M.; Mitani, N.; Nakamura, Y.; Mori, K.; Tanizawa, Y. The anti-apoptotic role of the unfolded protein response in Bcr-Abl-positive leukemia cells. Leuk. Res. 2009, 33, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, G.M.; Zheng, S.; Leclerc, G.J.; DeSalvo, J.; Swords, R.T.; Barredo, J.C. The NEDD8-activating enzyme inhibitor pevonedistat activates the eIF2α and mTOR pathways inducing UPR-mediated cell death in acute lymphoblastic leukemia. Leuk. Res. 2016, 50, 1–10. [Google Scholar] [CrossRef]
- Huiting, L.N.; Samaha, Y.; Zhang, G.L.; Roderick, J.E.; Li, B.; Anderson, N.M.; Wang, Y.W.; Wang, L.; Laroche, F.; Choi, J.W.; et al. UFD1 contributes to MYC-mediated leukemia aggressiveness through suppression of the proapoptotic unfolded protein response. Leukemia 2018, 32, 2339–2351. [Google Scholar] [CrossRef]
- Wilhelm, T.; Bick, F.; Peters, K.; Mohta, V.; Tirosh, B.; Patterson, J.B.; Kharabi-Masouleh, B.; Huber, M. Infliction of proteotoxic stresses by impairment of the unfolded protein response or proteasomal inhibition as a therapeutic strategy for mast cell leukemia. Oncotarget 2018, 9, 2984–3000. [Google Scholar] [CrossRef]
- Vieri, M.; Preisinger, C.; Schemionek, M.; Salimi, A.; Patterson, J.B.; Samali, A.; Brummendorf, T.H.; Appelmann, I.; Kharabi Masouleh, B. Targeting of BCR-ABL1 and IRE1α induces synthetic lethality in Philadelphia-positive acute lymphoblastic leukemia. Carcinogenesis 2020, bgaa095. [Google Scholar] [CrossRef]
- Jang, J.E.; Eom, J.I.; Jeung, H.K.; Chung, H.; Kim, Y.R.; Kim, J.S.; Cheong, J.W.; Min, Y.H. PERK/NRF2 and autophagy form a resistance mechanism against G9a inhibition in leukemia stem cells. J. Exp. Clin. Cancer Res. CR 2020, 39, 66. [Google Scholar] [CrossRef]
- Nguyen, T.K.; Grant, S. Dinaciclib (SCH727965) inhibits the unfolded protein response through a CDK1- and 5-dependent mechanism. Mol. Cancer Ther. 2014, 13, 662–674. [Google Scholar] [CrossRef]
- Evangelisti, C.; Evangelisti, C.; Teti, G.; Chiarini, F.; Falconi, M.; Melchionda, F.; Pession, A.; Bertaina, A.; Locatelli, F.; McCubrey, J.A.; et al. Assessment of the effect of sphingosine kinase inhibitors on apoptosis, unfolded protein response and autophagy of T-cell acute lymphoblastic leukemia cells; indications for novel therapeutics. Oncotarget 2014, 5, 7886–7901. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, M.; Mayo, M.; Dash, R.; Sokhi, U.K.; Dmitriev, I.P.; Sarkar, D.; Dent, P.; Curiel, D.T.; Fisher, P.B.; Grant, S. Melanoma differentiation associated gene-7/interleukin-24 potently induces apoptosis in human myeloid leukemia cells through a process regulated by endoplasmic reticulum stress. Mol. Pharm. 2010, 78, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, M.; Davis, E.M.; Crabtree, T.R.; Habibi, J.R.; Nguyen, T.K.; Dent, P.; Grant, S. The kinase inhibitor sorafenib induces cell death through a process involving induction of endoplasmic reticulum stress. Mol. Cell. Biol. 2007, 27, 5499–5513. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Wang, X.Y.; Zhou, Z.W.; Bai, H.; Shi, L.; Yang, Y.X.; Zhou, S.F.; Zhang, X.C. The combination of digoxin and GSK2606414 exerts synergistic anticancer activity against leukemia in vitro and in vivo. Biofactors 2017, 43, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, R.; Müller, G.A.; Rathore, M.S.; Mishra, D.P.; Dihazi, H. Anti-Leukemic Activity of Shikonin: Role of ERP57 in Shikonin Induced Apoptosis in Acute Myeloid Leukemia. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharm. 2016, 39, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Bi, C.; Cheong, L.L.; Mahara, S.; Liu, S.C.; Tay, K.G.; Koh, T.L.; Yu, Q.; Chng, W.J. The histone methyltransferase inhibitor, DZNep, up-regulates TXNIP, increases ROS production, and targets leukemia cells in AML. Blood 2011, 118, 2830–2839. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhang, Y.; Wark, L.; Ortiz, E.; Lim, S.; He, H.; Wang, W.; Medeiros, D.; Lin, D. Wolfberry Water Soluble Phytochemicals Down-Regulate ER Stress Biomarkers and Modulate Multiple Signaling Pathways Leading to Inhibition of Proliferation and Induction of Apoptosis in Jurkat Cells. J. Nutr. Food Sci. 2011, S2. [Google Scholar] [CrossRef]
- Leclerc, G.M.; Leclerc, G.J.; Kuznetsov, J.N.; DeSalvo, J.; Barredo, J.C. Metformin induces apoptosis through AMPK-dependent inhibition of UPR signaling in ALL lymphoblasts. PLoS ONE 2013, 8, e74420. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Yi, Z.; Zhang, Y.; Ma, Z.; Zhu, Y.; Gao, J. Low dose of 2-deoxy-D-glucose kills acute lymphoblastic leukemia cells and reverses glucocorticoid resistance via N-linked glycosylation inhibition under normoxia. Oncotarget 2017, 8, 30978–30991. [Google Scholar] [CrossRef] [PubMed]
- DeSalvo, J.; Kuznetsov, J.N.; Du, J.; Leclerc, G.M.; Leclerc, G.J.; Lampidis, T.J.; Barredo, J.C. Inhibition of Akt potentiates 2-DG-induced apoptosis via downregulation of UPR in acute lymphoblastic leukemia. Mol. Cancer Res. MCR 2012, 10, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Li, F.; Shi, K.; Wu, P.; An, J.; Yang, Y.; Xu, C. Involvement of p38 in signal switching from autophagy to apoptosis via the PERK/eIF2α/ATF4 axis in selenite-treated NB4 cells. Cell Death Dis. 2014, 5, e1270. [Google Scholar] [CrossRef] [PubMed]
- Rong, C.; Wei, W.; Yu-Hong, T. Asperuloside exhibits a novel anti-leukemic activity by triggering ER stress-regulated apoptosis via targeting GRP78. Biomed. Pharm. Biomed. Pharm. 2020, 125, 109819. [Google Scholar] [CrossRef] [PubMed]
- Maioral, M.F.; Stefanes, N.M.; Neuenfeldt, P.D.; Chiaradia-Delatorre, L.D.; Nunes, R.J.; Santos-Silva, M.C. Aldehyde biphenyl chalcones induce immunogenic apoptotic-like cell death and are promising new safe compounds against a wide range of hematologic cancers. Future Med. Chem. 2020, 12, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Xin, W.; Na, L.; Juan, L.; Lin, Z.; Lingyun, H.; Ai, F.; Zhonglin, W.; Yawen, W. Prolonged unfolded protein reaction is involved in the induction of CML cell death upon oprozomib treatment. Cancer Sci. 2020, 112, 133–143. [Google Scholar] [CrossRef]
- El Dor, M.; Dakik, H.; Polomski, M.; Haudebourg, E.; Brachet, M.; Gouilleux, F.; Prié, G.; Zibara, K.; Mazurier, F. VAS3947 Induces UPR-Mediated Apoptosis through Cysteine Thiol Alkylation in AML Cell Lines. Int. J. Mol. Sci. 2020, 21, 5470. [Google Scholar] [CrossRef]
- Hu, X.; Cai, J.; Zhu, J.; Lang, W.; Zhong, J.; Zhong, H.; Chen, F. Arsenic trioxide potentiates Gilteritinib-induced apoptosis in FLT3-ITD positive leukemic cells via IRE1a-JNK-mediated endoplasmic reticulum stress. Cancer Cell Int. 2020, 20, 250. [Google Scholar] [CrossRef]
- Chu, X.; Zhong, L.; Yu, L.; Xiong, L.; Li, J.; Dan, W.; Ye, J.; Liu, C.; Luo, X.; Liu, B. GSK-J4 induces cell cycle arrest and apoptosis via ER stress and the synergism between GSK-J4 and decitabine in acute myeloid leukemia KG-1a cells. Cancer Cell Int. 2020, 20, 209. [Google Scholar] [CrossRef]
- Bian, T.; Tagmount, A.; Vulpe, C.; Vijendra, K.C.; Xing, C. CXL146, a Novel 4H-Chromene Derivative, Targets GRP78 to Selectively Eliminate Multidrug-Resistant Cancer Cells. Mol. Pharm. 2020, 97, 402–408. [Google Scholar] [CrossRef]
- Matsumoto, T.; Jimi, S.; Migita, K.; Terada, K.; Mori, M.; Takamatsu, Y.; Suzumiya, J.; Hara, S. FF-10501 induces caspase-8-mediated apoptotic and endoplasmic reticulum stress-mediated necrotic cell death in hematological malignant cells. Int. J. Hematol. 2019, 110, 606–617. [Google Scholar] [CrossRef]
- Masciarelli, S.; Capuano, E.; Ottone, T.; Divona, M.; Lavorgna, S.; Liccardo, F.; Śniegocka, M.; Travaglini, S.; Noguera, N.I.; Picardi, A.; et al. Retinoic acid synergizes with the unfolded protein response and oxidative stress to induce cell death in FLT3-ITD+ AML. Blood Adv. 2019, 3, 4155–4160. [Google Scholar] [CrossRef]
- Masciarelli, S.; Capuano, E.; Ottone, T.; Divona, M.; De Panfilis, S.; Banella, C.; Noguera, N.I.; Picardi, A.; Fontemaggi, G.; Blandino, G.; et al. Retinoic acid and arsenic trioxide sensitize acute promyelocytic leukemia cells to ER stress. Leukemia 2018, 32, 285–294. [Google Scholar] [CrossRef]
- Mallick, D.J.; Soderquist, R.S.; Bates, D.; Eastman, A. Confounding off-target effects of BH3 mimetics at commonly used concentrations: MIM1, UMI-77, and A-1210477. Cell Death Dis. 2019, 10, 185. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.C.; Peng, S.F.; Lai, K.C.; Liao, C.L.; Huang, Y.P.; Lin, C.C.; Lin, M.L.; Liu, K.C.; Tsai, C.C.; Ma, Y.S.; et al. Genistein induces apoptosis in vitro and has antitumor activity against human leukemia HL-60 cancer cell xenograft growth in vivo. Environ. Toxicol. 2019, 34, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, G.; Wu, W.; Yao, S.; Han, X.; He, D.; He, J.; Zheng, G.; Zhao, Y.; Cai, Z.; et al. Camalexin Induces Apoptosis via the ROS-ER Stress-Mitochondrial Apoptosis Pathway in AML Cells. Oxidative Med. Cell. Longev. 2018, 2018, 7426950. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wu, J.; Sheng, L. Ibrutinib improves the development of acute lymphoblastic leukemia by activating endoplasmic reticulum stress-induced cell death. Pharmazie 2018, 73, 294–299. [Google Scholar] [CrossRef]
- Mazumder, A.; Lee, J.Y.; Talhi, O.; Cerella, C.; Chateauvieux, S.; Gaigneaux, A.; Hong, C.R.; Kang, H.J.; Lee, Y.; Kim, K.W.; et al. Hydroxycoumarin OT-55 kills CML cells alone or in synergy with imatinib or Synribo: Involvement of ER stress and DAMP release. Cancer Lett. 2018, 438, 197–218. [Google Scholar] [CrossRef]
- Florean, C.; Kim, K.R.; Schnekenburger, M.; Kim, H.J.; Moriou, C.; Debitus, C.; Dicato, M.; Al-Mourabit, A.; Han, B.W.; Diederich, M. Synergistic AML Cell Death Induction by Marine Cytotoxin (+)-1(R), 6(S), 1′(R), 6′(S), 11(R), 17(S)-Fistularin-3 and Bcl-2 Inhibitor Venetoclax. Mar. Drugs 2018, 16, 518. [Google Scholar] [CrossRef]
- Wang, X.Y.; Zhang, X.H.; Peng, L.; Liu, Z.; Yang, Y.X.; He, Z.X.; Dang, H.W.; Zhou, S.F. Bardoxolone methyl (CDDO-Me or RTA402) induces cell cycle arrest, apoptosis and autophagy via PI3K/Akt/mTOR and p38 MAPK/Erk1/2 signaling pathways in K562 cells. Am. J. Trans. Res. 2017, 9, 4652–4672. [Google Scholar]
- Mitsuhashi, Y.; Furusawa, Y.; Aradate, T.; Zhao, Q.L.; Moniruzzaman, R.; Kanamori, M.; Noguchi, K.; Kondo, T. 3-O-trans-p-coumaroyl-alphitolic acid, a triterpenoid from Zizyphus jujuba, leads to apoptotic cell death in human leukemia cells through reactive oxygen species production and activation of the unfolded protein response. PLoS ONE 2017, 12, e0183712. [Google Scholar] [CrossRef]
- Meier-Stephenson, V.; Riemer, J.; Narendran, A. The HIV protease inhibitor, nelfinavir, as a novel therapeutic approach for the treatment of refractory pediatric leukemia. Oncotargets 2017, 10, 2581–2593. [Google Scholar] [CrossRef]
- Mahoney, E.; Maddocks, K.; Flynn, J.; Jones, J.; Cole, S.L.; Zhang, X.; Byrd, J.C.; Johnson, A.J. Identification of endoplasmic reticulum stress-inducing agents by antagonizing autophagy: A new potential strategy for identification of anti-cancer therapeutics in B-cell malignancies. Leuk. Lymphoma 2013, 54, 2685–2692. [Google Scholar] [CrossRef] [PubMed]
- Gugliotta, G.; Sudo, M.; Cao, Q.; Lin, D.C.; Sun, H.; Takao, S.; Le Moigne, R.; Rolfe, M.; Gery, S.; Müschen, M.; et al. Valosin-Containing Protein/p97 as a Novel Therapeutic Target in Acute Lymphoblastic Leukemia. Neoplasia 2017, 19, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Tsitsipatis, D.; Jayavelu, A.K.; Müller, J.P.; Bauer, R.; Schmidt-Arras, D.; Mahboobi, S.; Schnöder, T.M.; Heidel, F.; Böhmer, F.D. Synergistic killing of FLT3ITD-positive AML cells by combined inhibition of tyrosine-kinase activity and N-glycosylation. Oncotarget 2017, 8, 26613–26624. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.F.; Seo, E.J.; Klauck, S.M.; Efferth, T. Cryptotanshinone deregulates unfolded protein response and eukaryotic initiation factor signaling in acute lymphoblastic leukemia cells. Phytomed. Int. J. Phytother. Phytopharm. 2016, 23, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Q.L.; Nong, X.H.; Zhang, X.Y.; Xu, X.Y.; Qi, S.H.; Wang, Y.F. Oxalicumone A, a new dihydrothiophene-condensed sulfur chromone induces apoptosis in leukemia cells through endoplasmic reticulum stress pathway. Eur. J. Pharm. 2016, 783, 47–55. [Google Scholar] [CrossRef]
- Lamothe, B.; Wierda, W.G.; Keating, M.J.; Gandhi, V. Carfilzomib Triggers Cell Death in Chronic Lymphocytic Leukemia by Inducing Proapoptotic and Endoplasmic Reticulum Stress Responses. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 4712–4726. [Google Scholar] [CrossRef]
- Zhou, L.; Jiang, L.; Xu, M.; Liu, Q.; Gao, N.; Li, P.; Liu, E.H. Miltirone exhibits antileukemic activity by ROS-mediated endoplasmic reticulum stress and mitochondrial dysfunction pathways. Sci. Rep. 2016, 6, 20585. [Google Scholar] [CrossRef]
- Wang, F.F.; Liu, M.Z.; Sui, Y.; Cao, Q.; Yan, B.; Jin, M.L.; Mo, X. Deficiency of SUMO-specific protease 1 induces arsenic trioxide-mediated apoptosis by regulating XBP1 activity in human acute promyelocytic leukemia. Oncol. Lett. 2016, 12, 3755–3762. [Google Scholar] [CrossRef]
- Chen, J.; Wei, H.; Xie, B.; Wang, B.; Cheng, J.; Cheng, J. Endoplasmic reticulum stress contributes to arsenic trioxide-induced apoptosis in drug-sensitive and -resistant leukemia cells. Leuk. Res. 2012, 36, 1526–1535. [Google Scholar] [CrossRef]
- Rosilio, C.; Nebout, M.; Imbert, V.; Griessinger, E.; Neffati, Z.; Benadiba, J.; Hagenbeek, T.; Spits, H.; Reverso, J.; Ambrosetti, D.; et al. L-type amino-acid transporter 1 (LAT1): A therapeutic target supporting growth and survival of T-cell lymphoblastic lymphoma/T-cell acute lymphoblastic leukemia. Leukemia 2015, 29, 1253–1266. [Google Scholar] [CrossRef]
- Hu, C.; Xu, M.; Qin, R.; Chen, W.; Xu, X. Wogonin induces apoptosis and endoplasmic reticulum stress in HL-60 leukemia cells through inhibition of the PI3K-AKT signaling pathway. Oncol. Rep. 2015, 33, 3146–3154. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Joo, J.H.; Ueda, E.; Bortner, C.D.; Yang, X.P.; Liao, G.; Jetten, A.M. Farnesol activates the intrinsic pathway of apoptosis and the ATF4-ATF3-CHOP cascade of ER stress in human T lymphoblastic leukemia Molt4 cells. Biochem. Pharm. 2015, 97, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Cagnetta, A.; Caffa, I.; Acharya, C.; Soncini, D.; Acharya, P.; Adamia, S.; Pierri, I.; Bergamaschi, M.; Garuti, A.; Fraternali, G.; et al. APO866 Increases Antitumor Activity of Cyclosporin-A by Inducing Mitochondrial and Endoplasmic Reticulum Stress in Leukemia Cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 3934–3945. [Google Scholar] [CrossRef] [PubMed]
- Takenokuchi, M.; Miyamoto, K.; Saigo, K.; Taniguchi, T. Bortezomib Causes ER Stress-related Death of Acute Promyelocytic Leukemia Cells Through Excessive Accumulation of PML-RARA. Anticancer Res. 2015, 35, 3307–3316. [Google Scholar] [PubMed]
- Buontempo, F.; Orsini, E.; Martins, L.R.; Antunes, I.; Lonetti, A.; Chiarini, F.; Tabellini, G.; Evangelisti, C.; Evangelisti, C.; Melchionda, F.; et al. Cytotoxic activity of the casein kinase 2 inhibitor CX-4945 against T-cell acute lymphoblastic leukemia: Targeting the unfolded protein response signaling. Leukemia 2014, 28, 543–553. [Google Scholar] [CrossRef]
- Winter, E.; Dal Pizzol, C.; Filippin-Monteiro, F.B.; Brondani, P.; Silva, A.M.; Silva, A.H.; Bonacorso, H.G.; Martins, M.A.; Zanatta, N.; Creczynski-Pasa, T.B. Antitumoral activity of a trichloromethyl pyrimidine analogue: Molecular cross-talk between intrinsic and extrinsic apoptosis. Chem. Res. Toxicol. 2014, 27, 1040–1049. [Google Scholar] [CrossRef]
- Winter, E.; Chiaradia, L.D.; Silva, A.H.; Nunes, R.J.; Yunes, R.A.; Creczynski-Pasa, T.B. Involvement of extrinsic and intrinsic apoptotic pathways together with endoplasmic reticulum stress in cell death induced by naphthylchalcones in a leukemic cell line: Advantages of multi-target action. Toxicol. Vitr. 2014, 28, 769–777. [Google Scholar] [CrossRef]
- Soderquist, R.; Pletnev, A.A.; Danilov, A.V.; Eastman, A. The putative BH3 mimetic S1 sensitizes leukemia to ABT-737 by increasing reactive oxygen species, inducing endoplasmic reticulum stress, and upregulating the BH3-only protein NOXA. Apoptosis Int. J. Program. Cell Death 2014, 19, 201–209. [Google Scholar] [CrossRef]
- Soderquist, R.S.; Danilov, A.V.; Eastman, A. Gossypol increases expression of the pro-apoptotic BH3-only protein NOXA through a novel mechanism involving phospholipase A2, cytoplasmic calcium, and endoplasmic reticulum stress. J. Biol. Chem. 2014, 289, 16190–16199. [Google Scholar] [CrossRef]
- Li, H.; Chang, G.; Wang, J.; Wang, L.; Jin, W.; Lin, Y.; Yan, Y.; Wang, R.; Gao, W.; Ma, L.; et al. Cariporide sensitizes leukemic cells to tumor necrosis factor related apoptosis-inducing ligand by up-regulation of death receptor 5 via endoplasmic reticulum stress-CCAAT/enhancer binding protein homologous protein dependent mechanism. Leuk. Lymphoma 2014, 55, 2135–2140. [Google Scholar] [CrossRef]
- Fiskus, W.; Saba, N.; Shen, M.; Ghias, M.; Liu, J.; Gupta, S.D.; Chauhan, L.; Rao, R.; Gunewardena, S.; Schorno, K.; et al. Auranofin induces lethal oxidative and endoplasmic reticulum stress and exerts potent preclinical activity against chronic lymphocytic leukemia. Cancer Res. 2014, 74, 2520–2532. [Google Scholar] [CrossRef] [PubMed]
- Bortolozzi, R.; Viola, G.; Porcù, E.; Consolaro, F.; Marzano, C.; Pellei, M.; Gandin, V.; Basso, G. A novel copper(I) complex induces ER-stress-mediated apoptosis and sensitizes B-acute lymphoblastic leukemia cells to chemotherapeutic agents. Oncotarget 2014, 5, 5978–5991. [Google Scholar] [CrossRef] [PubMed]
- Rosati, E.; Sabatini, R.; De Falco, F.; Del Papa, B.; Falzetti, F.; Di Ianni, M.; Cavalli, L.; Fettucciari, K.; Bartoli, A.; Screpanti, I.; et al. γ-Secretase inhibitor I induces apoptosis in chronic lymphocytic leukemia cells by proteasome inhibition, endoplasmic reticulum stress increase and notch down-regulation. Int. J. Cancer 2013, 132, 1940–1953. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.C.; Chang, C.L.; Yu, C.S.; Chen, P.Y.; Yang, J.S.; Ji, B.C.; Lin, T.P.; Chiu, C.F.; Yeh, S.P.; Huang, Y.P.; et al. Induction of apoptosis by curcumin in murine myelomonocytic leukemia WEHI-3 cells is mediated via endoplasmic reticulum stress and mitochondria-dependent pathways. Environ. Toxicol. 2013, 28, 255–266. [Google Scholar] [CrossRef]
- Ng, A.P.; Chng, W.J.; Khan, M. Curcumin sensitizes acute promyelocytic leukemia cells to unfolded protein response-induced apoptosis by blocking the loss of misfolded N-CoR protein. Mol. Cancer Res. MCR 2011, 9, 878–888. [Google Scholar] [CrossRef]
- Pae, H.O.; Jeong, S.O.; Jeong, G.S.; Kim, K.M.; Kim, H.S.; Kim, S.A.; Kim, Y.C.; Kang, S.D.; Kim, B.N.; Chung, H.T. Curcumin induces pro-apoptotic endoplasmic reticulum stress in human leukemia HL-60 cells. Biochem. Biophys. Res. Commun. 2007, 353, 1040–1045. [Google Scholar] [CrossRef]
- de Sá Bacelar, T.; da Silva, A.J.; Costa, P.R.; Rumjanek, V.M. The pterocarpanquinone LQB 118 induces apoptosis in tumor cells through the intrinsic pathway and the endoplasmic reticulum stress pathway. Anti-Cancer Drugs 2013, 24, 73–83. [Google Scholar] [CrossRef]
- Mahoney, E.; Byrd, J.C.; Johnson, A.J. Autophagy and ER stress play an essential role in the mechanism of action and drug resistance of the cyclin-dependent kinase inhibitor flavopiridol. Autophagy 2013, 9, 434–435. [Google Scholar] [CrossRef]
- Yu, C.S.; Huang, A.C.; Yang, J.S.; Yu, C.C.; Lin, C.C.; Chung, H.K.; Huang, Y.P.; Chueh, F.S.; Chung, J.G. Safrole induces G0/G1 phase arrest via inhibition of cyclin E and provokes apoptosis through endoplasmic reticulum stress and mitochondrion-dependent pathways in human leukemia HL-60 cells. Anticancer Res. 2012, 32, 1671–1679. [Google Scholar]
- Penna, F.; Pin, F.; Costamagna, D.; Reffo, P.; Baccino, F.M.; Bonelli, G.; Costelli, P. Caspase 2 activation and ER stress drive rapid Jurkat cell apoptosis by clofibrate. PLoS ONE 2012, 7, e45327. [Google Scholar] [CrossRef]
- Park, Y.K.; Do, Y.R.; Jang, B.C. Apoptosis of K562 leukemia cells by Abnobaviscum F®, a European mistletoe extract. Oncol. Rep. 2012, 28, 2227–2232. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.C.; Yang, J.S.; Chiang, J.H.; Hour, M.J.; Lin, K.L.; Lin, J.J.; Huang, W.W.; Tsuzuki, M.; Lee, T.H.; Chung, J.G. Novel quinazolinone MJ-29 triggers endoplasmic reticulum stress and intrinsic apoptosis in murine leukemia WEHI-3 cells and inhibits leukemic mice. PLoS ONE 2012, 7, e36831. [Google Scholar] [CrossRef] [PubMed]
- Pattacini, L.; Mancini, M.; Mazzacurati, L.; Brusa, G.; Benvenuti, M.; Martinelli, G.; Baccarani, M.; Santucci, M.A. Endoplasmic reticulum stress initiates apoptotic death induced by STI571 inhibition of p210 bcr-abl tyrosine kinase. Leuk. Res. 2004, 28, 191–202. [Google Scholar] [CrossRef]
- Chueh, F.S.; Hsiao, Y.T.; Chang, S.J.; Wu, P.P.; Yang, J.S.; Lin, J.J.; Chung, J.G.; Lai, T.Y. Glycyrrhizic acid induces apoptosis in WEHI-3 mouse leukemia cells through the caspase- and mitochondria-dependent pathways. Oncol. Rep. 2012, 28, 2069–2076. [Google Scholar] [CrossRef]
- Lin, J.J.; Hsu, H.Y.; Yang, J.S.; Lu, K.W.; Wu, R.S.; Wu, K.C.; Lai, T.Y.; Chen, P.Y.; Ma, C.Y.; Wood, W.G.; et al. Molecular evidence of anti-leukemia activity of gypenosides on human myeloid leukemia HL-60 cells in vitro and in vivo using a HL-60 cells murine xenograft model. Phytomedicine Int. J. Phytother. Phytopharm. 2011, 18, 1075–1085. [Google Scholar] [CrossRef]
- Kuznetsov, J.N.; Leclerc, G.J.; Leclerc, G.M.; Barredo, J.C. AMPK and Akt determine apoptotic cell death following perturbations of one-carbon metabolism by regulating ER stress in acute lymphoblastic leukemia. Mol. Cancer Ther. 2011, 10, 437–447. [Google Scholar] [CrossRef]
- Chang, Y.C.; Lai, T.Y.; Yu, C.S.; Chen, H.Y.; Yang, J.S.; Chueh, F.S.; Lu, C.C.; Chiang, J.H.; Huang, W.W.; Ma, C.Y.; et al. Emodin Induces Apoptotic Death in Murine Myelomonocytic Leukemia WEHI-3 Cells In Vitro and Enhances Phagocytosis in Leukemia Mice In Vivo. Evid. Based Complementary Altern. Med. ECAM 2011, 2011, 523596. [Google Scholar] [CrossRef]
- Anshu, A.; Thomas, S.; Agarwal, P.; Ibarra-Rivera, T.R.; Pirrung, M.C.; Schönthal, A.H. Novel proteasome-inhibitory syrbactin analogs inducing endoplasmic reticulum stress and apoptosis in hematological tumor cell lines. Biochem. Pharmacol. 2011, 82, 600–609. [Google Scholar] [CrossRef]
- Albershardt, T.C.; Salerni, B.L.; Soderquist, R.S.; Bates, D.J.; Pletnev, A.A.; Kisselev, A.F.; Eastman, A. Multiple BH3 mimetics antagonize antiapoptotic MCL1 protein by inducing the endoplasmic reticulum stress response and up-regulating BH3-only protein NOXA. J. Biol. Chem. 2011, 286, 24882–24895. [Google Scholar] [CrossRef]
- Wagh, V.; Mishra, P.; Thakkar, A.; Shinde, V.; Sharma, S.; Padigaru, M.; Joshi, K. Antitumor activity of NPB001-05, an orally active inhibitor of Bcr-Abl tyrosine kinase. Front. Biosci. 2011, 3, 1349–1364. [Google Scholar] [CrossRef]
- Yaari-Stark, S.; Shaked, M.; Nevo-Caspi, Y.; Jacob-Hircsh, J.; Shamir, R.; Rechavi, G.; Kloog, Y. Ras inhibits endoplasmic reticulum stress in human cancer cells with amplified Myc. Int. J. Cancer 2010, 126, 2268–2281. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.W.; Ali, M.; Wood, T.E.; Wong, D.; Maclean, N.; Wang, X.; Gronda, M.; Skrtic, M.; Li, X.; Hurren, R.; et al. The ubiquitin-activating enzyme E1 as a therapeutic target for the treatment of leukemia and multiple myeloma. Blood 2010, 115, 2251–2259. [Google Scholar] [CrossRef] [PubMed]
- Popovic, S.; Baskic, D.; Djurdjevic, P.; Zelen, I.; Mitrovic, M.; Nikolic, I.; Avramovic, D.; Radenkovic, M.; Arsenijevic, N. Endoplasmic reticulum stress associated with caspases-4 and -2 mediates korbazol-induced B-chronic lymphocytic leukemia cell apoptosis. J. B.U. Off. J. Balk. Union Oncol. 2010, 15, 783–790. [Google Scholar]
- Lust, S.; Vanhoecke, B.; Van Gele, M.; Philippé, J.; Bracke, M.; Offner, F. The flavonoid tangeretin activates the unfolded protein response and synergizes with imatinib in the erythroleukemia cell line K562. Mol. Nutr. Food Res. 2010, 54, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Cherla, R.P.; Lentz, E.K.; Leyva-Illades, D.; Tesh, V.L. Signaling through C/EBP homologous protein and death receptor 5 and calpain activation differentially regulate THP-1 cell maturation-dependent apoptosis induced by Shiga toxin type 1. Infect. Immun. 2010, 78, 3378–3391. [Google Scholar] [CrossRef]
- Slagsvold, J.E.; Pettersen, C.H.; Follestad, T.; Krokan, H.E.; Schønberg, S.A. The antiproliferative effect of EPA in HL60 cells is mediated by alterations in calcium homeostasis. Lipids 2009, 44, 103–113. [Google Scholar] [CrossRef]
- Lust, S.; Vanhoecke, B.; Van Gelle, M.; Boelens, J.; van Melckebeke, H.; Kaileh, M.; Vanden Berghe, W.; Haegeman, G.; Philippé, J.; Bracke, M.; et al. Xanthohumol activates the proapoptotic arm of the unfolded protein response in chronic lymphocytic leukemia. Anticancer Res. 2009, 29, 3797–3805. [Google Scholar]
- Huang, W.C.; Lin, Y.S.; Chen, C.L.; Wang, C.Y.; Chiu, W.H.; Lin, C.F. Glycogen synthase kinase-3beta mediates endoplasmic reticulum stress-induced lysosomal apoptosis in leukemia. J. Pharmacol. Exp. Ther. 2009, 329, 524–531. [Google Scholar] [CrossRef]
- Zhang, Q.Y.; Mao, J.H.; Liu, P.; Huang, Q.H.; Lu, J.; Xie, Y.Y.; Weng, L.; Zhang, Y.; Chen, Q.; Chen, S.J.; et al. A systems biology understanding of the synergistic effects of arsenic sulfide and Imatinib in BCR/ABL-associated leukemia. Proc. Natl. Acad. Sci. USA 2009, 106, 3378–3383. [Google Scholar] [CrossRef]
- Wang, K.; Fang, H.; Xiao, D.; Zhu, X.; He, M.; Pan, X.; Shi, J.; Zhang, H.; Jia, X.; Du, Y.; et al. Converting redox signaling to apoptotic activities by stress-responsive regulators HSF1 and NRF2 in fenretinide treated cancer cells. PLoS ONE 2009, 4, e7538. [Google Scholar] [CrossRef]
- Lai, W.L.; Wong, N.S. The PERK/eIF2 alpha signaling pathway of Unfolded Protein Response is essential for N-(4-hydroxyphenyl)retinamide (4HPR)-induced cytotoxicity in cancer cells. Exp. Cell Res. 2008, 314, 1667–1682. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Manevich, Y.; He, L.; Xiong, Y.; Bowers, R.R., Jr.; Hutchens, S.; Tew, K.D. Nitrosative stress-induced s-glutathionylation of protein disulfide isomerase leads to activation of the unfolded protein response. Cancer Res. 2009, 69, 7626–7634. [Google Scholar] [CrossRef] [PubMed]
- Dodo, K.; Minato, T.; Noguchi-Yachide, T.; Suganuma, M.; Hashimoto, Y. Antiproliferative and apoptosis-inducing activities of alkyl gallate and gallamide derivatives related to (-)-epigallocatechin gallate. Bioorganic Med. Chem. 2008, 16, 7975–7982. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xia, X.; Ke, Y.; Nie, H.; Smith, M.A.; Zhu, X. Trichosanthin induced apoptosis in HL-60 cells via mitochondrial and endoplasmic reticulum stress signaling pathways. Biochim. Biophys. Acta 2007, 1770, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Jun, D.Y.; Kim, J.S.; Park, H.S.; Han, C.R.; Fang, Z.; Woo, M.H.; Rhee, I.K.; Kim, Y.H. Apoptogenic activity of auraptene of Zanthoxylum schinifolium toward human acute leukemia Jurkat T cells is associated with ER stress-mediated caspase-8 activation that stimulates mitochondria-dependent or -independent caspase cascade. Carcinogenesis 2007, 28, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Anding, A.L.; Chapman, J.S.; Barnett, D.W.; Curley, R.W., Jr.; Clagett-Dame, M. The unhydrolyzable fenretinide analogue 4-hydroxybenzylretinone induces the proapoptotic genes GADD153 (CHOP) and Bcl-2-binding component 3 (PUMA) and apoptosis that is caspase- dependent and independent of the retinoic acid receptor. Cancer Res. 2007, 67, 6270–6277. [Google Scholar] [CrossRef]
- Yanamandra, N.; Colaco, N.M.; Parquet, N.A.; Buzzeo, R.W.; Boulware, D.; Wright, G.; Perez, L.E.; Dalton, W.S.; Beaupre, D.M. Tipifarnib and bortezomib are synergistic and overcome cell adhesion-mediated drug resistance in multiple myeloma and acute myeloid leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 591–599. [Google Scholar] [CrossRef]
- Ng, A.P.; Howe Fong, J.; Sijin Nin, D.; Hirpara, J.L.; Asou, N.; Chen, C.S.; Pervaiz, S.; Khan, M. Cleavage of misfolded nuclear receptor corepressor confers resistance to unfolded protein response-induced apoptosis. Cancer Res. 2006, 66, 9903–9912. [Google Scholar] [CrossRef]
- Feng, X.Q.; You, Y.; Xiao, J.; Zou, P. Thapsigargin-induced apoptosis of K562 cells and its mechanism. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2006, 14, 25–30. [Google Scholar]
- Feng, X.Q.; Xiao, J.; Nie, S.M.; Liu, F.; You, Y.; Zou, P. Expression and significance of melanoma antigen gene-3 in endoplasmic reticulum stress-induced apoptosis of K562 cells. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2005, 13, 741–745. [Google Scholar]
- Du, Y.; Wang, K.; Fang, H.; Li, J.; Xiao, D.; Zheng, P.; Chen, Y.; Fan, H.; Pan, X.; Zhao, C.; et al. Coordination of intrinsic, extrinsic, and endoplasmic reticulum-mediated apoptosis by imatinib mesylate combined with arsenic trioxide in chronic myeloid leukemia. Blood 2006, 107, 1582–1590. [Google Scholar] [CrossRef] [PubMed]
- Anether, G.; Tinhofer, I.; Senfter, M.; Greil, R. Tetrocarcin-A—Induced ER stress mediates apoptosis in B-CLL cells via a Bcl-2--independent pathway. Blood 2003, 101, 4561–4568. [Google Scholar] [CrossRef] [PubMed]
- Tinhofer, I.; Anether, G.; Senfter, M.; Pfaller, K.; Bernhard, D.; Hara, M.; Greil, R. Stressful death of T-ALL tumor cells after treatment with the anti-tumor agent Tetrocarcin-A. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2002, 16, 1295–1297. [Google Scholar] [CrossRef]
- Walczak, A.; Gradzik, K.; Kabzinski, J. The Role of the ER-Induced UPR Pathway and the Efficacy of Its Inhibitors and Inducers in the Inhibition of Tumor Progression. Oxidative Med. Cell. Longev. 2019, 2019, 5729710. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Molecule | Chemical Nature | Pubchem Compound CID | Target | Type of Leukemia | Proposed Role of ER Stress/UPR Activation | Mainly Implicated Pathways/Effectors | Ref. |
|---|---|---|---|---|---|---|---|
| BIX-01294 | 25150857 | Histone methyltransferase G9A | AML | Adaptive | PERK (prosurvival, via NRF2) | [170] | |
| SCH727965 (Dinaciclib) | 46926350 | CDKs 1,2,5 and 9 | AML, CML, T-ALL | Adaptive | XBP1s | [171] | |
| Ski, ROMe | 16760659 | spingosine kinases 1 and 2 | T-ALL | Adaptive | unclear | [172] | |
| MDA-7/IL-24) | cytokine | - | n.d. | AML, APL | Adaptive | GRP78/Bip, IRE1α, GADD34 | [173] |
| sorafenib | multikinase inhibitor | 216239 | MEK/ERK pathway | U937 cell line | Adaptive | PERK | [174] |
| Digoxine | cardiac glycoside | 2724385 | Na+/K+ ATPase | K562 (erythroleukemic) and THP-1 (acute monocytic leukemia) cell lines | Adaptive | PERK, IRE1α | [175] |
| Shikonin | naphthoquinone | 479503 | pyruvate kinase-M2 (PKM2), proteasome inhibition, NFkB, Thioredoxin reductase (TrxR1) | HL60 | Adaptive | ERP57 and Calreticulin | [176] |
| 3-deazaneplanocin A (DZNeP) | cyclopentenyl analog of 3-deazaadenosine | 73087 | histone methyltransferase | MV4-11, MOLM-14, Mono-Mac-1, THP-1, HL60 and KG-1 AML cell lines | Adaptive | GRP78, GRP94, PERK, PDIA isoform 3, 4, and 5 | [177] |
| wolfberry phytochemicals | n.d. | - | n.d. | Jurkat cell line | Adaptive | all UPR pathways | [178] |
| Imatinib | 5291 | tyrosine kinases inhibitor | CML/LAMA-84 CML cell line and murine myeloid progenitor primary cells | The constitutive activation of PERK in CML cells protects from imatinib treatment | PERK | [162] | |
| Metformin | 4091 | multiple, see PMID: 28776086 | T-ALL, B-ALL | Switch form adaptive to terminal | IRE1α, CHOP | [179] | |
| Nilotinib + MKC8866 | 644241 | Tyrosine kinases (Nilotinib); IRE1α (MKC8866) | ALL (Ph+) | Switch form adaptive to terminal | IRE1α (cytoprotective); PERK and ATF6α (cytotoxic) | [169] | |
| 2-deoxy-D-glucose | glucose analog | 108223 | n.d. | ALL Cell Lines | Switch form adaptive to terminal | GRP78/Bip, CHOP | [180,181] |
| Selenite | sodium selenite | 24934 | n.d. | NB4 cell line (APL) | Switch form adaptive to terminal | PERK/eIF2α/ATF4 | [182] |
| Asperuloside | iridoid glycoside | 84298 | n.d. | cell lines HL60 and U937, primary leukemic cells | Terminal: apoptosis induction | all UPR pathways | [183] |
| JA3 & JA7 | Aldehyde biphenyl chalcones | 134820953 | n.d. | (AML); T-ALL; CML | Terminal: immunogenic apoptosis-like cell death | CHOP; PERK | [184] |
| Oprozomib | tripeptide analog of carfilzomib | 25067547 | immunoproteasome subunit β5i/LMP7 (ubiquitin–proteasome pathway) | CML | Terminal: apoptosis induction | PERK, IRE1α (via ASK/JNK/Bim) | [185] |
| VAS3947 | given in [186] fig 1a | 7471335 | NADPH oxidases | AML | Terminal: apoptosis induction | IRE1α, PERK | [186] |
| Arsenic trioxide + Gilteritinib | 14888/49803313 | FLT3 (for Gilteritinib) | AML (FLT3-ITD) | Terminal: apoptosis induction | IRE1α | [187] | |
| GSK-J4 | 71729975 | H3K27me3 demethylase | AML | Terminal: apoptosis induction | PKCα; Bcl2 phosphorylation | [188] | |
| CXL146 | 4H-chromene derivative | - | AML (or APL): HL60; CML | Terminal: apoptosis induction | PERK, IRE1α, ATF6α | [189] | |
| FF-10501 | given in paper | 124343 | inosine monophosphate deshydrogenase | AML | Terminal: necrotic cell death | CHOP | [190] |
| Retinoic acid+Tunicamycin+ arsenic trioxide | 444795/11104835 | n.d. | AML | Terminal: Cytotoxic UPR | CHOP, XBP1s | [191] | |
| [Retinoic acid or arsenic trioxide] + tunicamycin | 444795/11104835 | n.d. | APL | Terminal: Cytotoxic UPR | [192] | ||
| MIM1 and UMI-77 | 135691163/992586 | n.d. | AML; T-ALL | Terminal: Cytotoxic UPR | NOXA | [193] | |
| PFR | peptide | - | n.d. | AML | Terminal: necroptosis | [193] | |
| Genistein | Isoflavone | 5280961 | n.d. | AML or APL (HL60 cell line) | Terminal: Cytotoxic UPR | [194] | |
| Camalexin | Phytoalexin (structure given in paper) | 636970 | n.d. | AML | Terminal: apoptosis induction | PERK, CHOP | [195] |
| Ibrutinib (PCI-32765) | 24821094 | Bruton’s tyrosine kinase | B-ALL | Terminal: apoptosis induction | ATF4; CHOP | [196] | |
| OT-55 | bis-coumarine derivative | - | n.d. | CML | Terminal: Immunogenic cell death induction; apoptosis induction | not well documented | [197] |
| RS-F3 | fistularin-3 stereoisomer | - | AML | Terminal: Cytotoxic UPR (assumed) | PERK; XBP1s; CHOP | [198] | |
| Bardoxolone methyl (CDDO-Me) | triterpenoid | 400769 | Nrf2 and NF-κB | Chronic myeloid leukemia, K562 cell line | Terminal: apoptosis induction | PERK, IRE1α, CHOP | [199] |
| 3-O-trans-p-coumaroyl-alphitolic acid (3OTPCA) | triterpenoid | - | n.d. | U937, Molt-4 and Jurkat cell lines. | Terminal: apoptosis induction | XBP-1 and CHOP | [200] |
| Nelfinavir | 64143 | HIV protease inhibitors | T-ALL, B-ALL, and AML; CLL primary leukemic cells | Terminal: apoptosis induction; In CLL, contributes to the induction of cell death in choroquine treated cells | CHOP | [201,202] | |
| CB-5083 | 1-[4-(benzylamino)-5H,7H,8H-pyrano[4,3-d]pyrimidin-2-yl]-2-methyl-1H-indole-4-carboxamide | 439268 | Valosin-Containing Protein/p97 | B-ALL Cell Lines (BALL1, REH, NALM6, OP1, ALL-PO, 697, RS4;11, BV173, SEM, and SUPB15) | Terminal: apoptosis induction | all UPR pathways | [203] |
| Tunicamycin ± Quizartinib (AC220) | AC220 | 11104835 + 24889392 | FLT3 | AML (FLT3-ITD) | Terminal: apoptosis induction | PERK, CHOP | [204] |
| Cryptotanshinone | lipophilic diterpene quinone | 160254 | n.d. | CCRF-CEM cell line (ALL) | Terminal: apoptosis induction | IRE1α-XBP1, PERK-eIF2α-ATF4 | [205] |
| Oxalicumone A | dihydrothiophene-condensed sulfur chromone | 90676613 | n.d. | KG-1a, HL60, U937, and K562 cell lines (AML) | Terminal: apoptosis induction | IRE1α-XBP1, PERK--CHOP | [206] |
| Pevonedistat (MLN4924) | adenosine sulfamate analog | 16720766 | NEDD8-activating enzyme | T-ALL (CCRF-CEM, Jurkat) and B-ALL (REH, NALM6, SupB15) cell lines | Terminal: apoptosis induction | all UPR pathways | [166] |
| Carfilzomib (PR-171) | tetrapeptide epoxyketone | 11556711 | ubiquitin–proteasome pathway | CLL MEC1 and MEC2 cell lines and primary leukemic cells | Terminal: apoptosis induction | ATF4, CHOP | [207] |
| Miltirone | abietane-type norditerpenoid quinone | 160142 | n.d. | Jurkat, U937, AML and ALL primary leukemic cells | Terminal: apoptosis induction | PERK | [208] |
| Arsenic trioxide | 14888 | n.d. | NB4 cell line (AML)/CML | Terminal: apoptosis induction | IRE1α-XBP1/GRP78/Bip, CHOP, Xbp1 (unspliced…) | [209,210] | |
| JPH203 | O-[(5-Amino-2-phenyl-7-benzoxazolyl)methyl]-3,5-dichloro-L-tyrosine dihydrochloride | 24853505 | LAT1 (L-type amino-acid transporter 1) | Ke37, DND41, Sil-ALL, Peer, Molt-16, Jurkat and SupT1 T-ALL cell lines | Terminal: apoptosis induction | CHOP | [211] |
| Wogonin | 5,7-dihydroxy-8-methoxyflavone | 5281703 | n.d. | HL-60 cell line. | Terminal: apoptosis induction | all UPR pathways-CHOP | [212] |
| Farnesol | acyclic sesquiterpene alcohol | 445070 | n.d. | Molt4 T-ALL cell line | Terminal: apoptosis induction | PERK-eIF2α-ATF3/4 | [213] |
| APO866 | (E)-N-[4-(1-benzoylpiperidin-4-yl)butyl]-3-pyridin-3-ylprop-2-enamide | 6914657 | nicotinamide phosphoribosyltransferase (NAMPT) | OCI/AML2, OCI/AML3, HL-60, HEL, KG1a, SET1, MV4-11, MEC.1, MEC.2, LAMA-84 cell lines and B-CLL and AML primary leukemic cells | Terminal: apoptosis induction | IRE1α-CHOP | [214] |
| Bortezomib | boronic acid | 387447 | proteasome (26S) | NB4 cell line (APL) | Terminal: apoptosis induction | Nd | [215] |
| CX-4945 | 5-(3-chloroanilino)benzo[c][2,6]naphthyridine-8-carboxylic acid | 24748573 | casein kinase 2 | T-ALL cell lines and primary cells | Terminal: apoptosis induction | GRP78/BIP-IRE1α-CHOP | [216] |
| compound 3 (Pyrimidine analogue) | 1-(5,5,5-trichloro-2-methoxy-4-oxopenten-2-yl)-4-trichloromethyl-pyrimidin-2(1H)-one | - | n.d. | L1210, CEM, JURKAT cell line (ALL) | Terminal: apoptosis induction | CHOP and caspase-12 | [217] |
| R7, R13 | Naphtylchalcones | n.d. | murine lymphoblastic leukemia | Terminal: apoptosis induction (assumed) | CHOP | [218] | |
| S1 (BH mimetic) | APL | Terminal: cytotoxic through NOXA induction | PERK; XBP1s, NOXA | [219] | |||
| Gossypol (BH3 mimetic) | polyphenol | 3503 | phospholipase A2 | AML, APL | Terminal: cytotoxic through NOXA induction | PERK; NOXA | [220] |
| Cariporide | 151172 | Na + H+ exchanger 1 (NHE1) | CML, APL, T-ALL | Terminal: sensitizes to extrinsic, TRAIL-induced, apoptosis | CHOP | [221] | |
| Auranofin | 6333901 | thioredoxin reductase | CLL | Terminal: contributes to the induction of cell death in treated cells | PERK; XBP1s, CHOP | [222] | |
| [Cu(thp)4][PF6] | phosphine copper(I) complex | n.d. | B-ALL | Terminal: apoptosis induction | Xbp1s, CHOP | [223] | |
| Z-Leu-Leu-Nle-CHO | leupeptin analog | - | γ-Secretase | CLL primary leukemic cells | Terminal: apoptosis induction | IRE1α, CHOP | [224] |
| curcumin | Diferuloylmethane | 969516 | n.d. | WEHI-3 myelomonocytic leukemia cell line/NB4 and UF-1 APL cell lines/HL60 cell line | Terminal: apoptosis induction | IRE1α, ATF6α, CHOP/PERK, CHOP, ASK | [225,226,227] |
| LQB 118 | pterocarpanquinone | 46233300 | n.d. | K562 and Jurkat cell lines | Terminal: apoptosis induction (assumed) | caspase 12 | [228] |
| Flavopiridol | Flavonoid alkaloid | 5287969 | CDKs inhibitor | CLL primary leukemic cells | Terminal: contributes to the induction of cell death in choroquine treated cells | IRE1α/XBP1 and CHOP | [229] |
| Safrole | 5144 | AML (HL-60) | Terminal: apoptosis induction (assumed) | ATF6α, CHOP | [230] | ||
| Clofibrate | 2796 | peroxisome proliferator-activated receptor (PPAR) alpha | T-ALL | Terminal: apoptosis induction (assumed) | ASAPK/JNK | [231] | |
| Abnobaviscum F ® | Mitletoe aqueous extract | 135343633 | n.d. | CML | Terminal: contribution to the induction of cell death in treated cells (assumed) | GRP78/Bip, CHOP | [232] |
| MJ-29 | Quinazolinone | - | n.d. | murine myelomonocytic leukemia | Terminal: apoptosis induction | GRP78/Bip, CHOP, PERK | [233] |
| Imatinib (STI571) | 5291 | BCR-ABL tyrosine kinase | Terminal: apoptosis induction | not well documented | [234] | ||
| glycyrrhizic acid | 14982 | n.d. | murine myelomonocytic leukemia | Terminal: contributes to the induction of cell death in treated cells | GRP78/Bip, CHOP | [235] | |
| Gypenosides | - | n.d | HL-60 AML cell line | Terminal: apoptosis induction | ATF6α and ATF4 | [236] | |
| AICAr (+ methotrexate) | 5-aminoimidazole-4-carboxamide (AICA) riboside | 266934 | n.d | Nalm6 and CCRF-CEM cell lines (ALL) | Terminal: apoptosis induction | CHOP (C/EPB homologous protein) | [237] |
| Emodin | 6-methyl-1,3,8-trihydroxyanthraquinone | 3220 | n.d. | WEHI-3 murine myelomonocytic leukemia cell line | Terminal: apoptosis induction (assumed) | n.d. | [238] |
| Syrbactin | azamacrocyclic product | - | proteasome (26S) | REH ALL cell line | Terminal: apoptosis induction | CHOP (C/EPB homologous protein) | [239] |
| ABT-737 and GX15-070 | BH3 mimetics | 11228183/46930997 | BCL2 family proteins | Jurkat, NB4 and K562 cell lines | Terminal: Cytotoxic UPR | ATF4, ATF3, CHOP and NOXA, | [240] |
| NPB001-05 | n.d. | - | BCR-ABL | K562 cell line | Terminal: apoptosis induction (assumed) | not well documented | [241] |
| Ras inhibitor farnesylthiosalicylic acid (FTS, Salirasib) | 2-[[(2E,6E)-3,7,11-trimethyl-2,6,10-dodecatrien-1-yl]thio]-benzamide | 5469318 | RAS | K562 cell line | Terminal: Cytotoxic UPR | not well documented | [242] |
| PYZD-4409 | 3,5-dioxopyrazolidine compound, 1-(3-chloro-4-fluorophenyl)-4-[(5-nitro-2-furyl)methylene]-3,5-pyrazolidinedione | 60111983 | ubiquitin-activating enzyme UBA1 | K562, NB4, THP1, and U937 cell lines and AML primary leukemic cells | Terminal: apoptosis induction | PERK, CHOP, ATF4 | [243] |
| Korbazol | n.d. | - | n.d. | CLL primary leukemic cells | Terminal: apoptosis induction (assumed) | n.d. | [244] |
| Polymethoxyflavone tangeretin (TAN) | Flavonoids | - | n.d. | K562 cell line | Terminal: apoptosis induction | IRE1α, PERK, CHOP | [245] |
| Shiga toxine type 1 (Stx1) | n.d. | - | ribosomes (protein synthesis) | THP-1 cell line | Terminal: apoptosis induction | CHOP, TNF-related apoptosis-inducing ligand (TRAIL), DR5 and calpain | [246] |
| Eicosapentaenoic acid | 446284 | n.d. | HL60 (AML or APL) | Terminal: apoptosis induction (assumed) | PERK | [247] | |
| Xanthohumol | prenylated chalcone | 639665 | n.d. | CLL (patient samples) | Terminal: apoptosis induction | PERK, CHOP | [248] |
| Tunicamycin (UPR inducer) | 11104835 | N-acetylglucosamine phophotransferase | AML (U937 and HL60) | Terminal: cytotoxic through induction of lysosomal apoptotic pathway | GRP78/Bip, CHOP | [249] | |
| arsenic sulfide | [As4S4 (AS)] | 61569 | n.d. | BCR/ABL-positive K562 cell line | Terminal: apoptosis induction | not well documented | [250] |
| Fenretinide | synthetic retinoid derivative (related to vitamin A) | 5288209 | n.d. | NB4, U937 and HL60 cell lines | Terminal: apoptosis induction | PERK/eIF2α-CHOP (C/EPB homologous protein) | [251,252] |
| PABA/NO | O2-[2,4-dinitro-5-(N-methyl-N-4-carboxyphenylamino)phenyl]1-(N,N-dimethylamino)diazen-1-ium-1,2-diolate | - | PDI | HL60 cell line | Terminal: Cytotoxic UPR | CHOP (C/EPB homologous protein) | [253] |
| alkyl gallate and gallamide derivatives | - | n.d. | HL60 cell line | Terminal: apoptosis induction | not well documented | [254] | |
| Trichosanthin | type I ribosome-inactivating protein | 596174 | Ribosomes | HL60 cell line | Terminal: apoptosis induction | CHOP (C/EPB homologous protein) | [255] |
| auraptene | monoterpene coumarin ether | 1550607 | n.d. | Jurkat cell line | Terminal: apoptosis induction | Caspase 8 | [256] |
| 4-hydroxybenzylretinone | fenretinide analogue/synthetic retinoid derivative (related to vitamin A) | - | n.d. | HL60 cell line | Terminal: Cytotoxic UPR | CHOP (C/EPB homologous protein) | [257] |
| tipifarnib combined with bortezomib | quinolone and boronic acid | 159324/387447 | Farnesyltransferase Inhibiteur and 26 s proteasome inhibitor | KG-1, and U937 cell lines | Terminal: Cytotoxic UPR | not well documented | [258] |
| AEBSF | 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride | 186136 | serine protease inhibitor | NB4 cel line | Terminal: Cytotoxic UPR | not well documented | [259] |
| Thapsigargin (UPR inducer) | sesquiterpene lactone | 446378 | sarco/endoplasmic reticulum Ca++ ATPase | K562 cell line | Terminal: apoptosis induction | not well documented | [260,261] |
| arsenic trioxide (ATO) + kinase inhibitor imatinib mesylate (STI571) | 14888/5291 | BCR-ABL tyrosine kinase | K562 cell line and CML primary leukemic cells | Terminal: apoptosis induction | not well documented | [262] | |
| Tetrocarcin-A | 54681516 | n.d. | CLL/T-ALL | Terminal: apoptosis induction | not well documented | [263,264] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Féral, K.; Jaud, M.; Philippe, C.; Di Bella, D.; Pyronnet, S.; Rouault-Pierre, K.; Mazzolini, L.; Touriol, C. ER Stress and Unfolded Protein Response in Leukemia: Friend, Foe, or Both? Biomolecules 2021, 11, 199. https://doi.org/10.3390/biom11020199
Féral K, Jaud M, Philippe C, Di Bella D, Pyronnet S, Rouault-Pierre K, Mazzolini L, Touriol C. ER Stress and Unfolded Protein Response in Leukemia: Friend, Foe, or Both? Biomolecules. 2021; 11(2):199. https://doi.org/10.3390/biom11020199
Chicago/Turabian StyleFéral, Kelly, Manon Jaud, Céline Philippe, Doriana Di Bella, Stéphane Pyronnet, Kevin Rouault-Pierre, Laurent Mazzolini, and Christian Touriol. 2021. "ER Stress and Unfolded Protein Response in Leukemia: Friend, Foe, or Both?" Biomolecules 11, no. 2: 199. https://doi.org/10.3390/biom11020199
APA StyleFéral, K., Jaud, M., Philippe, C., Di Bella, D., Pyronnet, S., Rouault-Pierre, K., Mazzolini, L., & Touriol, C. (2021). ER Stress and Unfolded Protein Response in Leukemia: Friend, Foe, or Both? Biomolecules, 11(2), 199. https://doi.org/10.3390/biom11020199