
Transcriptome Analysis Reveals Altered Inflammatory Pathway in an Inducible Glial Cell Model of Myotonic Dystrophy Type 1

 , , , ,
, , , ,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Generation of MIO-M1 DM1 Cell Model
2.3. Genotyping
2.4. RNA Isolation, Retrotranscription and Polymerase Chain Reaction (RT-PCR)
2.5. RNA Fluorescence In Situ Hybridization (RNA-FISH) and Immunofluorescence
2.6. Microarray Processing
2.7. Microarrays Data Analysis
2.8. Functional Genomic Analysis
2.9. TaqMan Validation Assays
2.10. Statistical Analysis
3. Results
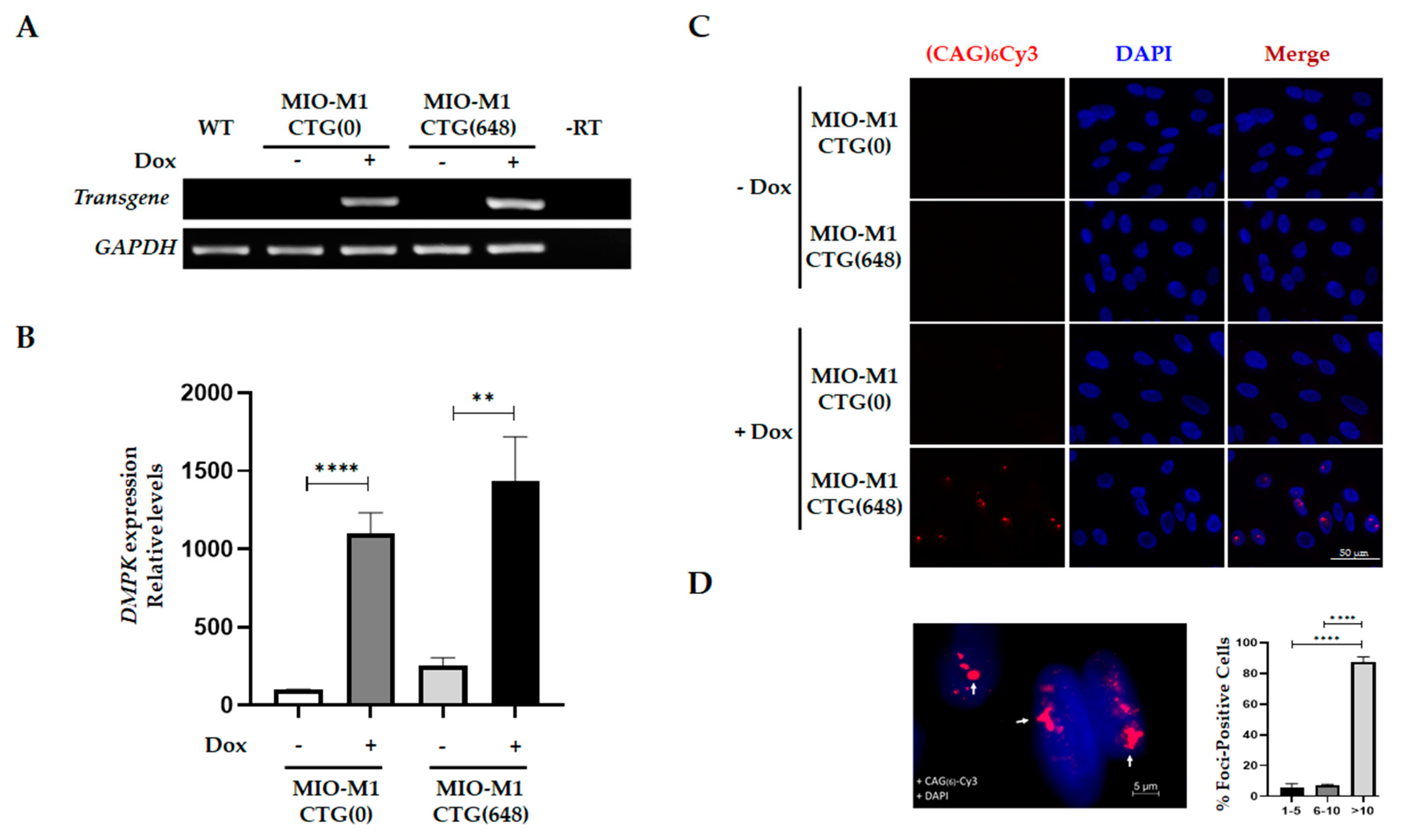
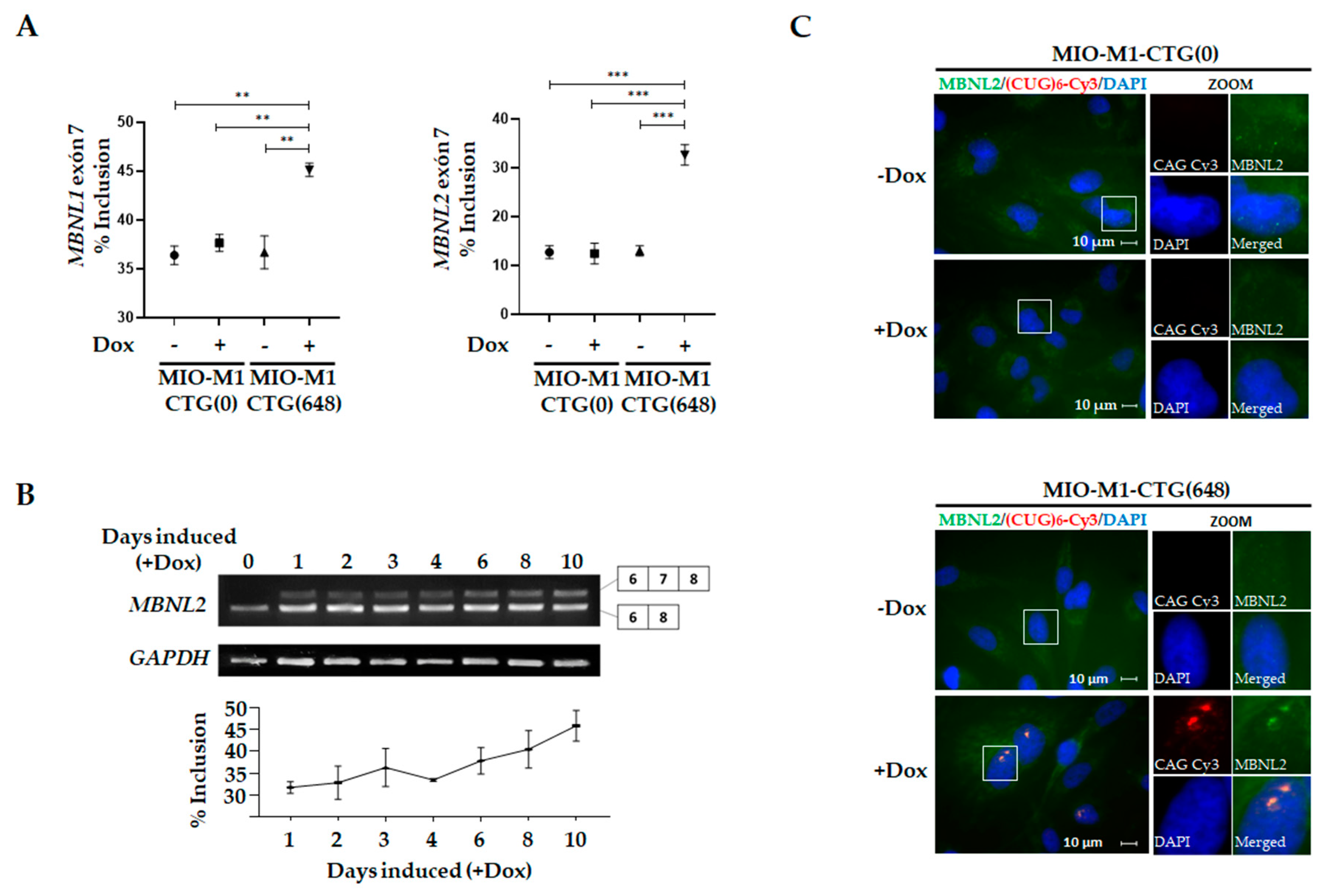
3.1. Generation and Characterization of a MIO-M1 Glial Cell-based Model for DM1
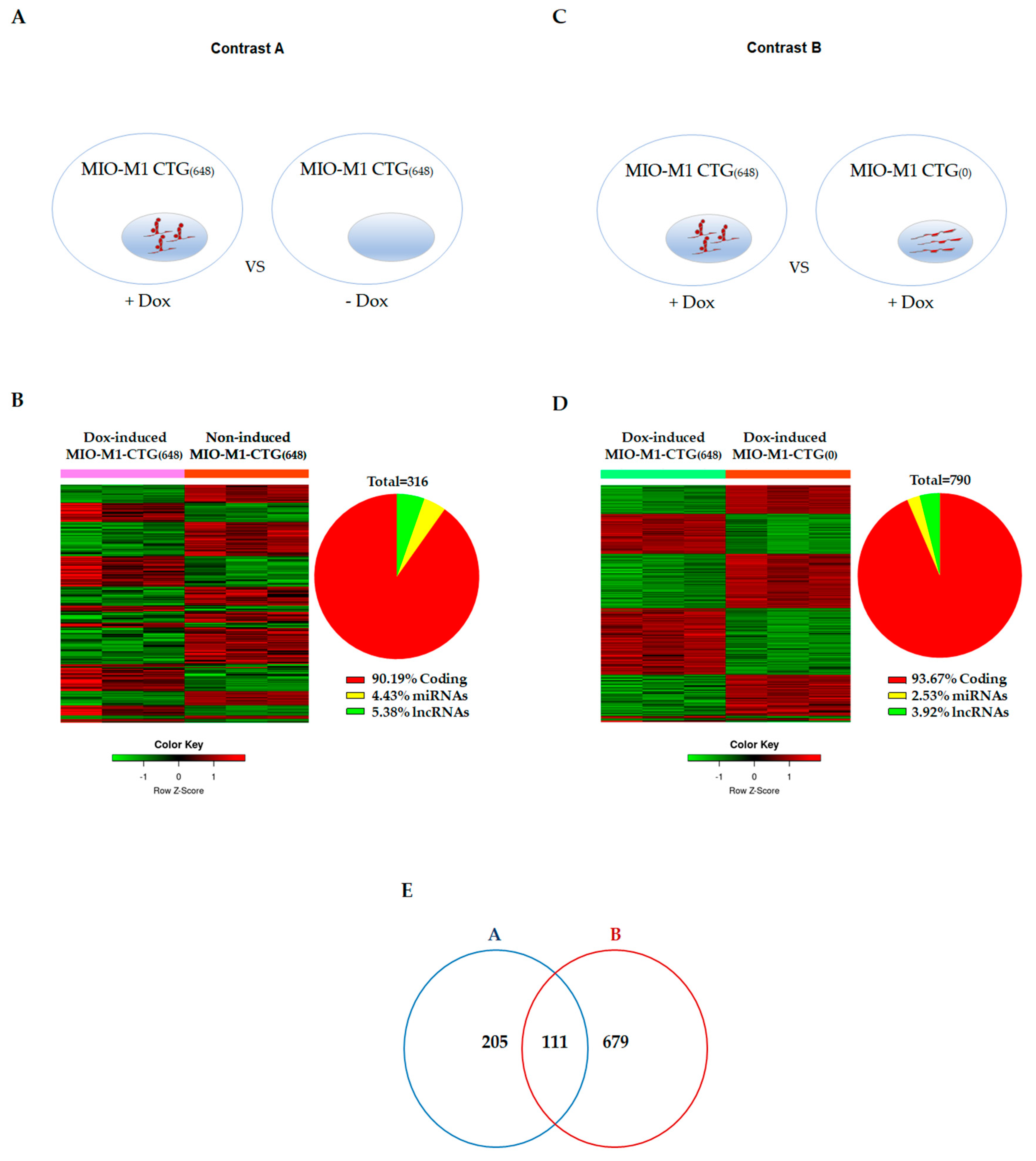
3.2. Effects of DM1 Mutation on Global Gene Expression in MIO-M1-CTG(648) Cells
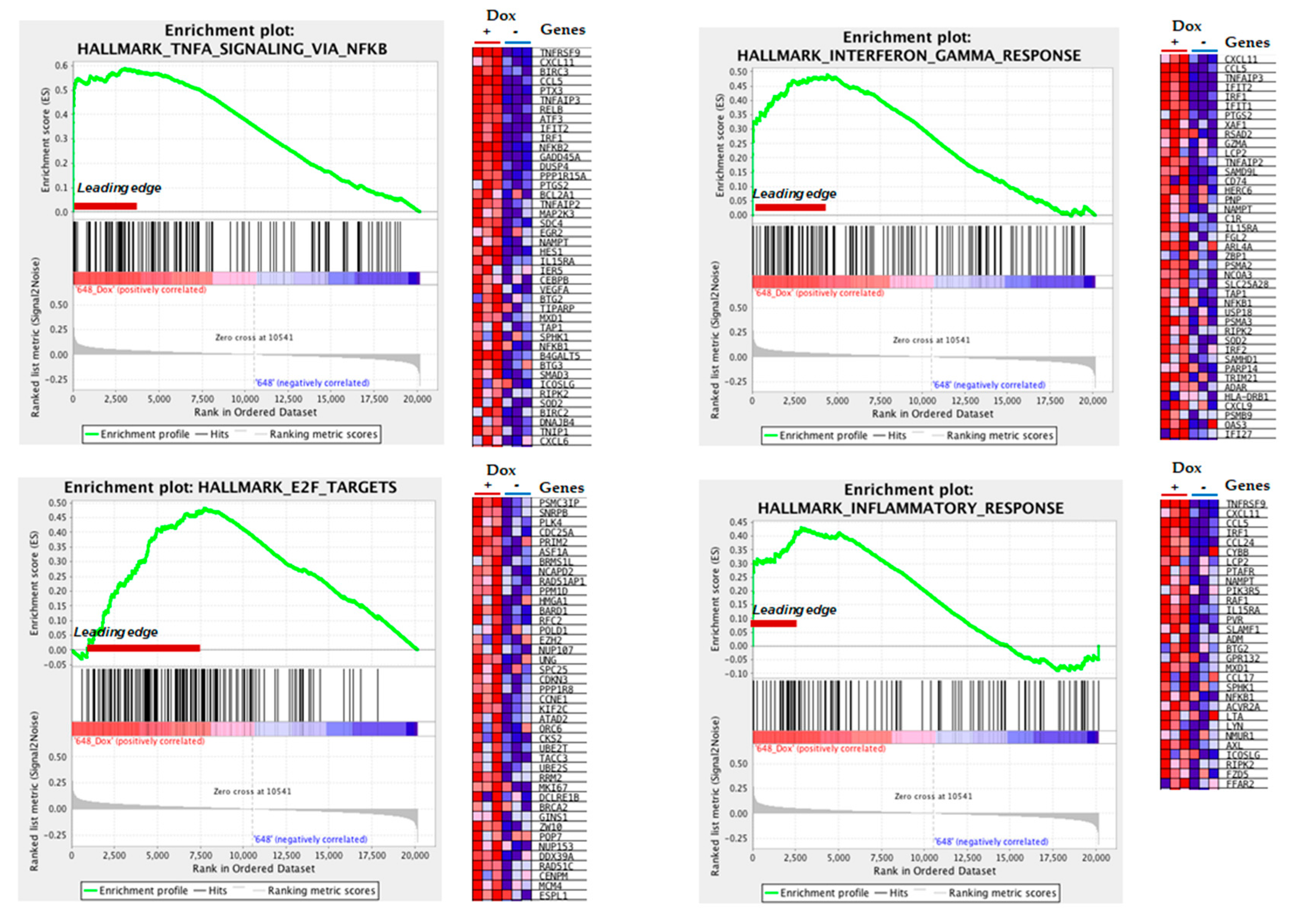
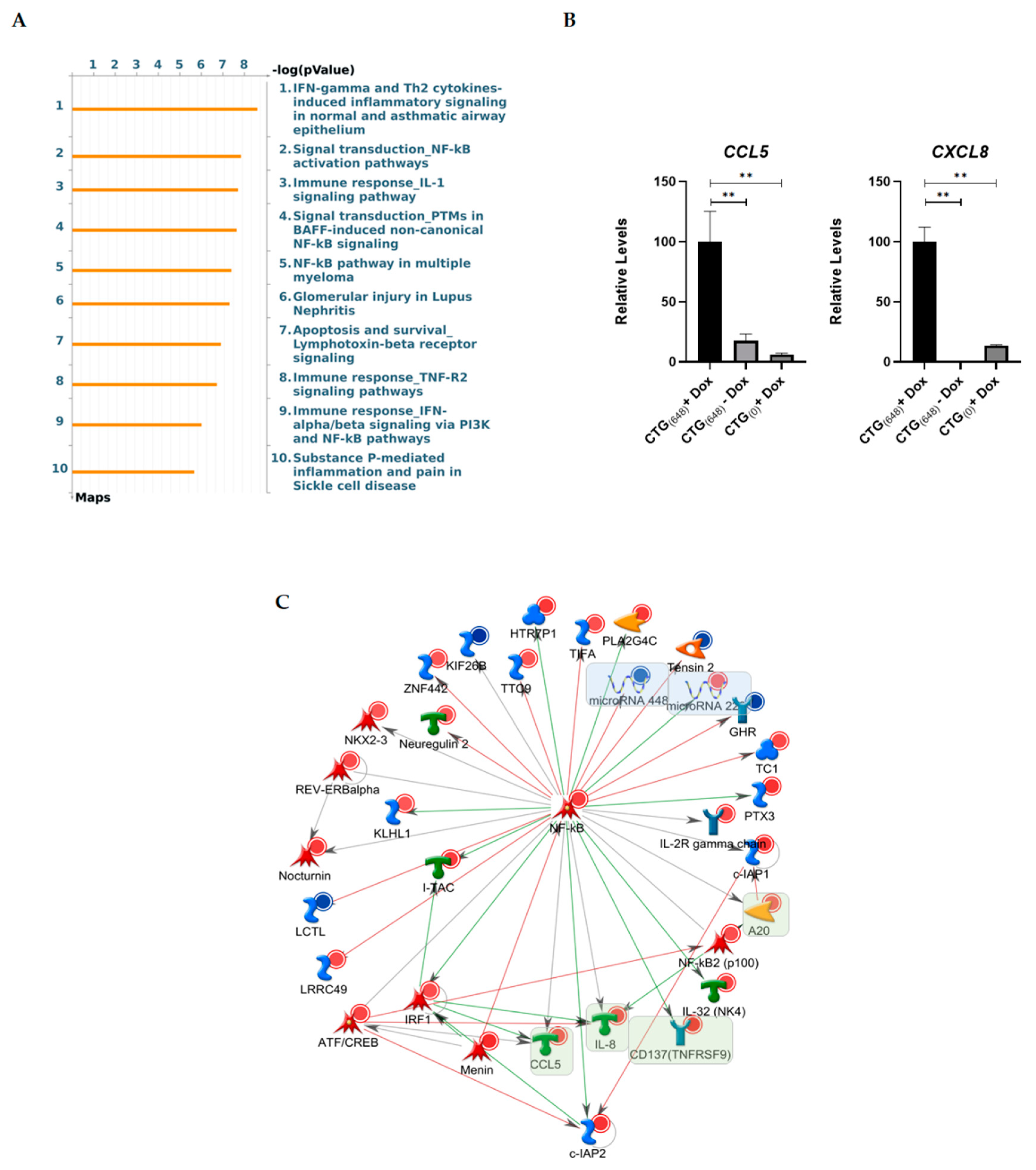
3.3. Gene Ontology Analysis Reveals Altered Inflammatory Processes in MIO-M1-CTG(648) Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Theadom, A.; Rodrigues, M.; Roxburgh, R.; Balalla, S.; Higgins, C.; Bhattacharjee, R.; Jones, K.; Krishnamurthi, R.; Feigin, V. Prevalence of muscular dystrophies: A systematic literature review. Neuroepidemiology 2014, 43, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.P.; Hudson, T.; et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 1992, 69, 385. [Google Scholar] [CrossRef]
- Fu, Y.H.; Pizzuti, A.; Fenwick, R.G.; King, J., Jr.; Rajnarayan, S.; Dunne, P.W.; Dubel, J.; Nasser, G.A.; Ashizawa, T.; de Jong, P.; et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 1992, 255, 1256–1258. [Google Scholar] [CrossRef]
- Mahadevan, M.; Tsilfidis, C.; Sabourin, L.; Shutler, G.; Amemiya, C.; Jansen, G.; Neville, C.; Narang, M.; Barcelo, J.; O’Hoy, K.; et al. Myotonic dystrophy mutation: An unstable CTG repeat in the 3′ untranslated region of the gene. Science 1992, 255, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Udd, B.; Krahe, R. The myotonic dystrophies: Molecular, clinical, and therapeutic challenges. Lancet Neurol. 2012, 11, 891–905. [Google Scholar] [CrossRef]
- Dogan, C.; De Antonio, M.; Hamroun, D.; Varet, H.; Fabbro, M.; Rougier, F.; Amarof, K.; Arne Bes, M.C.; Bedat-Millet, A.L.; Behin, A.; et al. Gender as a Modifying Factor Influencing Myotonic Dystrophy Type 1 Phenotype Severity and Mortality: A Nationwide Multiple Databases Cross-Sectional Observational Study. PLoS ONE 2016, 11, e0148264. [Google Scholar] [CrossRef] [PubMed]
- Gourdon, G.; Meola, G. Myotonic Dystrophies: State of the Art of New Therapeutic Developments for the CNS. Front. Cell Neurosci. 2017, 11, 101. [Google Scholar] [CrossRef]
- Angeard, N.; Jacquette, A.; Gargiulo, M.; Radvanyi, H.; Moutier, S.; Eymard, B.; Heron, D. A new window on neurocognitive dysfunction in the childhood form of myotonic dystrophy type 1 (DM1). Neuromuscul. Disord. 2011, 21, 468–476. [Google Scholar] [CrossRef]
- Douniol, M.; Jacquette, A.; Cohen, D.; Bodeau, N.; Rachidi, L.; Angeard, N.; Cuisset, J.M.; Vallee, L.; Eymard, B.; Plaza, M.; et al. Psychiatric and cognitive phenotype of childhood myotonic dystrophy type 1. Dev. Med. Child Neurol. 2012, 54, 905–911. [Google Scholar] [CrossRef]
- Bertrand, J.A.; Jean, S.; Laberge, L.; Gagnon, C.; Mathieu, J.; Gagnon, J.F.; Richer, L. Psychological characteristics of patients with myotonic dystrophy type 1. Acta Neurol. Scand. 2015, 132, 49–58. [Google Scholar] [CrossRef]
- Callus, E.; Bertoldo, E.G.; Beretta, M.; Boveri, S.; Cardani, R.; Fossati, B.; Brigonzi, E.; Meola, G. Neuropsychological and Psychological Functioning Aspects in Myotonic Dystrophy Type 1 Patients in Italy. Front. Neurol. 2018, 9, 751. [Google Scholar] [CrossRef] [PubMed]
- Rakocevic-Stojanovic, V.; Peric, S.; Madzarevic, R.; Dobricic, V.; Ralic, V.; Ilic, V.; Basta, I.; Nikolic, A.; Stefanova, E. Significant impact of behavioral and cognitive impairment on quality of life in patients with myotonic dystrophy type 1. Clin. Neurol. Neurosurg. 2014, 126, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.M.; McCurrach, M.E.; Taneja, K.L.; Singer, R.H.; Housman, D.E. Expansion of a CUG trinucleotide repeat in the 3′ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc. Natl. Acad. Sci. USA 1997, 94, 7388–7393. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Mankodi, A.; Swanson, M.S.; Moxley, R.T.; Thornton, C.A. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet. 2004, 13, 3079–3088. [Google Scholar] [CrossRef] [PubMed]
- Taneja, K.L.; McCurrach, M.; Schalling, M.; Housman, D.; Singer, R.H. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J. Cell Biol. 1995, 128, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Braz, S.O.; Acquaire, J.; Gourdon, G.; Gomes-Pereira, M. Of Mice and Men: Advances in the Understanding of Neuromuscular Aspects of Myotonic Dystrophy. Front. Neurol. 2018, 9, 519. [Google Scholar] [CrossRef]
- Meola, G.; Cardani, R. Myotonic dystrophies: An update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim. Biophys. Acta 2015, 1852, 594–606. [Google Scholar] [CrossRef]
- Moxley, R.T.; Hilbert, J.E.; Meola, G. The myotonic dystrophies. In Rosenerg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease, 6th ed.; Rosenberg, R.N., Pascual, J.M., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 491–510. [Google Scholar]
- Sicot, G.; Gourdon, G.; Gomes-Pereira, M. Myotonic dystrophy, when simple repeats reveal complex pathogenic entities: New findings and future challenges. Hum. Mol. Genet. 2011, 20, R116–R123. [Google Scholar] [CrossRef]
- Sznajder, L.J.; Swanson, M.S. Short Tandem Repeat Expansions and RNA-Mediated Pathogenesis in Myotonic Dystrophy. Int. J. Mol. Sci. 2019, 20, 3365. [Google Scholar] [CrossRef]
- Cho, D.H.; Thienes, C.P.; Mahoney, S.E.; Analau, E.; Filippova, G.N.; Tapscott, S.J. Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Mol. Cell 2005, 20, 483–489. [Google Scholar] [CrossRef]
- Nguyen, L.; Cleary, J.D.; Ranum, L.P.W. Repeat-Associated Non-ATG Translation: Molecular Mechanisms and Contribution to Neurological Disease. Annu. Rev. Neurosci. 2019, 42, 227–247. [Google Scholar] [CrossRef] [PubMed]
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.; et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA 2011, 108, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.W.; Urbinati, C.R.; Teng-Umnuay, P.; Stenberg, M.G.; Byrne, B.J.; Thornton, C.A.; Swanson, M.S. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000, 19, 4439–4448. [Google Scholar] [CrossRef] [PubMed]
- Timchenko, N.A.; Cai, Z.J.; Welm, A.L.; Reddy, S.; Ashizawa, T.; Timchenko, L.T. RNA CUG repeats sequester CUGBP1 and alter protein levels and activity of CUGBP1. J. Biol. Chem. 2001, 276, 7820–7826. [Google Scholar] [CrossRef]
- Hernandez-Hernandez, O.; Guiraud-Dogan, C.; Sicot, G.; Huguet, A.; Luilier, S.; Steidl, E.; Saenger, S.; Marciniak, E.; Obriot, H.; Chevarin, C.; et al. Myotonic dystrophy CTG expansion affects synaptic vesicle proteins, neurotransmission and mouse behaviour. Brain 2013, 136 (Pt 3), 957–970. [Google Scholar] [CrossRef]
- Suenaga, K.; Lee, K.Y.; Nakamori, M.; Tatsumi, Y.; Takahashi, M.P.; Fujimura, H.; Jinnai, K.; Yoshikawa, H.; Du, H.; Ares, M., Jr.; et al. Muscleblind-like 1 knockout mice reveal novel splicing defects in the myotonic dystrophy brain. PLoS ONE 2012, 7, e33218. [Google Scholar] [CrossRef]
- Allen, N.J.; Lyons, D.A. Glia as architects of central nervous system formation and function. Science 2018, 362, 181–185. [Google Scholar] [CrossRef]
- Oude Ophuis, R.J.; Mulders, S.A.; van Herpen, R.E.; van de Vorstenbosch, R.; Wieringa, B.; Wansink, D.G. DMPK protein isoforms are differentially expressed in myogenic and neural cell lineages. Muscle Nerve 2009, 40, 545–555. [Google Scholar] [CrossRef]
- Sicot, G.; Servais, L.; Dinca, D.M.; Leroy, A.; Prigogine, C.; Medja, F.; Braz, S.O.; Huguet-Lachon, A.; Chhuon, C.; Nicole, A.; et al. Downregulation of the Glial GLT1 Glutamate Transporter and Purkinje Cell Dysfunction in a Mouse Model of Myotonic Dystrophy. Cell Rep. 2017, 19, 2718–2729. [Google Scholar] [CrossRef]
- Limb, G.A.; Salt, T.E.; Munro, P.M.; Moss, S.E.; Khaw, P.T. In vitro characterization of a spontaneously immortalized human Muller cell line (MIO-M1). Investig. Ophthalmol. Vis. Sci. 2002, 43, 864–869. [Google Scholar]
- Ho, T.H.; Savkur, R.S.; Poulos, M.G.; Mancini, M.A.; Swanson, M.S.; Cooper, T.A. Colocalization of muscleblind with RNA foci is separable from mis-regulation of alternative splicing in myotonic dystrophy. J. Cell Sci. 2005, 118 (Pt 13), 2923–2933. [Google Scholar] [CrossRef]
- Murillo-Melo, N.M.; Marquez-Quiroz, L.C.; Gomez, R.; Orozco, L.; Mendoza-Caamal, E.; Tapia-Guerrero, Y.S.; Camacho-Mejorado, R.; Cortes, H.; Lopez-Reyes, A.; Santana, C.; et al. Origin of the myotonic dystrophy type 1 mutation in Mexican population and influence of Amerindian ancestry on CTG repeat allelic distribution. Neuromuscul. Disord. 2017, 27, 1106–1114. [Google Scholar] [CrossRef] [PubMed]
- Tome, S.; Nicole, A.; Gomes-Pereira, M.; Gourdon, G. Non-radioactive detection of trinucleotide repeat size variability. PLoS Curr. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Bolstad, B.M.; Collin, F.; Cope, L.M.; Hobbs, B.; Speed, T.P. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003, 31, e15. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, Article3. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Tokar, T.; Pastrello, C.; Rossos, A.E.M.; Abovsky, M.; Hauschild, A.C.; Tsay, M.; Lu, R.; Jurisica, I. mirDIP 4.1-integrative database of human microRNA target predictions. Nucleic Acids Res. 2018, 46, D360–D370. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Charizanis, K.; Lee, K.Y.; Batra, R.; Goodwin, M.; Zhang, C.; Yuan, Y.; Shiue, L.; Cline, M.; Scotti, M.M.; Xia, G.; et al. Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron 2012, 75, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Hernandez, O.; Sicot, G.; Dinca, D.M.; Huguet, A.; Nicole, A.; Buee, L.; Munnich, A.; Sergeant, N.; Gourdon, G.; Gomes-Pereira, M. Synaptic protein dysregulation in myotonic dystrophy type 1, Disease neuropathogenesis beyond missplicing. Rare Dis. 2013, 1, e25553. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.Y.; Lin, Y.M.; Wang, L.H.; Kuo, T.Y.; Cheng, S.J.; Wang, G.S. Reduced cytoplasmic MBNL1 is an early event in a brain-specific mouse model of myotonic dystrophy. Hum. Mol. Genet. 2017, 26, 2247–2257. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Weber, A.; Wasiliew, P.; Kracht, M. Interleukin-1 (IL-1) pathway. Sci. Signal. 2010, 3, cm1. [Google Scholar] [CrossRef]
- Yamazaki, K.; Gohda, J.; Kanayama, A.; Miyamoto, Y.; Sakurai, H.; Yamamoto, M.; Akira, S.; Hayashi, H.; Su, B.; Inoue, J. Two mechanistically and temporally distinct NF-kappaB activation pathways in IL-1 signaling. Sci. Signal. 2009, 2, ra66. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Lopez, A.L.; Hernandez-Castillo, J.; Hernandez-Kelly, L.C.; Olivares-Banuelos, T.N.; Ortega, A. Fluoride Exposure Affects Glutamine Uptake in Muller Glia Cells. Neurotox Res. 2020, 38, 765–774. [Google Scholar] [CrossRef]
- Lawrence, J.M.; Singhal, S.; Bhatia, B.; Keegan, D.J.; Reh, T.A.; Luthert, P.J.; Khaw, P.T.; Limb, G.A. MIO-M1 cells and similar muller glial cell lines derived from adult human retina exhibit neural stem cell characteristics. Stem Cells 2007, 25, 2033–2043. [Google Scholar] [CrossRef]
- Wang, E.T.; Treacy, D.; Eichinger, K.; Struck, A.; Estabrook, J.; Olafson, H.; Wang, T.T.; Bhatt, K.; Westbrook, T.; Sedehizadeh, S.; et al. Transcriptome alterations in myotonic dystrophy skeletal muscle and heart. Hum. Mol. Genet. 2019, 28, 1312–1321. [Google Scholar] [CrossRef]
- Wang, E.T.; Ward, A.J.; Cherone, J.M.; Giudice, J.; Wang, T.T.; Treacy, D.J.; Lambert, N.J.; Freese, P.; Saxena, T.; Cooper, T.A.; et al. Antagonistic regulation of mRNA expression and splicing by CELF and MBNL proteins. Genome Res. 2015, 25, 858–871. [Google Scholar] [CrossRef]
- Lopez Castel, A.; Overby, S.J.; Artero, R. MicroRNA-Based Therapeutic Perspectives in Myotonic Dystrophy. Int. J. Mol. Sci. 2019, 20, 5600. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, J.D.; Lott, M.C.; Russell, S.L.; Moulton, V.; Sanderson, J.; Wormstone, I.M.; Broadway, D.C. Activation of the innate immune response and interferon signalling in myotonic dystrophy type 1 and type 2 cataracts. Hum. Mol. Genet. 2012, 21, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.; Beffy, P.; Del Carratore, R.; Falleni, A.; Pretini, V.; D’Aurizio, R.; Botta, A.; Evangelista, M.; Stoccoro, A.; Coppede, F.; et al. Activation of the interferon type I response rather than autophagy contributes to myogenesis inhibition in congenital DM1 myoblasts. Cell Death Dis. 2018, 9, 1071. [Google Scholar] [CrossRef] [PubMed]
- Todd, P.K.; Ackall, F.Y.; Hur, J.; Sharma, K.; Paulson, H.L.; Dowling, J.J. Transcriptional changes and developmental abnormalities in a zebrafish model of myotonic dystrophy type 1. Dis. Models Mech. 2014, 7, 143–155. [Google Scholar] [CrossRef]
- Fernandez-Real, J.M.; Molina, A.; Broch, M.; Ricart, W.; Gutierrez, C.; Casamitjana, R.; Vendrell, J.; Soler, J.; Gomez-Saez, J.M. Tumor necrosis factor system activity is associated with insulin resistance and dyslipidemia in myotonic dystrophy. Diabetes 1999, 48, 1108–1112. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.; Carlstrom, K.; Ahren, B.; Cederquist, K.; Krylborg, E.; Forsberg, H.; Olsson, T. Abnormal cytokine and adrenocortical hormone regulation in myotonic dystrophy. J. Clin. Endocrinol. Metab. 2000, 85, 3169–3176. [Google Scholar] [CrossRef]
- Mammarella, A.; Ferroni, P.; Paradiso, M.; Martini, F.; Paoletti, V.; Morino, S.; Antonini, G.; Gazzaniga, P.P.; Musca, A.; Basili, S. Tumor necrosis factor-alpha and myocardial function in patients with myotonic dystrophy type 1. J. Neurol. Sci. 2002, 201, 59–64. [Google Scholar] [CrossRef]
- Hohlfeld, R.; Kerschensteiner, M.; Meinl, E. Dual role of inflammation in CNS disease. Neurology 2007, 68 (Suppl. 3), S58–S63, discussion S91–S56. [Google Scholar] [CrossRef]
- Blank, T.; Detje, C.N.; Spiess, A.; Hagemeyer, N.; Brendecke, S.M.; Wolfart, J.; Staszewski, O.; Zoller, T.; Papageorgiou, I.; Schneider, J.; et al. Brain Endothelial- and Epithelial-Specific Interferon Receptor Chain 1 Drives Virus-Induced Sickness Behavior and Cognitive Impairment. Immunity 2016, 44, 901–912. [Google Scholar] [CrossRef]
- Marciniak, E.; Faivre, E.; Dutar, P.; Alves Pires, C.; Demeyer, D.; Caillierez, R.; Laloux, C.; Buee, L.; Blum, D.; Humez, S. The Chemokine MIP-1alpha/CCL3 impairs mouse hippocampal synaptic transmission, plasticity and memory. Sci. Rep. 2015, 5, 15862. [Google Scholar] [CrossRef]
- Callewaere, C.; Banisadr, G.; Rostene, W.; Parsadaniantz, S.M. Chemokines and chemokine receptors in the brain: Implication in neuroendocrine regulation. J. Mol. Endocrinol. 2007, 38, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Hooton, J.; Schaefer, C.A.; Zhang, H.; Petkova, E.; Babulas, V.; Perrin, M.; Gorman, J.M.; Susser, E.S. Elevated maternal interleukin-8 levels and risk of schizophrenia in adult offspring. Am. J. Psychiatry 2004, 161, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Leighton, S.P.; Nerurkar, L.; Krishnadas, R.; Johnman, C.; Graham, G.J.; Cavanagh, J. Chemokines in depression in health and in inflammatory illness: A systematic review and meta-analysis. Mol. Psychiatry 2018, 23, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Ellman, L.M.; Deicken, R.F.; Vinogradov, S.; Kremen, W.S.; Poole, J.H.; Kern, D.M.; Tsai, W.Y.; Schaefer, C.A.; Brown, A.S. Structural brain alterations in schizophrenia following fetal exposure to the inflammatory cytokine interleukin-8. Schizophr. Res. 2010, 121, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Pittaluga, A. CCL5-Glutamate Cross-Talk in Astrocyte-Neuron Communication in Multiple Sclerosis. Front. Immunol. 2017, 8, 1079. [Google Scholar] [CrossRef]
- Stuart, M.J.; Singhal, G.; Baune, B.T. Systematic Review of the Neurobiological Relevance of Chemokines to Psychiatric Disorders. Front. Cell Neurosci. 2015, 9, 357. [Google Scholar] [CrossRef]
- Barnes, D.A.; Huston, M.; Holmes, R.; Benveniste, E.N.; Yong, V.W.; Scholz, P.; Perez, H.D. Induction of RANTES expression by astrocytes and astrocytoma cell lines. J. Neuroimmunol. 1996, 71, 207–214. [Google Scholar] [CrossRef]
- Mennicken, F.; Maki, R.; de Souza, E.B.; Quirion, R. Chemokines and chemokine receptors in the CNS: A possible role in neuroinflammation and patterning. Trends Pharm. Sci. 1999, 20, 73–78. [Google Scholar] [CrossRef]
- Reali, C.; Curto, M.; Sogos, V.; Scintu, F.; Pauly, S.; Schwarz, H.; Gremo, F. Expression of CD137 and its ligand in human neurons, astrocytes, and microglia: Modulation by FGF-2. J. NeuroSci. Res. 2003, 74, 67–73. [Google Scholar] [CrossRef]
- Yeo, Y.A.; Martinez Gomez, J.M.; Croxford, J.L.; Gasser, S.; Ling, E.A.; Schwarz, H. CD137 ligand activated microglia induces oligodendrocyte apoptosis via reactive oxygen species. J. Neuroinflamm. 2012, 9, 173. [Google Scholar] [CrossRef]
- Voet, S.; Mc Guire, C.; Hagemeyer, N.; Martens, A.; Schroeder, A.; Wieghofer, P.; Daems, C.; Staszewski, O.; Vande Walle, L.; Jordao, M.J.C.; et al. A20 critically controls microglia activation and inhibits inflammasome-dependent neuroinflammation. Nat. Commun. 2018, 9, 2036. [Google Scholar] [CrossRef] [PubMed]
- Ramon-Duaso, C.; Gener, T.; Consegal, M.; Fernandez-Aviles, C.; Gallego, J.J.; Castarlenas, L.; Swanson, M.S.; de la Torre, R.; Maldonado, R.; Puig, M.V.; et al. Methylphenidate Attenuates the Cognitive and Mood Alterations Observed in Mbnl2 Knockout Mice and Reduces Microglia Overexpression. Cereb. Cortex 2019, 29, 2978–2997. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Shen, X.; Chen, X.; Liang, R.; Azares, A.R.; Liu, Y. Celf1 regulates cell cycle and is partially responsible for defective myoblast differentiation in myotonic dystrophy RNA toxicity. Biochim. Biophys. Acta 2015, 1852, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Kaltschmidt, B.; Widera, D.; Kaltschmidt, C. Signaling via NF-kappaB in the nervous system. Biochim. Biophys. Acta 2005, 1745, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Ledoux, A.C.; Perkins, N.D. NF-kappaB and the cell cycle. Biochem. Soc. Trans. 2014, 42, 76–81. [Google Scholar] [CrossRef]
- Li, Q.; Verma, I.M. NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef]
- Mattson, M.P. NF-kappaB in the survival and plasticity of neurons. Neurochem. Res. 2005, 30, 883–893. [Google Scholar] [CrossRef]
- Sen, R.; Baltimore, D. Inducibility of kappa immunoglobulin enhancer-binding protein Nf-kappa B by a posttranslational mechanism. Cell 1986, 47, 921–928. [Google Scholar] [CrossRef]
- Wang, T.; Wang, J. K63-linked polyubiquitination of IRF1, An essential step in the IL-1 signaling cascade. Cell Mol. Immunol. 2014, 11, 407–409. [Google Scholar] [CrossRef]
- Newton, R.; Holden, N.S.; Catley, M.C.; Oyelusi, W.; Leigh, R.; Proud, D.; Barnes, P.J. Repression of inflammatory gene expression in human pulmonary epithelial cells by small-molecule IkappaB kinase inhibitors. J. Pharm. Exp. 2007, 321, 734–742. [Google Scholar] [CrossRef]
- Sizemore, N.; Lerner, N.; Dombrowski, N.; Sakurai, H.; Stark, G.R. Distinct roles of the Ikappa B kinase alpha and beta subunits in liberating nuclear factor kappa B (NF-kappa B) from Ikappa B and in phosphorylating the p65 subunit of NF-kappa B. J. Biol. Chem. 2002, 277, 3863–3869. [Google Scholar] [CrossRef] [PubMed]
- Koehorst, E.; Ballester-Lopez, A.; Arechavala-Gomeza, V.; Martinez-Pineiro, A.; Nogales-Gadea, G. The Biomarker Potential of miRNAs in Myotonic Dystrophy Type, I. J. Clin. Med. 2020, 9, 3939. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Zhu, X.; Sun, W.; Huo, L.; Chen, W.; Wang, H.; Liu, B.; Pan, P. Investigation of the Mol.ecular mechanisms underlying myotonic dystrophy types 1 and 2 cataracts using microRNAtarget gene networks. Mol. Med. Rep. 2017, 16, 3737–3744. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Xu, F.; Li, M.; Wu, S.; Zhang, J.; Wang, A.; Xu, L.; Liu, Y.; Zhu, G. miR-322/-503 rescues myoblast defects in myotonic dystrophy type 1 cell model by targeting CUG repeats. Cell Death Dis. 2020, 11, 891. [Google Scholar] [CrossRef] [PubMed]
- Cappella, M.; Perfetti, A.; Cardinali, B.; Garcia-Manteiga, J.M.; Carrara, M.; Provenzano, C.; Fuschi, P.; Cardani, R.; Renna, L.V.; Meola, G.; et al. High-throughput analysis of the RNA-induced silencing complex in myotonic dystrophy type 1 patients identifies the dysregulation of miR-29c and its target ASB2. Cell Death Dis. 2018, 9, 729. [Google Scholar] [CrossRef] [PubMed]
- Decembrini, S.; Bressan, D.; Vignali, R.; Pitto, L.; Mariotti, S.; Rainaldi, G.; Wang, X.; Evangelista, M.; Barsacchi, G.; Cremisi, F. MicroRNAs couple cell fate and developmental timing in retina. Proc. Natl. Acad. Sci. USA 2009, 106, 21179–21184. [Google Scholar] [CrossRef]
- Kapsimali, M.; Kloosterman, W.P.; de Bruijn, E.; Rosa, F.; Plasterk, R.H.; Wilson, S.W. MicroRNAs show a wide diversity of expression profiles in the developing and mature central nervous system. Genome Biol. 2007, 8, R173. [Google Scholar] [CrossRef]
- Zheng, P.; Bin, H.; Chen, W. Inhibition of microRNA-103a inhibits the activation of astrocytes in hippocampus tissues and improves the pathological injury of neurons of epilepsy rats by regulating BDNF. Cancer Cell Int. 2019, 19, 109. [Google Scholar] [CrossRef]
- Kondo, M.; Yamada, H.; Munetsuna, E.; Yamazaki, M.; Hatta, T.; Iwahara, A.; Ohashi, K.; Ishikawa, H.; Tsuboi, Y.; Inoue, T.; et al. Associations of serum microRNA-20a, -27a, and -103a with cognitive function in a Japanese population: The Yakumo study. Arch. Gerontol. Geriatr. 2019, 82, 155–160. [Google Scholar] [CrossRef]
- Boissonneault, V.; Plante, I.; Rivest, S.; Provost, P. MicroRNA-298 and microRNA-328 regulate expression of mouse beta-amyloid precursor protein-converting enzyme 1. J. Biol. Chem. 2009, 284, 1971–1981. [Google Scholar] [CrossRef]
- Lukiw, W.J. Micro-RNA speciation in fetal, adult and Alzheimer’s disease hippocampus. Neuroreport 2007, 18, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Marti, E.; Pantano, L.; Banez-Coronel, M.; Llorens, F.; Minones-Moyano, E.; Porta, S.; Sumoy, L.; Ferrer, I.; Estivill, X. A myriad of miRNA variants in control and Huntington’s disease brain regions detected by massively parallel sequencing. Nucleic Acids Res. 2010, 38, 7219–7235. [Google Scholar] [CrossRef] [PubMed]
- Packer, A.N.; Xing, Y.; Harper, S.Q.; Jones, L.; Davidson, B.L. The bifunctional microRNA miR-9/miR-9* regulates REST and CoREST and is downregulated in Huntington’s disease. J. Neurosci. 2008, 28, 14341–14346. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C. Natural compounds that modulate BACE1-processing of amyloid-beta precursor protein in Alzheimer’s disease. Discov. Med. 2012, 14, 189–197. [Google Scholar] [PubMed]
- Ding, J.; Xu, Z.; Zhang, Y.; Tan, C.; Hu, W.; Wang, M.; Xu, Y.; Tang, J. Exosome-mediated miR-222 transferring: An insight into NF-kappaB-mediated breast cancer metastasis. Exp. Cell Res. 2018, 369, 129–138. [Google Scholar] [CrossRef]
- Galardi, S.; Mercatelli, N.; Farace, M.G.; Ciafre, S.A. NF-kB and c-Jun induce the expression of the oncogenic miR-221 and miR-222 in prostate carcinoma and glioblastoma cells. Nucleic Acids Res. 2011, 39, 3892–3902. [Google Scholar] [CrossRef]
- Li, Q.Q.; Chen, Z.Q.; Cao, X.X.; Xu, J.D.; Xu, J.W.; Chen, Y.Y.; Wang, W.J.; Chen, Q.; Tang, F.; Liu, X.P.; et al. Involvement of NF-kappaB/miR-448 regulatory feedback loop in chemotherapy-induced epithelial-mesenchymal transition of breast cancer cells. Cell Death Differ. 2011, 18, 16–25. [Google Scholar] [CrossRef]
- Ying, S.Y.; Chang, D.C.; Miller, J.D.; Lin, S.L. MicroRNA protocols. Perspectives. Methods Mol. Biol. 2006, 342, 351–358. [Google Scholar]
- Pasquinelli, A.E. MicroRNAs and their targets: Recognition, regulation and an emerging reciprocal relationship. Nat. Rev. Genet. 2012, 13, 271–282. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contrast A | Contrast B | ||||
|---|---|---|---|---|---|
| Gene Symbol | Fold Change | p Value | Gene Symbol | Fold Change | p Value |
| CXCL10 | 4.66 | 4.49 × 10−9 | CXCL10 | 9.19 | 6.24 × 10−11 |
| TNFRSF9 | 4.63 | 2.51 × 10−8 | CCL5 | 5.04 | 1.86 × 10−7 |
| CXCL8 | 3.44 | 1.51 × 10−6 | SCG2 | 4.32 | 3.26 × 10−8 |
| CCL5 | 3.10 | 8.12 × 10−6 | TNFRSF9 | 4.12 | 6.13 × 10−8 |
| SCG2 | 3.10 | 7.30 × 10−7 | ATF3 | 3.82 | 1.61 × 10−7 |
| PTX3 | 2.79 | 1.47 × 10−6 | CXCL11 | 3.35 | 1.21 × 10−5 |
| MIR4288 | 2.78 | 1.43 × 10−5 | RFPL4AL1 | 2.93 | 1.57 × 10−3 |
| BIRC3 | 2.68 | 6.22 × 10−6 | PLA2G4C | 2.84 | 1.59 × 10−7 |
| PLA2G4C | 2.59 | 4.50 × 10−7 | LINC00707 | 2.78 | 5.88 × 10−6 |
| HTR7P1 | 2.58 | 2.53 × 10−6 | MIR4288 | 2.76 | 1.53 × 10−5 |
| HHAT | −1.56 | 6.74 × 10−3 | LINC00673 | −2.04 | 1.03 × 10−6 |
| SCD | −1.66 | 9.01 × 10−4 | PLEKHS1 | −2.06 | 8.13 × 10−6 |
| PLXDC1 | −1.66 | 5.11 × 10−5 | LRMDA | −2.07 | 1.47 × 10−5 |
| C3AR1 | −1.74 | 9.89 × 10−5 | PLXDC1 | −2.10 | 1.14 × 10−6 |
| KIF26B | −1.76 | 3.89 × 10−4 | TSPAN18 | −2.12 | 1.36 × 10−5 |
| APLN | −1.77 | 1.68 × 10−4 | SV2A | −2.25 | 5.14 × 10−7 |
| SCG5 | −1.79 | 4.25 × 10−5 | APLN | −2.28 | 4.76 × 10−6 |
| DCN | −2.02 | 3.28 × 10−3 | COL11A1 | −2.59 | 8.67 × 10−7 |
| PLEKHS1 | −2.12 | 5.69 × 10−6 | KIF26B | −2.74 | 1.63 × 10−6 |
| TSPAN18 | −2.27 | 5.60 × 10−8 | DCN | −3.28 | 4.51 × 10−5 |
| Gene | Contrast A | Contrast B | ||
|---|---|---|---|---|
| FC RT-qPCR | FC MA | FC RT-qPCR | FC MA | |
| CXCL10 | 25.2 (1.0 × 10−4) | 4.66 (4.4 × 10−9) | 228.2 (1.0 × 10−4) | 9.19 (6.2 × 10−11) |
| TNFRSF9 | 11.7 (2.1 × 10−3) | 4.63 (2.5 × 10−8) | 10.2 (2.4 × 10−3) | 4.12 (6.1 × 10−8) |
| TNFAIP3 | 4.1 (1.7 × 10−2) | 2.53 (1.6 × 10−7) | 2.2 (4.0 × 10−2) | 1.97 (4.8 × 10−6) |
| PLXDC1 | −5.8 (9.8 × 10−3) | −1.66 (5.1 × 10−5) | −2.4 (3.3 × 10−2) | −2.10 (1.1 × 10−6) |
| TSPAN18 | −4.1 (1.0 × 10−4) | −2.27 (5.6 × 10−6) | −2.3 (2.3 × 10−2) | −2.12 (1.3 × 10−5) |
| Resource | Category | Name | Genes | ||
|---|---|---|---|---|---|
| Size | NES | ||||
| GSEA | H | TNF-α signaling pathway | TNFRSF9, CXCL11, BRIC3, CCL5, PTX3, TNFAIP3, RELB, ATF3, IFIT2, IRF1, NFKB2 | 131 | 2.43 |
| BP | Chemokine mediated signaling pathway | CCR4, CXCL11, CCL5, CCL24, GPR75, MPL, CXCR3, CCL17, CCR6 | 25 | 2.30 | |
| CC | Spliceosomal complex | PRPF39, SNRPB, SNRNP48, XAB2, PTBP2, ARSR2, LUC7L, BUD31, AQR, ZCCHC8 | 137 | 1.91 | |
| MF | Chemokine receptor binding | CXCL10, CXCL11, CCL5 | 24 | 2.02 | |
| TFT | GGGNNTTTCC_NFκB_Q6_01 | CXCL10, TNFRSF9, BIRC3, CCL5, RELB, NFKB2, WNT10A, POUF2F3, CCND2 | 85 | 2.09 | |
| KP | Chemokine signaling pathway | CCR4, CXCL10, CXCL11, CCL5, PLCB4, TIAM1HCK, ADCY4, PIK3R5, RAF1, PIK3CD | 118 | 1.89 | |
| % | pval | ||||
| DAVID | BP | Inflammatory response | CCL5, CXCL10, CCR4, CXCL11, CXCL8, RELB, TNFAIP3, TNFRSF9, C3AR1, IRGM, NFKB2, PTX3, PLA2G4C, SCG2 | 12.1 | 2.9 × 10−7 |
| CC | Extracellular space | CCL5, CXCL11, CXCL8, TNFRSF9, APLN, CST7, DCN, GHR, ITGAM, IFNA14, IL32, NRG2, PTX3, PLXDC1, SCG2, SERPIND1 | 16.0 | 1.3 × 10−3 | |
| MF | RNA Pol II regulatory region sequence-specific | RELB, SOX13, ATF3, DLX2, NR1D1 | 4.7 | 1.9 × 10−2 | |
| KP | Cytokine-cytokine receptor interaction | CCL5, CCR4, CXCL10, CXCL11, CXCL8, TNFRSF9, GHR, IFNA14, IL2RG | 7.5 | 7.9 × 10−4 | |
| KPA | KH p val | Union p val | |||
| KPM | Immune response IL-1 signaling pathway | CXCL8, CCL5, CXCL10, MAP3K14, MAPK14, JNK, IRAK1, RELA, IRF1, MAP3K8, NFKB2, CIAP2, IFNB. | 2.9 × 10−7 | 1.1 × 10−12 | |
| PaPM | NF-kB pathway in multiple myeloma | CIAP2, MAP3K14, IKKA, NFKB2, RELB, CYLD, IKB | 1.5 × 10−4 | 5.0 × 10−9 | |
| PhPM | Immune response TNFR2 signaling pathways | IKKA, RELA, TRAF1, IKB, NFKB, CIAP2, MAP3K14, IKKA, NFKB2, RELB, MAPK8-10, BCL-XL, C-JUN | 7.3 × 10−7 | 4.7 × 10−11 |
| miRNA | FC | Target Genes | Biological Process | FDR |
|---|---|---|---|---|
| miR-4288 | 2.79 | Up-regulated: SYTL2, NRG2 | Not determined | - |
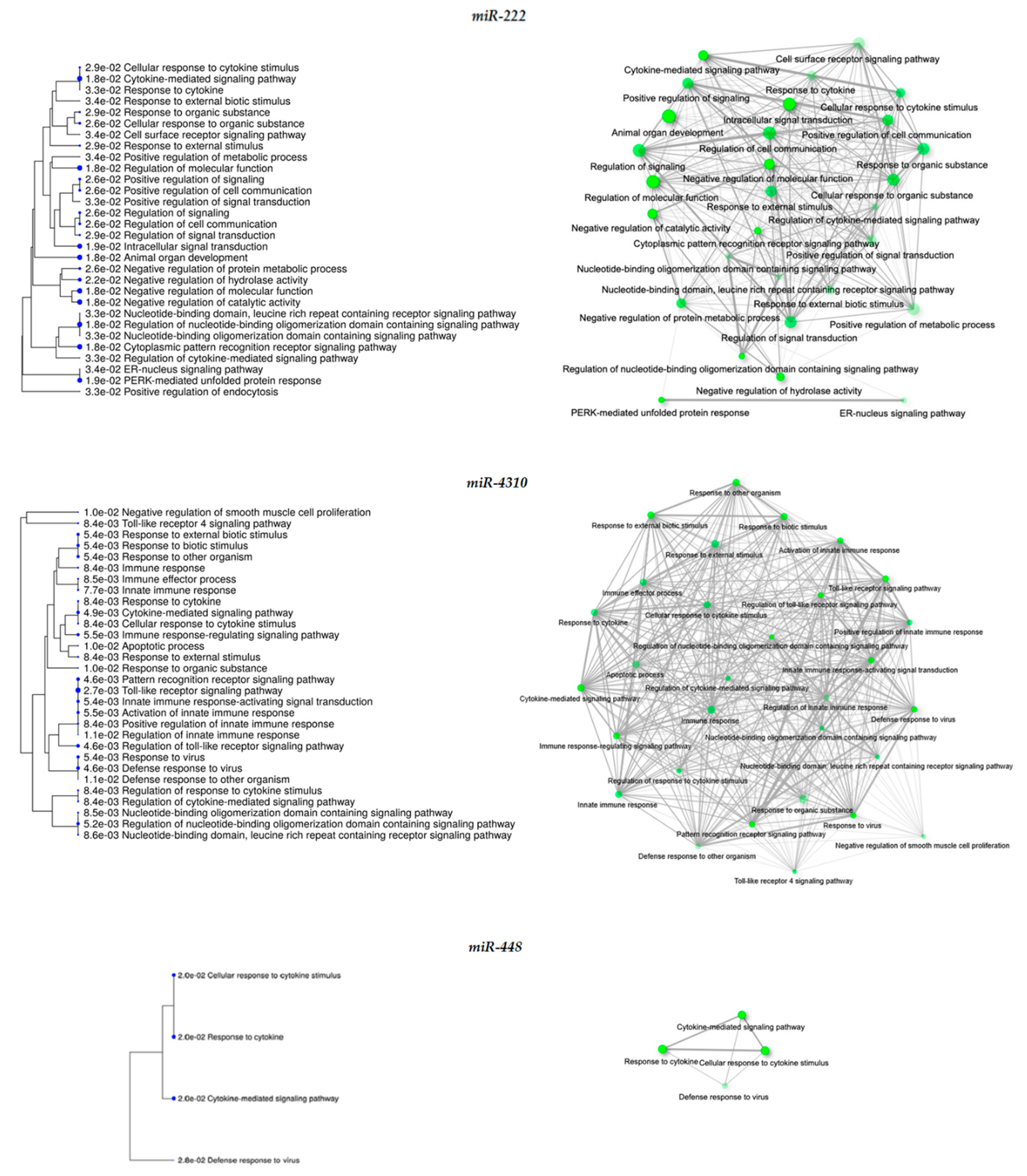
| miR-222 | 2.37 | Up-regulated: ATF3, BAMBI, BIRC3, CXCL11, IFIT2, IL2RG, KIF26B, KLHL1, LRRC49, PPP1R15A, PTX3, SERPIND1, SH3RF2, SNAPC1, SOX13, TUFA, TNFAIP3, TTC9, TUF1 | Cytokine-mediated signaling pathway Cytoplasmic pattern recognition receptor signaling pathway Regulation of molecular function Negative regulation of catalytic activity Intracellular signal transduction | 1.8 × 10−2 1.8 × 10−2 1.8 × 10−2 1.8 × 10−2 1.9 × 10−2 |
| Down-regulated: APLN, CD24, COL11A1, GHR, HHAT, PLEKHS1, PLXDC1, PSD3, SCG5, SV2A, SCD | ||||
| miR-103a1 | 2.04 | Up-regulated: ATF3, CSRNP1, CYHR1, DLX2, DUSP4, HSPA4L, IRF1, MYLK3, PLA2G4C, PTX3, SERPIND1, SH3RF2, SYTL2, TIFA, TMEM45B, TNFAIP3, TTC9, TUFT1 | Animal organ development Animal organ morphogenesis Skeletal system morphogenesis Skeletal system development Anatomical structure morphogenesis Regulation of signal transduction | 1.3 × 10−3 3.9 × 10−3 4.0 × 10−3 1.3 × 10−2 1.6 × 10−2 1.9 × 10−2 |
| Down-regulated: APLN, CD24, COL11A1, DCN, EYA1, GHR, HHAT, KIF26B, PLEKHS1, PLXDC1, PSD3, SCD, SV2A, TSPAN18 | ||||
| miR-298 | 1.91 | Up-regulated: BAMBI, CCL5, CSRNP1, DUSP4, HSPA4L, KLHL1, MYLK3, NR1D1, PLCB4, SCG2, SNAPC1, SOX13, SYTL5, TNFAIP3, TUFT1 | Anatomical structure morphogenesis Negative regulation of signal transduction Regulation of cell communication Regulation of signaling Regulation of chronic inflammatory response | 2.8 × 10−3 2.8 × 10−3 2.8 × 10−3 2.8 × 10−3 2.8 × 10−3 |
| Down-regulated: APLN, CD24, DCN, GHR, PLXDC1, PSD3, SCD, SCG5, TSPAN18 | ||||
| miR-4310 | 1.48 | Up-regulated: ATF3, BIRC3, IFIT2, IRF1, ITGAM, KCTD16, PTX3, SNAPC1, SYTL5, TNFAIP3, TTC9 | Toll-like receptor signaling pathway Cytokine-mediated signaling pathway Activation of innate immune response Cellular response to cytokine stimulus | 2.7 × 10−3 4.9 × 10−3 5.5 × 10−3 8.4 × 10−3 |
| Down-regulated: APLN, CD24, PLEKHS1, PLXDC1, PSD3, SCD, SV2A | ||||
| miR-448 | −1.43 | Up-regulated: CXCL8, DHRS2, HSPA4L, IFIT2, IRF1 TNFRSF9, KCTD16, LRRC49, MYLK3, NRG2, PLCB4, SGIP1, SH3RF2, SNAPC1, SOX13, TNFAIP3, TTC9, TUFT1, ZNF620 | Cellular response to cytokine stimulus Response to cytokine Cytokine-mediate signaling pathway Defense response to virus | 2.0 × 10−2 2.0 × 10−2 2.0 × 10−2 2.8 × 10−2 |
| Down-regulated: EYA1, GHR, PSD, SCD3, SCG5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azotla-Vilchis, C.N.; Sanchez-Celis, D.; Agonizantes-Juárez, L.E.; Suárez-Sánchez, R.; Hernández-Hernández, J.M.; Peña, J.; Vázquez-Santillán, K.; Leyva-García, N.; Ortega, A.; Maldonado, V.; et al. Transcriptome Analysis Reveals Altered Inflammatory Pathway in an Inducible Glial Cell Model of Myotonic Dystrophy Type 1. Biomolecules 2021, 11, 159. https://doi.org/10.3390/biom11020159
Azotla-Vilchis CN, Sanchez-Celis D, Agonizantes-Juárez LE, Suárez-Sánchez R, Hernández-Hernández JM, Peña J, Vázquez-Santillán K, Leyva-García N, Ortega A, Maldonado V, et al. Transcriptome Analysis Reveals Altered Inflammatory Pathway in an Inducible Glial Cell Model of Myotonic Dystrophy Type 1. Biomolecules. 2021; 11(2):159. https://doi.org/10.3390/biom11020159
Chicago/Turabian StyleAzotla-Vilchis, Cuauhtli N., Daniel Sanchez-Celis, Luis E. Agonizantes-Juárez, Rocío Suárez-Sánchez, J. Manuel Hernández-Hernández, Jorge Peña, Karla Vázquez-Santillán, Norberto Leyva-García, Arturo Ortega, Vilma Maldonado, and et al. 2021. "Transcriptome Analysis Reveals Altered Inflammatory Pathway in an Inducible Glial Cell Model of Myotonic Dystrophy Type 1" Biomolecules 11, no. 2: 159. https://doi.org/10.3390/biom11020159
APA StyleAzotla-Vilchis, C. N., Sanchez-Celis, D., Agonizantes-Juárez, L. E., Suárez-Sánchez, R., Hernández-Hernández, J. M., Peña, J., Vázquez-Santillán, K., Leyva-García, N., Ortega, A., Maldonado, V., Rangel, C., Magaña, J. J., Cisneros, B., & Hernández-Hernández, O. (2021). Transcriptome Analysis Reveals Altered Inflammatory Pathway in an Inducible Glial Cell Model of Myotonic Dystrophy Type 1. Biomolecules, 11(2), 159. https://doi.org/10.3390/biom11020159